
Abstract
Connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH) is a progressive and high-risk subtype of PAH, with outcomes generally worse than those seen in idiopathic PAH. Early recognition and treatment are essential for improving survival, yet early-stage CTD-PAH remains challenging to identify, particularly for non-specialist clinicians. The 2022 ESC/ERS guidelines introduced several key updates that support an earlier diagnosis and more targeted management. These include a revised echocardiographic threshold for pulmonary hypertension probability (tricuspid regurgitation velocity >2.8 m/s), a lowered hemodynamic definition of PAH (mean pulmonary artery pressure >20 mmHg and pulmonary vascular resistance >2 WU), and a preference for annual screening using the DETECT algorithm in asymptomatic systemic sclerosis (SSc) patients. Additionally, novel therapeutic targets such as the activin/TGF-β pathway have been incorporated into updated treatment algorithms. Although CTD-PAH remains associated with worse outcomes than idiopathic PAH, recent advances in screening, risk assessment, and targeted therapies have begun to improve the trajectory of the disease. Early detection, personalized treatment, and comprehensive care are now key to transforming this high-risk condition into a more manageable one.
Introduction
Pulmonary arterial hypertension (PAH) is a progressive condition characterized by increased pulmonary artery pressure and elevated pulmonary vascular resistance (PVR), leading to right ventricular (RV) failure. Historically, PAH was associated with poor survival; however, over recent decades, advances in pulmonary vasodilator therapies have improved patient outcomes, extending life expectancy and enhancing quality of life across both physical and symptom-related domains.1–5 PAH is classified into five groups by the World Health Organization (WHO), with WHO Group I encompassing various PAH causes, including connective tissue disease (CTD)-associated PAH (CTD-PAH; Table 1). 6 CTD-PAH is a complex condition observed in patients with systemic sclerosis (SSc), systemic lupus erythematosus (SLE), mixed connective tissue disease (MCTD), primary Sjögren’s disease, and idiopathic inflammatory myopathies (IIMs), each with unique pathophysiologies and management challenges. While rheumatoid arthritis (RA) was previously considered a potential cause of PAH, the current evidence does not strongly support an association between RA and PAH development. 7 CTD-PAH is currently associated with worse outcomes than idiopathic PAH. However, recent advances in screening, risk assessment, and targeted therapies have begun to improve the trajectory of the disease.8,9 Here, a comprehensive yet focused overview of CTD-PAH is provided for nonexpert clinicians, offering key insights into its diagnosis and management for patients with each disease while highlighting recent updates.
World Health Organization (WHO) groups of pulmonary hypertension (PH).

Scleroderma and PAH
SSc, also known as scleroderma, is a complex and heterogeneous auto-immune disorder characterized by endothelial dysfunction, fibroblast dysregulation, and an abnormal immune response, resulting in progressive skin and internal organ fibrosis. SSc can be classified into limited or diffuse forms, depending on the extent of skin involvement. 10 However, SSc extends beyond the skin, affecting vital organs such as the heart, lungs, gastrointestinal organs, and kidneys, where its progression can cause severe complications and significant morbidity.
Cardiac involvement in patients with SSc is common, with an estimated prevalence of clinically overt disease ranging from 7% to 39%. 11 Pulmonary involvement is equally prominent, with interstitial lung disease (ILD) observed in more than 90% of patients during autopsy. 12 ILD and PAH are the leading causes of morbidity and mortality in SSc patients. Notably, women are disproportionately affected by CTD-PAH, including SSc-PAH, reflecting the overall sex bias seen in auto-immune diseases. 13
SSc patients are susceptible to developing PH across all five WHO PH groups (Table 1). 14 Within WHO Group 1, SSc-PAH results from the direct involvement of the pulmonary arteries, with a subset of patients developing pulmonary veno-occlusive disease (PVOD), a rare and challenging form of PAH that further complicates management. In addition to Group 1 PAH, SSc can contribute to PH through multiple mechanisms. SSc may lead to left-sided heart dysfunction, resulting in heart failure with a preserved or reduced ejection fraction (HFpEF or HFrEF, respectively), which falls under Group 2 PH. Additionally, SSc-related ILD can contribute to PH through Group 3 mechanisms. The above are the more frequently encountered subtypes in clinical practice. Less common PH subtypes in SSc include chronic thromboembolic PH (CTEPH; Group 4), reflecting the heightened thrombotic risk associated with SSc. In addition, SSc-related end-stage renal disease may be accompanied by PH classified under Group 5.
Among the predictors of the PAH risk is the presence of telangiectasias. Shah and colleagues reported a strong association between increased telangiectasias in SSc patients and pulmonary vascular disease, suggesting that telangiectasias may serve as a potential marker for the PAH risk. 15
In addition, the antibody profile of SSc patients provides valuable insights into the PAH risk stratification. Anti-U3 RNP and anticentromere antibody positivity are associated with a greater risk of developing PAH. 16 Conversely, anti-U1 RNP-positive patients who develop PAH tend to have better outcomes than those who are negative for this antibody (Table 2). 17
Summary of the prevalence, risk factors, and prognostic markers of CTD-PAH.
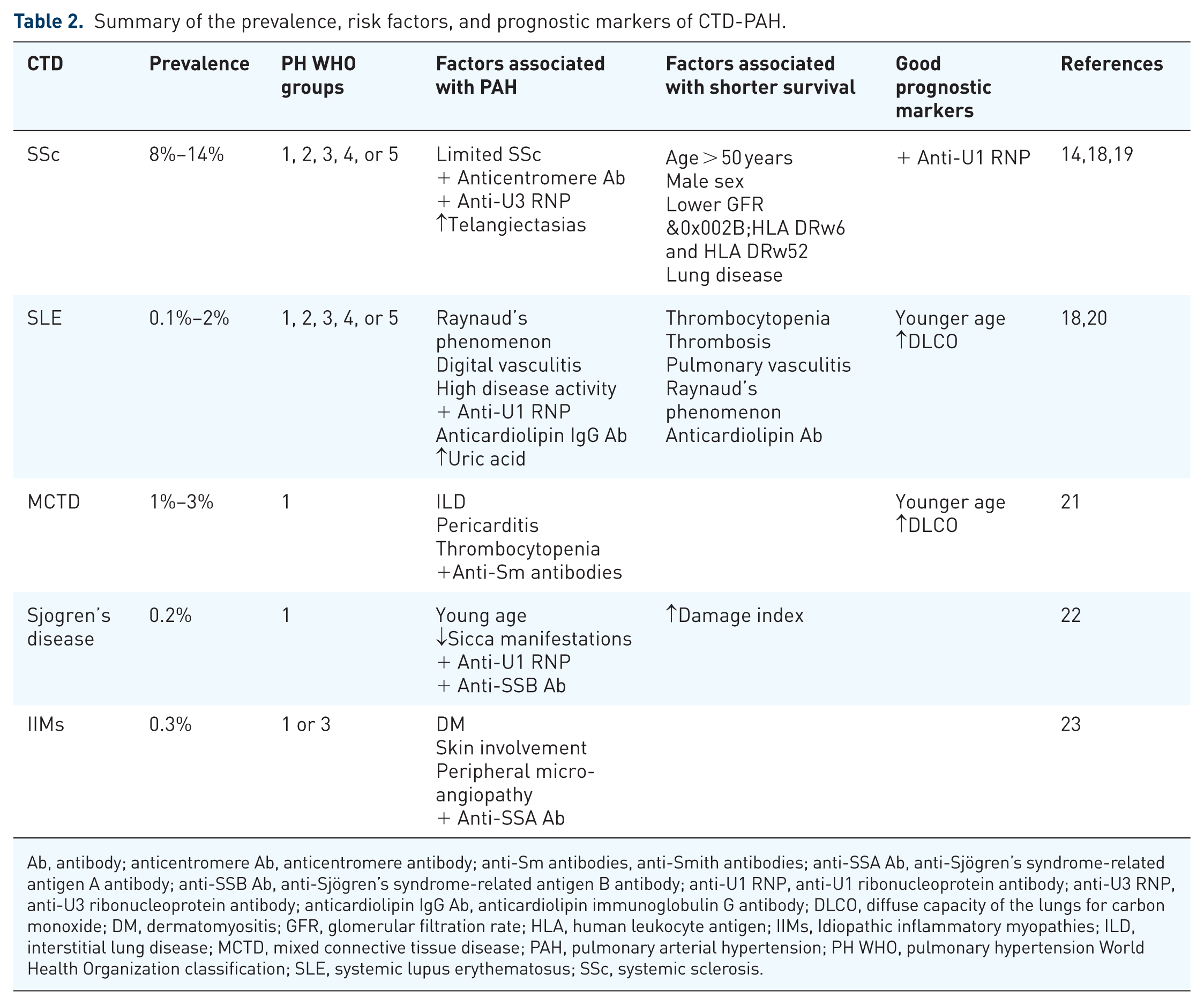
Ab, antibody; anticentromere Ab, anticentromere antibody; anti-Sm antibodies, anti-Smith antibodies; anti-SSA Ab, anti-Sjögren’s syndrome-related antigen A antibody; anti-SSB Ab, anti-Sjögren’s syndrome-related antigen B antibody; anti-U1 RNP, anti-U1 ribonucleoprotein antibody; anti-U3 RNP, anti-U3 ribonucleoprotein antibody; anticardiolipin IgG Ab, anticardiolipin immunoglobulin G antibody; DLCO, diffuse capacity of the lungs for carbon monoxide; DM, dermatomyositis; GFR, glomerular filtration rate; HLA, human leukocyte antigen; IIMs, Idiopathic inflammatory myopathies; ILD, interstitial lung disease; MCTD, mixed connective tissue disease; PAH, pulmonary arterial hypertension; PH WHO, pulmonary hypertension World Health Organization classification; SLE, systemic lupus erythematosus; SSc, systemic sclerosis.
Despite advances in PAH management, the outcomes of SSc-PAH patients remain worse than those of patients with other forms of CTD-PAH or idiopathic PAH (IPAH).16,24,25 Poor prognostic factors for SSc-PAH include an age >50 years, male sex, comorbid heart or lung disease, advanced New York Heart Association (NYHA) functional class (FC) III/IV status, and RV failure. However, outcomes have improved over time.26,27 In 1996, patients with SSc-PAH had a median survival of 1 year, but the outcomes improved. From 1999–2010 to 2010–2021, the 5-year transplantation-free survival rose from 37% to 60%, with the median survival increasing to 4–8 years.8,9 However, similar gains have not been observed in patients with PH caused by lung or heart disease (WHO Groups 2 and 3), and survival remains worse for SSc patients with respiratory-associated PH.8,9
Systemic lupus erythematosus and PAH
SLE is a chronic auto-immune disease that can affect nearly any organ system, and patients present with a wide spectrum of clinical manifestations. SLE patients commonly experience arthritis or arthralgias, mucocutaneous lesions, alopecia, oral ulcers, Raynaud’s phenomenon, nephritis, gastrointestinal symptoms, neuropsychiatric disturbances, and hematological abnormalities. 28 Pulmonary involvement occurs in 50%–70% of SLE patients, with PAH being one of the most concerning complications. 29 Cardiovascular disease is also common in SLE patients. The prevalence of heart failure in SLE patients is estimated to range from 1% to 10%, similar to that in SSc-PAH patients. 14
PAH in SLE is a heterogeneous condition with distinct mechanisms. One subset is linked to antiphospholipid antibody syndrome (APS)-related thrombosis and CTEPH, another to SSc-like vasculopathy with anti-RNP positivity, and a third to immune-mediated vasculitis, which may be reversible with immunosuppression. 27
Several clinical and laboratory findings are strongly associated with PAH development in SLE patients. Raynaud’s phenomenon, digital vasculitis, pericardial effusion, and anti-U1 RNP and anticardiolipin IgG antibody positivity have all been linked to the PAH risk in observational studies.27,30–33 High pulmonary pressures on echocardiography have been observed in patients with longer disease durations, pericarditis, peripheral nervous system involvement, and anti-Sm and anticardiolipin antibody positivity (Table 2). 34 Interestingly, an elevated pulmonary pressure is more common in patients of African descent, highlighting the potential role of genetic or environmental factors. 30
Clustering analyses have provided further insights into auto-antibody patterns in SLE patients, typically revealing 3–5 distinct clusters. 35 One cluster often features patients with high disease activity, characterized by elevated anti-dsDNA antibody titers and a higher vasculitis incidence (vasculitic type). Another cluster includes patients with low disease activity but severe PAH (vasculopathic type), whereas a third cluster features patients with antiphospholipid antibodies, who are at increased risk of thromboembolic events.35,36 The overall 5-year survival rate for SLE-related PAH patients ranges from approximately 60% to 85%. 29
Mixed connective tissue disease and PAH
MCTD has clinical features that overlap with those of several CTDs, including SSc, SLE, RA, and PM/DM. MCTD is distinguished by anti-U1 RNP antibody positivity, which is one of the diagnostic criteria.21,37 Nearly any organ system can be affected by MCTD, although severe renal and central nervous system involvement is uncommon. 38
PAH in MCTD patients is more likely to fall under WHO Group 1 than Group 3 (Table 2). 39 Compared with MCTD patients without PAH, patients with MCTD-PAH tend to have more severe organ involvement and higher anti-U1 RNP titers. Interestingly, in a study by Vegh and colleagues, all PAH cases developed in MCTD patients at least 3 years after the MCTD diagnosis. 40 Survival rates for patients with MCTD-PAH are generally favorable compared with those for patients with SSc-PAH, with 1-, 3-, and 5-year survival rates of 96.6%, 70.2%, and 59.3%, respectively, according to the COMPERA registry. 41 A 2023 meta-analysis further indicated that factors such as ILD, age, and sex have minimal impacts on the survival of MCTD-PAH patients.
Sjögren’s disease and PAH
Sjögren’s disease is a systemic auto-immune disease that primarily affects the lacrimal and salivary glands, leading to dry eyes and mouth. It can occur as a primary condition or secondary to other CTDs, such as SLE or RA. 42 Although cardiac involvement is rare in patients with Sjögren’s disease, PAH can occur, particularly in those with primary Sjögren’s disease. 23 PH in patients with Sjögren’s disease is typically associated with either PAH (WHO Group 1) or Group 3 PH due to lung disease, such as ILD, which affects 9%–20% of patients with Sjögren’s disease.22,43
Although Sjögren’s-associated PAH is relatively rare, with a reported prevalence of 0.2%, it has been associated with the presence of antinuclear antibodies, anti-Ro/SSA antibodies, anti-RNP antibodies, rheumatoid factor, and hypergammaglobulinemia (Table 2). 44 The prognosis of patients with Sjögren’s-associated PAH is generally poor, with survival rates of 73% at 1 year and 66% at 3 years. However, a multicenter study in China revealed higher survival rates, with rates of 94%, 88.8%, and 79% at 1, 3, and 5 years, respectively, indicating that the outcomes may vary significantly by region. In this cohort, a low cardiac index and a high damage index, a scoring tool that quantifies cumulative, irreversible organ damage in Sjögren’s syndrome, were associated with worse outcomes.22,45
Idiopathic inflammatory myopathies and PAH
IIMs, including dermatomyositis (DM) and polymyositis (PM), are a group of auto-immune diseases characterized by immune-mediated muscle damage. 46 These conditions are associated with PH, particularly when combined with chronic respiratory conditions (WHO Group 3 PH; Table 1). IIMs are also associated with antisynthetase syndrome, which can lead to significant lung disease and subsequent PH (WHO Group 3).
PAH is less common in IIM patients than in those with other CTDs, but when it does occur, it is often linked to DM, skin involvement, and anti-SSA antibody positivity. 6 Interestingly, survival rates are better for patients with IIM-associated PAH than for those with other forms of CTD-PAH. 23 Data from the UK registry suggest that IIM-PAH patients have a 100% survival rate at 1 and 3 years, a stark contrast to the poorer outcomes observed for SSc-PAH patients. 47
Screening and diagnosis
Early PAH detection in CTD patients is crucial for improving outcomes. According to the updated ESC/ERS guidelines, an annual evaluation for PAH is recommended for asymptomatic patients with SSc and may be considered for patients with CTD with overlapping features of SSc, using a combination of clinical features, echocardiography, PFTs, and NT-proBNP to select patients with SSc for RHC (Figure 1). 6

Algorithm for screening and diagnosing PAH in SSc patients.
Another screening method that has been used is the DETECT algorithm, which is recommended for screening adults with SSc who have had the disease for >3 years, with a forced vital capacity (FVC) of ⩾40% and a diffusing capacity for carbon monoxide (DLCO) <60% to identify asymptomatic PAH patients.39–42 Coghlan and colleagues reported that the DETECT algorithm has 96% sensitivity, with a false-negative rate of only 4%, compared with a 29% missed diagnosis rate when echocardiographic screening thresholds alone are used.
For symptomatic SSc patients who experience exertional shortness of breath, fatigue, syncope, or palpitations, echocardiography serves as the initial diagnostic method. If the tricuspid regurgitation velocity (TRV) exceeds 2.8 m/s on an echocardiogram, RHC is advised due to an increased PAH risk (Figure 1).
If the findings are normal, further investigations should focus on identifying other potential causes, such as lung or heart disease. However, if no alternative cause is found and the symptoms persist, RHC should be considered, even if the echocardiography findings are unremarkable. 6
A CTD-PAH diagnosis requires confirmation by RHC, which reveals the following: mPAP > 20 mmHg, pulmonary artery wedge pressure (PAWP) ⩽15 mmHg, and PVR > 2 WU. After diagnosis, risk stratification via validated tools (e.g., the REVEAL risk score or the European three-strata model) is essential to guide therapy.22,39
Management and challenges
CTD-PAH management has advanced significantly in recent years, driven by improvements in risk stratification and targeted therapy development. Traditionally, treatment has focused on three key pathways: endothelin-1, nitric oxide, and prostacyclin (Figure 2). However, the addition of a fourth pathway targeting the activin/TGF-β signaling axis represents a significant breakthrough, addressing PAH proliferative mechanisms. Currently, treatments span all four pulmonary vasodilator pathways, with 15 available medications in various formulations, including oral, inhaled, subcutaneous, and intravenous formulations. 48

Available pulmonary vasodilator pathways, treatments, and their basic mechanisms.
The ESC/ERS guideline recommends performing an initial risk stratification at diagnosis, using validated risk assessment tools to classify patients into low-, intermediate-, or high-risk categories, as shown in the three-strata model outlined in Figure 3. 39 For those classified as low- to intermediate-risk, first-line oral therapy—typically a combination of phosphodiesterase type 5 (PDE5) inhibitors and endothelin receptor antagonists (ERAs)—is advised. 39 This approach is supported by findings from the AMBITION trial, in which 41% of CTD-PAH patients received combination therapy. The trial demonstrated a significantly lower risk of clinical failure among patients whose first-line treatment was dual ambrisentan and tadalafil therapy than among those who received either agent alone. 49 A post hoc analysis of the AMBITION trial further revealed that CTD-PAH patients experienced a prolonged time to clinical worsening and increased 6-minute walking distance (6MWD) with initial PDE5 inhibitor and ERA therapy, similar to IPAH patients. These findings reinforce the current consensus recommendations advocating for a standardized treatment approach, applying the same therapeutic algorithm to CTD-PAH as to other PAH subtypes. 50

Suggested 2024 treatment algorithm for PAH from the 7th world symposium on pulmonary hypertension.
During follow-up, further stratification into low-, intermediate–low-, intermediate–high-, and high-risk groups based on the NYHA FC, 6MWD, and NT-proBNP levels is advised. 39
In intermediate–low-risk patients, according to the GRIPHON study, adding selexipag may be considered. 51 The CTD-PAH patients composed the second-largest group after the IPAH patients in this study. Compared with the placebo, selexipag was associated with a reduced risk of the primary composite endpoint (death or PAH-related complications), including patients on stable background therapy. 51 Additionally, switching from PDE5 inhibitors (PDE5is) to riociguat may be an option, as supported by evidence from the REPLACE trial, which included 111 patients in the riociguat group that consisted of a slightly greater number of PAH-CTD patients (24 [22%] vs 19 [17%]) than the PDE5i group. 52 The trial resulted in a significant clinical improvement—as measured by the NYHA FC, 6MWD, and NT-proBNP levels—without clinical worsening at 24 weeks after transitioning to riociguat. 52 For high-risk patients, parenteral prostacyclin therapy remains the most effective treatment available, with strong evidence supporting its role in reducing mortality, particularly when administered in the intravenous form. 53 The 2024 World Symposium on PH introduced significant updates by simplifying the initial risk assessment of patients to at a high risk or not and recommending Sotatercept, an activin signaling inhibitor, for all intermediate–low to high-risk patients during follow-up (Figure 3). 48 This update marked a notable advancement in CTD-PAH treatment.5,48,54 Sotatercept increased the exercise capacity by ⩾40 m for patients on stable background therapy, as shown in the STELLER trial, which included 29 (17.8%) CTD-PAH patients. 5 A post hoc analysis of the trial revealed improvements in heart function, hemodynamics, and NT-proBNP levels. 55 In addition, the ZENITH trial was a phase III, randomized, placebo-controlled study evaluating Sotatercept in high-risk PAH patients, including 48 (27.9%) with CTD-PAH. Patients (WHO FC III/IV, REVEAL Lite 2 ⩾9) received Sotatercept or placebo every 21 days alongside stable dual or triple therapy. Sotatercept reduced the risk of the composite primary endpoint (death, transplant, or PAH-related hospitalization) by 76%, leading to early trial termination due to efficacy. 56 Although subgroup results for CTD-PAH were not separately reported, their inclusion and stratification support the relevance of findings to this population.
It is not uncommon to encounter patients with CTD complicated by ILD and PH. For these patients, there is fortunately an effective treatment option. The INCREASE trial demonstrated that inhaled treprostinil significantly improves exercise capacity, as measured by the 6MWT, and reduces NT-proBNP levels in patients with PH associated with ILD, including patients with CTD-ILD. 57 Based on these findings, the FDA approved inhaled treprostinil in 2021 for this indication, making it the only pulmonary vasodilator with proven efficacy in Group 3 PH, and an important treatment option for patients with CTD-PAH and lung involvement.
Regarding immunosuppression, Yasuoka et al. 58 retrospectively evaluated its role in CTD-PAH, including patients with SLE, MCTD, and primary Sjögren’s disease. A short-term treatment response, defined as improvement in NYHA FC at 3 months, was observed in 53% of patients. Independent predictors of response included a simultaneous diagnosis of PAH with CTD and the use of immunosuppressive therapy—most notably intravenous cyclophosphamide in combination with glucocorticoids. 58 Given the auto-immune nature of CTD-PAH, these findings highlight that immunosuppressive therapy may play a crucial role in certain subsets of patients. In particular, patients with SLE-PAH or MCTD-PAH may derive clinical benefit from a combination of glucocorticoids and cyclophosphamide, especially in the context of less advanced PAH.58–61 However, the survival benefits of intensive immunosuppression remain uncertain, largely due to the limited number of trials. Preclinical studies in rats have suggested a potential benefit of mycophenolate mofetil in PAH through its anti-inflammatory and anti-proliferative effects.62,63 In humans, multiple studies have demonstrated its efficacy in both renal and non-renal manifestations of SLE. 64 However, specific data regarding its role in SLE-associated PAH are lacking, with evidence limited to case reports. 65 In a 2025 retrospective study, Rodolfi et al. 66 reported that early initiation of immunosuppression within 5 years of SSc diagnosis did not delay PAH development or improve survival. However, when analyzing individual immunosuppressants, mycophenolate mofetil was associated with a lower risk of PAH (OR 0.12, 95% CI 0.01–0.98; p = 0.048), whereas hydroxychloroquine use was linked to reduced mortality (HR 0.04, 95% CI 0.06–0.37; p = 0.004). 66 Collectively, these data suggest that while certain CTD subsets such as SLE-PAH and MCTD-PAH may respond favorably to immunosuppression, SSc-PAH follows a different trajectory, with limited evidence showing benefit from such therapy. Accordingly, a proactive, multimodal approach that integrates immunosuppressive, anti-inflammatory, and vasodilator therapies is often advocated.67–69 Nevertheless, immunosuppressive therapy is not recommended for SSc-PAH, as underscored by the findings of Zamanian et al., 70 who demonstrated that B-cell depletion with rituximab was safe but did not improve outcomes compared with placebo in patients with SSc-PAH.
CTD-PAH management extends beyond disease severity, with challenges such as a poor vasodilator response, medication intolerance from GI involvement, and prevalent neuropsychiatric symptoms.71,72 These complexities demand a personalized, multidisciplinary approach. Future research should explore the role of immunosuppression in curing CTD-PAH beyond SSc, focusing on the optimal timing and predictors of the treatment response. Identifying reliable biomarkers—through auto-immune markers, multiomics, and advanced imaging—could refine patient selection and personalize therapy.
Conclusion
Although CTD-PAH remains associated with worse outcomes than idiopathic PAH, recent advances in screening, risk assessment, and targeted therapies have begun to improve the trajectory of the disease. Early detection, personalized treatment, and comprehensive care are now key to transforming this high-risk condition into a more manageable one.