
Abstract
Multiple medical therapies have been developed for the treatment of pulmonary arterial hypertension (PAH) over the last decade and a half. Unfortunately, none of these medications is curative and the majority of patients develop disease progression despite treatment. Presently available medications target one of three known pathways that have been implicated in disease pathogenesis. The multiplicity of pulmonary vascular abnormalities identified in PAH provides the rationale for a therapeutic strategy that targets more than one mechanism at a time. Although a handful of studies have demonstrated clinical improvement in PAH patients who have a second medication added to stable background therapy in a randomized, placebo-controlled fashion, it is unclear whether the derived benefit is due to the combination of two therapies or merely the response to the new agent. This review discusses the rationale for combination therapy, critically reviews the findings of presently completed combination studies and outlines the need for new studies that are better designed to determine whether combination therapy is more efficacious than single agent therapies for the treatment of PAH.
Introduction
Pulmonary arterial hypertension (PAH) is a pulmonary vasculopathy characterized by proliferation and hypertrophy of pulmonary vascular endothelial and smooth muscle cells, predominantly on the arterial side of the pulmonary circulation. These abnormalities of vascular cell growth lead to intimal and medial thickening of the smaller pulmonary resistance arteries and arterioles. In some vessels, these changes are so severe that they result in near obliteration of the vascular lumen. These diffuse vascular changes are often referred to as pulmonary vascular remodeling and result in narrowing of the pulmonary vascular lumen and an increase in resistance to blood flow through the lungs. As the disease progresses, the right ventricle becomes unable to compensate for the steady rise in afterload and begins to fail. Elevation in right-sided filling pressures leads to right ventricular dilation, decreased ejection fraction and eventually the inability to maintain adequate cardiac output.
In the early disease stage, most patients are asymptomatic at rest, but become short of breath with ordinary physical activity. As the disease advances, patients are able to do progressively less and develop signs of right heart failure such as peripheral edema and jugular venous distention. In its final stages, patients become severely debilitated and unable to perform any activity without dyspnea or chest pain. If left untreated, most patients progress to overt right heart failure and death within 3 years of diagnosis. Although PAH is a rare disease, occurring in less than 1/100,000 people annually, there are several forms of pulmonary hypertension (PH) that are encountered more frequently. The World Health Organization (WHO) categorizes the pulmonary hypertensive diseases into five groups (Table 1) [Simonneau et al. 2009]. The first group (WHO group 1) includes idiopathic PAH that is defined as PAH not explained by any other associated condition (formerly referred to as primary pulmonary hypertension) and heritable PAH, defined as patients in whom PAH occurs in multiple familial members or patients with PH who have one of the known genetic mutations associated with PAH. Within WHO group 1 PAH, is PH associated with drugs or toxins, connective tissue disease, HIV infection, portal hypertension, congenital left to right cardiac shunts, schistosomiasis and hemolytic anemias. Although these forms of PH were often referred to as secondary PH prior to the widespread adaptation of the WHO categories, patients with these associated forms of PH demonstrate a pulmonary vasculopathy and clinical course that closely resembles that of PAH and hence are now referred to as associated PAH (APAH). Persistent PH of the newborn is also included in WHO group 1 as are two highly unusual diseases of the pulmonary veins and capillaries, pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis, respectively. PAH is defined as a mean pulmonary artery (PA) pressure ≥25 mmHg at rest with a normal pulmonary venous pressure (PCWP ≤ 15 mmHg) [Badesch et al. 2009].
WHO categories of pulmonary hypertension.

Adapted from Simonneau et al. [2009].
The most common form of PH occurs as a result of left-sided heart failure or valvular heart disease. These conditions raise pulmonary arterial pressure predominantly by elevating pulmonary venous pressure and are referred to as PH owing to left heart disease (WHO group 2). PH is also prevalent in many chronic lung diseases such as COPD, interstitial lung disease and obstructive sleep apnea. These forms of PH tend to be mild and comprise WHO group 3. While most cases correlate with the extent of underlying lung disease there is a subgroup of patients in group 3 with PH out of proportion to their underlying lung disease. WHO group 4 is reserved for patients with pulmonary embolic disease, such as chronic thromboembolic pulmonary hypertension (CTEPH). The last grouping, WHO group 5, includes PH that has been described with a variety of miscellaneous diseases where the pathogenetic mechanism is unclear such as sarcoidosis, disorders of glycogen storage and thyroid disease. For this review, PAH refers to those types of PH in WHO group 1.
Diagnosis
Proper diagnosis of PAH requires accurate assessment of pressures in the pulmonary arterial and pulmonary venous bed, a technique that is best accomplished by pulmonary artery (PA) catheterization. Although echocardiography has been extremely helpful in identifying patients with elevated PA pressure, it is incapable of determining whether the elevation in pressure is due to an increase in precapillary resistance (PAH) or elevation of postcapillary pressure. It is also less accurate for measuring cardiac output than the thermodilution or Fick techniques used during catheterization. Once pulmonary hemodynamics are determined by PA catheterization, a variety of noninvasive techniques are often used to follow disease progression and response to therapy. The most commonly used tests are transthoracic echocardiography, WHO functional class, 6 minute walking distance (6MWD) and plasma brain natriuretic protein (BNP) levels.
Echocardiography is used routinely to estimate PA pressure and examine right ventricular dilation and function every 3–6 months. The WHO functional class is a modification of the New York Heart Association (NYHA) class developed for left-sided heart failure. Functional capacity is assessed by the patient’s subjective impression of their ability to tolerate different types of activity and assigned one of four levels of impairment. Patients who are asymptomatic and deny respiratory problems with normal physical activity are WHO functional class I. Class II patients complain of dyspnea with normal daily activities such as carrying packages or climbing the stairs. Patients who have dyspnea with less than normal daily activities such as walking or dressing are class III and those who are dyspneic with any activity or at rest are class IV. The 6MWD test is a standardized measurement of submaximal exercise. Healthy adults can normally walk 500 meters or greater on level ground in 6 minutes without a fall in arterial oxygenation. The 6MWD decreases steadily as PAH progresses such that most patients in WHO functional class IV are unable to walk 150 meters in 6 minutes and are often unable to complete the test without stopping to rest. In addition to distance and pulse oximetry, systemic blood pressure can be measured at the beginning and end of the test and usually rises in patients who are able to maintain adequate cardiac output. The degree of dyspnea is assessed at the end of the walk using the Borg dyspnea index. Plasma BNP levels are used as a biomarker of disease severity and rise as the right ventricle fails. Patients with a plasma BNP below 180 pg/ml and particularly those who have a decrease in BNP below 180 pg/ml within 6 months of starting therapy have a considerably better prognosis than those whose BNP level remains elevated [Nagaya et al. 2000].
Treatment
Presently available medical therapies for the treatment of PAH are targeted at one of three vascular biology pathways that have been identified as being abnormal. The first of these is the prostacyclin pathway, described and fully characterized by Sir John Vane in the late 1970s [Moncada et al. 1976]. Prostacyclin is a member of the prostaglandin family of peptides that are synthesized by arachidonic acid metabolism. Prostacyclin is synthesized in platelets and vascular endothelial cells and has potent vasodilatory and antimitogenic effects on vascular smooth muscle and anti-aggregation effects on platelets. The role of prostacyclin in the pathogenesis of PAH has been well described. Decreased expression of prostacyclin synthase, the major enzyme involved in the synthesis of prostacyclins has been described in the lungs of PAH patients and the ratio of circulating PGI2 to thromboxane is reduced in PAH, suggesting a state of relative PGI2 deficiency [Tuder et al. 1999; Christman et al. 1992]. Prostacyclin replacement therapy was initially attempted as a bridge to lung transplantation in patients with advanced disease, but its ability to stabilize disease progression and often improve functional capacity in these patients led to its development as a primary therapy. In 1995, the synthetic PGI2, epoprostenol became the first FDA approved treatment for PAH. However, the need for continuous intravenous infusion via central venous access has deterred its widespread acceptance as initial therapy for most patients. Two other prostanoids that are stable at room temperature and have considerably longer half-lives than epoprostenol have been developed for PAH therapy using alternative methods of administration. In the United States, both iloprost and treprostinil are approved for inhalation therapy and treprostinil is approved for subcutaneous as well as intravenous infusion. All three prostanoids have been shown to be efficacious at improving pulmonary hemodynamics, WHO functional class or 6MWD in PAH. Intravenous epoprostenol has also been shown to reduce 12-week mortality [Barst et al. 1996].
Increased pulmonary expression of the potent vasoconstrictor and smooth muscle mitogen, endothelin-1 (ET-1) has also been implicated in the pathogenesis of PAH. ET-1 is expressed at high levels in the pulmonary vascular lesions of PAH patients and circulating levels of ET-1 are increased [Giaid et al. 1993; Stewart et al. 1991]. Furthermore, plasma ET-1 levels correlate with the severity of PH [Rubens et al. 2001]. The biologic activity of ET-1 is mediated by two receptors, endothelin receptor A and receptor B (ETA and ETB). Both nonselective and ETA selective endothelin receptor antagonists (ERAs) have been developed for the treatment of PAH and both have shown to improve pulmonary hemodynamics, 6MWD and to delay time to clinical worsening [Galie et al. 2008; Rubin et al. 2002].
Nitric oxide and the natriuretic peptides play important roles in modulating pulmonary hypertensive and right ventricular hypertrophic responses. The biologic effects of these agents are mediated by increasing intracellular cGMP levels via binding to soluble guanylate cyclase (nitric oxide) or particulate guanylate cyclase (natriuretic peptide). Patients with PAH have been shown to have decreased expression of endothelial nitric oxide synthase (eNOS) in pulmonary arteries and increased expression of arginase, the enzyme that metabolizes the primary substrate for eNOS, L-arginine [Giaid and Saleh, 1995]. Although eNOS expression is increased in obliterative lesions of smaller pulmonary vessels [Xu et al. 2004], patients with PAH have been found to have decreased levels of exhaled NO, suggesting a pulmonary NO deficiency state [Kharitonov et al. 1997]. Circulating levels of atrial natriuretic peptide (ANP) and BNP are increased in patients with PAH, but the ratio of urinary cyclic GMP/BNP is markedly decreased suggesting down regulation of particulate guanylate cyclase [Charloux et al. 2006]. At the same time, the activity of phosphodiesterase type 5 (PDE5), the primary enzyme responsible for degradation of cyclic GMP, is increased in animal models of PH [Hanson et al. 1998]. Selective inhibitors of PDE-5 (PDEIs) such as sildenafil and tadalafil have been shown to be effective at lowering pulmonary vascular resistance (PVR) and improving 6MWD in patients with PAH [Galie et al. 2009, 2005].
Rationale for combination therapy
The major rationale for combining different therapies in the treatment of PAH is to attack the underlying disease process by as many pathways as possible. Prostacyclin replacement therapy, endothelin receptor antagonism and phosphodiesterase inhibition are all effective at improving pulmonary hemodynamics and functional capacity in PAH. It is therefore conceivable that using two or three of these different therapies together may have additive effects. It is also possible that combing these therapies may have synergistic effects, that is, one therapy may facilitate the action of another. For example, most of the vasodilatory effects of prostacyclins are mediated via elevation of intracellular cAMP levels. Cyclic GMP acts as a substrate for some of the same phosphodiesterases that metabolize cyclic AMP and elevation of intracellular cGMP by PDEIs may slow the metabolism of cAMP. Indeed, studies in the hypertrophied right ventricle of rats with monocrotaline-induced PH have shown that PDEIs result in elevation of the intracellular levels of both cGMP and cAMP and that the increase in right ventricular contractility in response to PDEIs is mediated by cAMP [Nagendran et al. 2007]. Thus, combining prostacyclin therapy with a PDEI may result in better elevation of both nucleotide levels. Similarly, nitric oxide and the natriuretic peptides have been shown to suppress synthesis of endothelin, likely via elevation of intracellular cyclic GMP levels [Emori et al. 1993; Boulanger and Luscher, 1990]. Thus, a combination of a PDE5 inhibitor and an ERA may be more effective at blunting the effects of endothelin than either agent alone.
Clinical trials of combination therapy
As new medications for PAH were developed, it became common practice to treat patients who were not responding to their initial therapy with a different agent. Rather than switching patients from one agent to another, physicians simply added a different agent to the background therapy that the patient was already taking. This practice most likely developed over the concern that most therapies for PAH take several months to reach their full effect making physicians hesitant to remove one therapy until it had been overlapped with the other for a sufficient period of time. At the same time, animal studies began to demonstrate additive effects of some pulmonary vasodilator agents when used in combination to blunt the development of experimental PH [Itoh et al. 2004]. To date, nearly all clinical trials of combination therapy have employed a strategy of adding a new agent or placebo to patients who failed to respond or began to deteriorate on their initial therapy (Table 2). Few studies have compared the efficacy of a single agent versus a combination of agents as initial therapy in PAH patients using a prospective parallel study design. Numerous obstacles have prevented the development of properly controlled trials of combination therapy for PAH, including limited numbers of newly diagnosed PAH patients, the cost of multiple drug regimes and the difficulty in getting different manufactures to collaborate on studies that examine the efficacy of one agent versus the other versus the two together.
Summary of major clinical trials of combination therapy.

PAH, pulmonary arterial hypertension; CTPEH, chronic thromboembolic hypertension; 6MWD, 6-minute walk distance; CPET, cardiopulmonary exercise testing; TPR, total pulmonary resistance; RV, right ventricular; PA, pulmonary artery; CI, cardiac index; PVR, pulmonary vascular resistance; NYHA, New York Heart Association.
Interpreting the beneficial effects of so-called ‘add-on’ therapies can be difficult. To properly evaluate the difference between combination and monotherapy, study protocols would need to randomize patients to one of three arms: (1) drug A + placebo, (2) drug B + placebo, or (3) drug A + drug B (Figure 1). This type of study protocol has not yet been employed in patients with PAH, although currently such studies are being initiated. Instead, clinical trials in PAH that have examined the effects of combination therapy have used a sequential add-on strategy. In this model, a clinically stable patient on one therapy (drug A) is continued on that therapy and randomized to receive a new drug (drug A + drug B) or a placebo (drug A + placebo) (Figure 1). However, in this model a beneficial effect in the combination therapy group does not exclude the possibility that the new drug was simply superior to first drug. The sequential add-on model also creates a potential selection bias in that patients who have not responded to their initial therapy may be more likely to volunteer for the study. Such selection bias is shown in Figure 2. If all patients have the same pathologic abnormalities responsible for their PAH, then the beneficial effect of combination therapy is likely to be due to the additive or synergistic effects of the combined medications (Figure 2A). However, if the pathogenic mechanisms of PAH differs between patients, then the initial therapy would select out only those patients who were responsive to that medication. Those not responsive to the initial medication may be more likely to have a second or third medication added to their original therapy. When new medications are added to the failed background therapy, patients who improve may simply be responding to the new medications and not the additive effects of the original and newly added agents (Figure 2B).
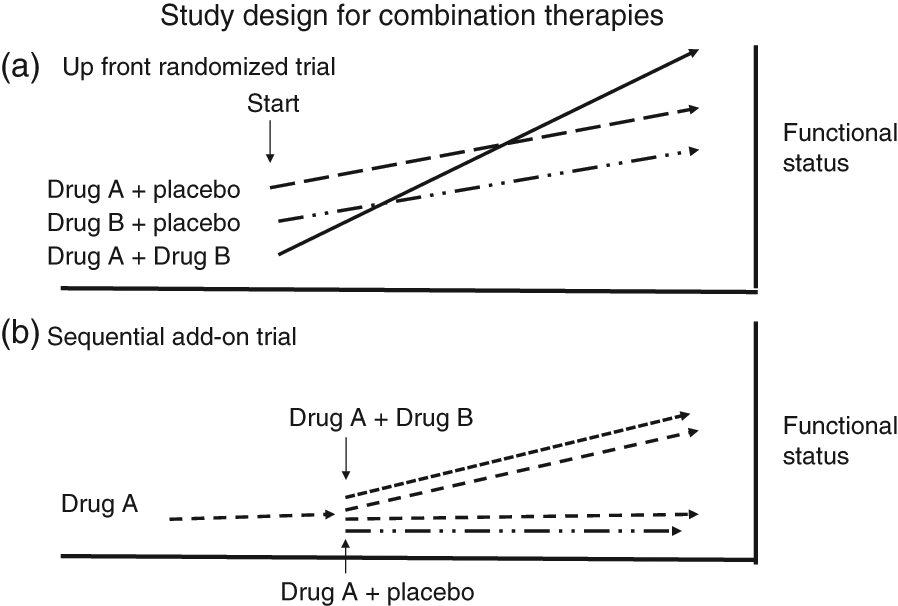
(A) Proper testing of the efficacy of a combination of two therapies requires three arms. One tests the efficacy of the first drug alone, the second arm tests the efficacy of the other agent alone and the third arm tests the efficacy of the two drugs together versus either of the agents alone. In order to blind the patient and investigators a placebo for each drug is required. (B) Study designs that add either a second drug or placebo after patients have already been treated with another agent cannot determine whether any improvement is due to the combination of the two medications or the second medication alone. See the text for a discussion. RCT, randomized controlled trial.
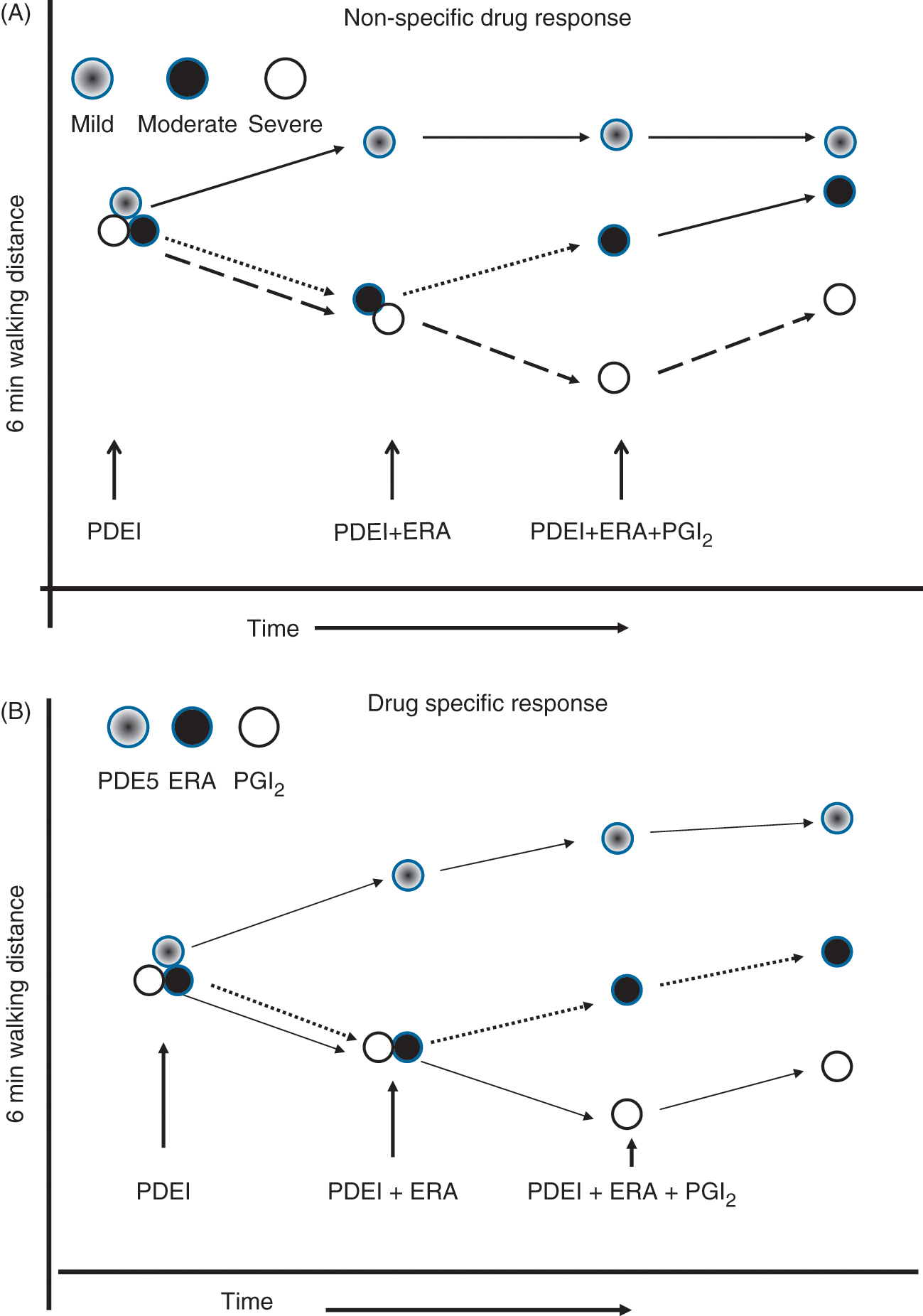
Combination therapy for PAH may be more effective than monotherapy because a combination of two or more medications have additive or synergistic effects or because using more agents increases the odds of exposing the patient to a therapy they are more responsive to. In (A), patients with mild disease may respond to monotherapy with any of the three drug classes, whereas patients with moderate disease may require the combined effects of two therapies and those with the most severe disease may require the combined effects of all three medications. However, if the study population consists of a mix of patients with different responses to each class of medication, then clinical improvement observed after addition of a new therapy may be due to the new therapy alone rather than the additive or synergistic effects of the combination. In the example illustrated in (B), each medication favorably affects a third of the study population. Treating the group with a combination of three medications results in a favorable response of the entire group, but no one in the group requires all three medications to have a favorable response. PDEI, phosphodiesterase inhibitor; ERA, endothelin receptor antagonist; PGI2; prostacyclin.
Combination therapy
Phosphodiesterase inhibitors and prostacyclin analogues
Proof of concept studies that examined the effects of elevating intracellular cAMP via prostanoid administration and cGMP via inhibition of phosphodiesterases were conducted in animal models of PH. Although the relevance of these experimental models of PH to human disease has been questioned, they remain a reasonable starting point for evaluating the efficacy of multidrug regimes. Itoh and colleagues demonstrated that both sildenafil and an oral prostacyclin analogue beraprost blunted the development of monocrotaline-induced PH in rats when either agent was used alone, but the combination of sildenafil and beraprost resulted in significantly greater reduction in right ventricular systolic pressure, right ventricular mass and pulmonary vascular remodeling [Itoh et al. 2004]. Interestingly, all 10 rats given the combination of sildenafil and beraprost were alive 6 weeks after monocrotaline while 1 died in the sildenafil alone group and 2 died in the beraprost alone group.
A year later, Ikeda and colleagues published the acute effects of combining sildenafil and beraprost in six patients with WHO class II or III PAH [Ikeda et al. 2005]. Patients were given beraprost on day 1 and the combination of sildenafil and beraprost on day 2. The reduction in mean PA pressure was twofold greater and the decrease in PVR was 1.6-fold greater with the combination of sildenafil plus beraprost than with beraprost alone. Furthermore, the pulmonary vasodilator effect of sildenafil plus beraprost was at least 2 hours longer than beraprost alone. There was no significant difference in cardiac output, right atrial pressure, or pulmonary wedge pressure with the use of monotherapy or combination therapy.
Earlier studies by Ghofrani and collegues were also able to demonstrate that sildenafil enhanced the acute pulmonary vasodilator effect of inhaled iloprost in 30 patients with severe PAH or CTEPH [Ghofrani et al. 2002]. Patients were randomized to receive 12.5 mg of oral sildenafil, 50 mg of sildenafil, 12.5 mg of sildenafil plus inhaled iloprost, or 50 mg of sildenafil plus inhaled iloprost. Either drug produced similar decreases in mean PA pressure and PVR when used alone, but the effect was additive when used together and sustained for a longer period of time.
In another study, sildenafil was found to improve functional capacity as assessed by 6MWD when added to a background of monotherapy with inhaled iloprost. In that study, 14/73 patients deteriorated on inhaled iloprost over an average of 18 months after initially improving during the first 3 months of iloprost. The addition of sildenafil to their regular iloprost treatments resulted in a mean increase in 6MWD of nearly 100 meters. This improvement was sustained for up to 12 months by the combination of both therapies [Ghofrani et al. 2003]. The largest study to examine the effect of any add-on therapy was the Pulmonary Arterial Hypertension Combination Study of Epoprostenol and Sildenafil (PACES). This double-blind, placebo-controlled, parallel group study randomized 267 patients who were on a stable dose of epoprostenol therapy for at least 3 months to receive placebo or sildenafil at 46 centers in 11 countries. The primary outcome was change in 6MWD. Time to clinical worsening, mean change from baseline to end of treatment in mean PA pressure, and change in Borg dyspnea scale were among the secondary endpoints. The study found an adjusted treatment difference of 28.8 meters in the change in 6MWD in favor of the epoprostenol plus sildenafil group versus the epoprostenol plus placebo group [Simonneau et al. 2008]. Patients who received sildenafil also had greater improvement in mean PA pressure and cardiac output and longer time to clinical worsening than those given placebo. This landmark study was notable for 2 reasons: (1) it was by far the largest study to evaluate the effect of adding one therapy to another in a randomized, double-blind placebo-controlled fashion and (2) patients were considered to be on maximal PAH therapy, i.e. high doses of prostanoid infusion therapy for an average of a year prior to the addition of sildenafil or placebo. Taken together, the results of these studies demonstrate that patients who have either deteriorated or who are no longer responding to therapy with inhaled or intravenous prostanoids may improve with the addition of a PDEI. Whether this occurs because of a favorable interaction between prostanoids and PDEI or because of patient response to PDEI alone is unclear. Further studies are needed to determine if the combination of prostanoid and PDEI given up front is superior to monotherapy with either agent alone.
Phosphodiesterase-5 inhibitors and endothelin receptor antagonists
From an ease of administration standpoint alone, the combination of an ERA and PDEI may be the best combination of drugs for treating PAH. These two classes of drugs are available in an oral preparation and many investigators as well as patients have hoped that using them in combination might potentiate their effects and obviate the need for continuous infusion therapy. Unfortunately, few studies have examined the combined effects of these agents. In a small nonrandomized observational study, 58 patients with functional class III or IV PAH were started on the nonselective ERA bosentan [Hoeper et al. 2004]. Nine of the 58 patients had no improvement or a decline in their 6MWD and cardiopulmonary exercise testing (CPET) after the first 3 months of therapy. These patients were subsequently given 25–50 mg of sildenafil in addition to bosentan. After 3 months of combined therapy, the 6MWD increased from 277 to 392 m (p = 0.007 compared with before the addition of sildenafil) and the improvement was sustained over an average of 9 months of follow up. Similar improvements were noted in CPET. Although the number of patients who were treated with the combination of bosentan and sildenafil was small, Benza recently published similar results in an abstract form using a somewhat different design [Benza, 2010]. In their study, 16/100 (16%) treatment-naive PAH patients treated with bosentan alone reached a predetermined 6MWD goal of >360 meters by week 16. Six other patients withdrew from the study. The remaining 78 were given sildenafil 20 mg tid in addition to bosentan. Twelve weeks later, 15/78 patients treated with the combination of bosentan and sildenafil were able to achieve a 6MWD >380 meters. Although it is possible that the improvement in 6MWD between week 16 and 28 was due to a delayed effect of bosentan alone, previous studies have suggested that the maximal effect of bosentan on 6MWD is achieved within the first 3-4 months of therapy [Rubin et al. 2002]. More information on the efficacy of combining an ERA and PDEI may be determined by the results of Compass 2, an ongoing randomized placebo controlled trial randomizing patients with WHO Group 1 PAH who are already on a stable dose of sildenafil to bosentan or placebo.
Prostacyclins and endothelin receptor antagonists
Several small studies have examined the combination of an ERA with oral, inhaled and intravenous prostacyclins. The first of these was the BREATHE-2 study that randomized 33 patients with PAH in a 2:1 ratio to bosentan or placebo 48 hours after starting therapy with intravenous epoprostenol [Humbert et al. 2004]. The primary outcome was change from baseline to week 16 in total pulmonary resistance (TPR) measured by PA catheterization. Secondary outcomes included change in 6MWD, dyspnea scores, and NYHA functional class. The results were inconclusive, due to the small number of patients, with a statistically nonsignificant greater reduction in TPR in combination therapy group compared with the epoprostenol + placebo group. There was also no statistically significant difference in 6MWD, dyspnea scores, and NYHA functional class between the two groups. One major potential concern however was that in the bosentan + epoprostenol group two patients died while receiving treatment and a third patient died after being withdrawn from the study for worsening PAH. In contrast, there were no deaths in the epoprostenol + placebo group. The authors attributed the deaths to worsening PAH and not a medication effect.
The second study was a prospective nonrandomized nonblinded study that examined the effect of adding bosentan to 16 patients with PAH or CTEPH who had evidence of disease progression during initial therapy with beraprost or inhaled or intravenous Iloprost [Seyfarth et al. 2005]. Patients were on a maximally tolerated dose of prostanoid for at least 3 months before study entry. Patients were then given bosentan in addition to their prostanoid therapy. A statistically significant improvement in right ventricular function index and 6MWD was found 6 months after starting bosentan and sustained for up to 22 months. Nine of the 16 patients showed an improvement in NYHA functional class.
The STEP study (Safety and pilot efficacy Trial in combination with bosentan for Evaluation in Pulmonary arterial hypertension) was a 12-week randomized, multicenter, double-blinded study of patients with NYHA functional class III or IV symptoms who were given iloprost or placebo after at least 16 weeks of bosentan. The primary endpoint was 6MWD. Secondary endpoints included change in NYHA functional class, Borg dyspnea score, and mean PA pressure by right heart catheterization. The study found no statistically significant difference in placebo adjusted 6MWD from baseline However, there was a strong trend toward improved walking distance (p = 0.051) and a significant improvement in NYHA functional class in the bosentan + iloprost group compared with the bosentan + placebo group. There was also a significant reduction in the mean PA pressure and delay in time to clinical worsening in the bosentan + iloporost group [McLaughlin et al. 2006].
A second randomized study comparing bosentan plus inhaled iloprost or placebo was also published in 2006. The COMBI Trial (Combination Therapy of Bosentan and aerosolized Iloprost in Idiopathic Pulmonary Arterial Hypertension), was a 12-week, phase IV, open-label, randomized, controlled multicenter German study. Only patients with IPAH who were functional class III were eligible for enrollment. The primary study endpoint was the change in 6MWD after 12 weeks. Secondary endpoints included changes in functional class and clinical worsening. The trial was terminated early after enrolling 40 patients when an interim analysis showed futility of the combination therapy with a mean decrease of 9 meters in 6MWD in the iloprost group compared with a 1 meter mean increase in the placebo group (p = 0.49). There was no statistical difference between the combination therapy and control group in terms of secondary outcomes [Hoeper et al. 2006].
In the most recently published clinical trial of combination therapy, TRIUMPH (Treprostinil Sodium Inhalation Used in the Management of Pulmonary Arterial Hypertension), 235 patients with WHO functional class III or IV PAH were randomized to receive either inhaled treprostinil or placebo four times daily. All patients were in a steady clinical state on background therapy of either bosentan or sildenafil. The primary endpoint was peak 6MWD at the end of the 12-week study period. Secondary endpoints included time to clinical worsening, Borg dyspnea score, and change in NYHA functional class. There was a statistically significant difference in change from baseline 6MWD of 19 meters that favored the group treated with inhaled treprostinil. There was, however, no difference in most of the secondary endpoints, including time to clinical worsening, Borg dyspnea score, and NYHA functional class between treatment groups [McLaughlin et al. 2010].
Ongoing clinical trials
None of the studies described above included an arm where the background therapy was stopped and patients were switched to the new medication alone. Thus, it is not possible to determine whether the beneficial effects observed were due to the new medication or a combination of the background therapy plus new medication. To properly compare the efficacy of combination therapy to therapy with a single agent, future studies will need to utilize a design such as that employed in the recently initiated AMBITION study (A Randomized, Multicenter Study of First-Line Ambrisentan and Tadalafil Combination Therapy in Subjects With Pulmonary Arterial Hypertension). This phase 4 prospective clinical trial randomly assigns patients into one of three arms using a double placebo: (1) tadalafil plus ambrisentan placebo, (2) ambrisentan plus tadalafil placebo, or (3) tadalafil plus ambrisentan. All patients are treatment naïve. The primary outcome is time to clinical failure. Secondary outcomes include changes in 6MWD, change in baseline WHO functional class, and change in Borg dyspnea scale. In previous studies, both drugs have resulted in similar placebo-adjusted improvement in 6MWD when used alone. It will be interesting to see whether these drugs have an additive or synergistic effect when used together as initial PAH treatment. Enrollment in the study is expected to be completed in August 2013 [ClinicalTrials.gov identifier: NCT01178073].
ATPAHSS (A Clinical Trial of Ambrisentan and Tadalafil in Pulmonary Arterial Hypertension Associated With Systemic Sclerosis) is a similar randomized, double-blind, parallel group study currently randomizing patients with PAH secondary to systemic sclerosis to the same three groups as in AMBITION. The primary endpoint is the effects of the various medications on right ventricular mass as assessed by cardiac MRI and PVR by right heart catheterization. Secondary endpoints are similar to those in AMBITION. The study is planned to be completed by July 2013 [ClinicalTrials.gov identifier: NCT01042158]. At least two other randomized placebo-controlled clinical trials for PAH combination therapy are in the planning stages [ClinicalTrials.gov identifier: NCT01178073].
Summary and conclusions
Combining presently available therapies to combat PAH by more than one pathogenic mechanism has some scientific rationale and considerable appeal. Unlike some other diseases, it is often difficult to reclaim ground lost in the fight against PAH. Using more than a single agent upfront increases the odds of the patient being treated with a drug to which they will respond and provides the possibility of an additive or synergistic effect of the two medications. However, little data are presently available to support the hypothesis that multiple drugs are better than one. Several placebo-controlled trials have shown improvement in functional capacity, hemodynamic measurements and time to clinical worsening in patients given a new PAH medication after failing to respond or deteriorating on their original therapy. However, it is not known whether the observed benefit was the result of the combined effects of the new therapy and the old or if similar results would have been obtained by simply switching from their initial therapy to the new one. Ongoing studies that have just begun enrolling patients will attempt to answer this question directly by comparing combination therapy to each arm of therapy individually. Until the results of these and other studies are known, the clinician is left with the results of previous studies that used the add-on design. These studies suggest that patients who fail to respond to one of the three classes of currently approved PAH therapies, prostacyclin, ERA or PDE, may benefit from the addition of a new agent from one of the remaining classes.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Dr Klinger receives grant support, consultant or speakers bureau fees from Actelion, Bayer, Gilead Sciences, Lung Rx, NIH-NHLBI, Pfizer, and United Therapeutics.