
Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease defined by an elevation in pulmonary arterial pressure that can lead to right heart failure and death. Ambrisentan is a selective endothelin receptor antagonist approved for the treatment of idiopathic, heritable PAH and connective tissue disease-associated PAH. Ambrisentan has been shown to improve exercise capacity and hemodynamics with an acceptable side-effect profile. It has also proven to be safely used in combination with other PAH-specific medications, especially with phosphodiesterase-5 inhibitors. In the recent randomized trial, AMBITION, it was shown that upfront combination therapy of ambrisentan and tadalafil significantly decreased the risk of clinical failure compared with monotherapy. This review describes the drug profile of ambrisentan and its safety and efficacy in the treatment of PAH.
Keywords
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease defined by elevated pulmonary arterial pressures and vascular resistance. As per the 5th World Symposium on Pulmonary Hypertension (held in France, 2013) Group 1 pulmonary hypertension consists of a disease state that shares similar pathologic, hemodynamic and treatment strategies (Table 1).1,2 This group includes idiopathic PAH (iPAH), heritable/familial PAH (FPAH), drug and toxin-induced PAH, PAH associated with connective tissue disease (CTD-PAH), HIV, portopulmonary hypertension (POPH), congenital heart disease and schistosomiasis. The clinical severity of PAH is classified using the World Health Organization functional class (WHO FC) system, ranging from I to IV, progressing from least to worse disease.
Classification of Pulmonary Hypertension Groups 1 – 5. Adapted from: Simonneau and colleagues. 2

PAH, pulmonary arterial hypertension.
The past 20 years have heralded a highly productive age of therapy for PAH. All of the approved PAH-specific medications target one of three main pathways: the prostacyclin pathway, the nitric oxide pathway and the endothelin pathway. 3
One of the most common oral medications for the treatment of Group 1 PAH are the endothelial receptor antagonists (ERAs). Bosentan (Tracleer®; Actelion Pharmaceuticals Ltd, Switzerland) was the first-in-class ERA approved in 2001 and is available as twice daily oral therapy for the treatment of WHO FC II−IV patients with idiopathic or heritable PAH, congenital heart disease with left-to-right shunts and CTD-PAH. Ambrisentan (Letaris®; Gilead, USA; Volbris®; GSK, United Kingdom, UK) was the second-in-class ERA approved by the United States (US) Food and Drug Administration (FDA) in 2007, as a once a day oral therapy for WHO Group 1 patients in patients with WHO FC II−III to improve exercise capacity and delay clinical worsening. Ambrisentan is the only ERA that has an indication for upfront combination therapy with tadalafil for Group 1 PAH. More recently macitentan (Opsumit®; Actelion) was US FDA-approved in 2013 as once daily oral medication for the treatment of PAH to delay disease progression and reduce hospitalizations.
There are subtle differences in the indications for ambrisentan in the US and the UK. Indications for use in the US and UK include WHO FC II−III symptoms. In the US the WHO Group 1 patients include idiopathic or heritable PAH and connective tissue disease-related PAH, while in the UK the label only includes Group 1 PAH patients with iPAH or CTD-PAH.
Endothelin and the pulmonary vasculature
Endothelin-1 is a potent pulmonary vasoconstrictor and smooth muscle mitogen that has been found to be overexpressed in PAH patients. 4 The receptors EndothelinA and EndothelinB (ETA and ETB, respectively) bind available endothelin, which results in adverse remodeling of the pulmonary arterial vasculature. Endothelin receptor antagonists can be described as either nonselective receptor antagonists (blocks both ETA and ETB receptors) such as bosentan and macitentan, or single receptor antagonists (blocks ETA only), such as ambrisentan. ETA receptors are primarily expressed in smooth muscle cells of the pulmonary arteries, airways, lung fibroblasts and cardiomyocytes. The ETB receptors predominantly reside in the pulmonary vascular endothelial cells with a lesser expression in pulmonary artery smooth muscle cells and perivascular fibroblasts. 5 A suspected benefit of the blockade of single receptor antagonists, such as ETA, may include the production of vasodilatory and anti-mitogenic substances activated through the ETB pathway; however, this is not quite clear. Additionally, data from animal models of PAH demonstrate that there in an imbalance in the ratio of ETA and ETB receptors with early induced expression of ETA and subsequent upregulation of ETB receptor of pulmonary artery smooth muscle cells, which contributes to the pathogenesis of PAH.5–7
Ambrisentan discovery
The initial discovery of the first ERA, bosentan, led to small molecule screening of the BASF Corporation’s (Ludwigshafen, Germany) chemical library, which produced compounds that selectively bound recombinant ETA.4,8 Simplification and further modification of the structures led to the production of darusentan (LU-13252) and ambrisentan (LU-208075) respectively.9,10
Pharmacodynamics and pharmacokinetics of ambrisentan
Ambrisentan is an orally active, diphenyl propanoic acid derivative, nonsulfonamide. It is a potent ERA with higher selectivity for the ETA receptor over the ETB receptor, with ranges of 29:1 to 4000:1 depending on the assay utilized. 11 Ambrisentan is rapidly absorbed into the systemic circulation with a half-life of approximately 15 h, which allows for a once daily dosing. 12 The mean terminal half-life is around 5 h 13 and may take up to 5 days to reach steady state. 14
The detailed pharmacologic aspects of ambrisentan have been previously reviewed in this journal and elsewhere.15,16 This review article will focus on the current clinical application of ambrisentan in PAH.
Clinical trials
Monotherapy
Table 2 summarizes the clinical trials using ambrisentan in PAH. We utilized PubMed for the literature search to find phase II and III randomized controlled clinical trials for ambrisentan in PAH.
Summary of the clinical trials using ambrisentan in PAH.
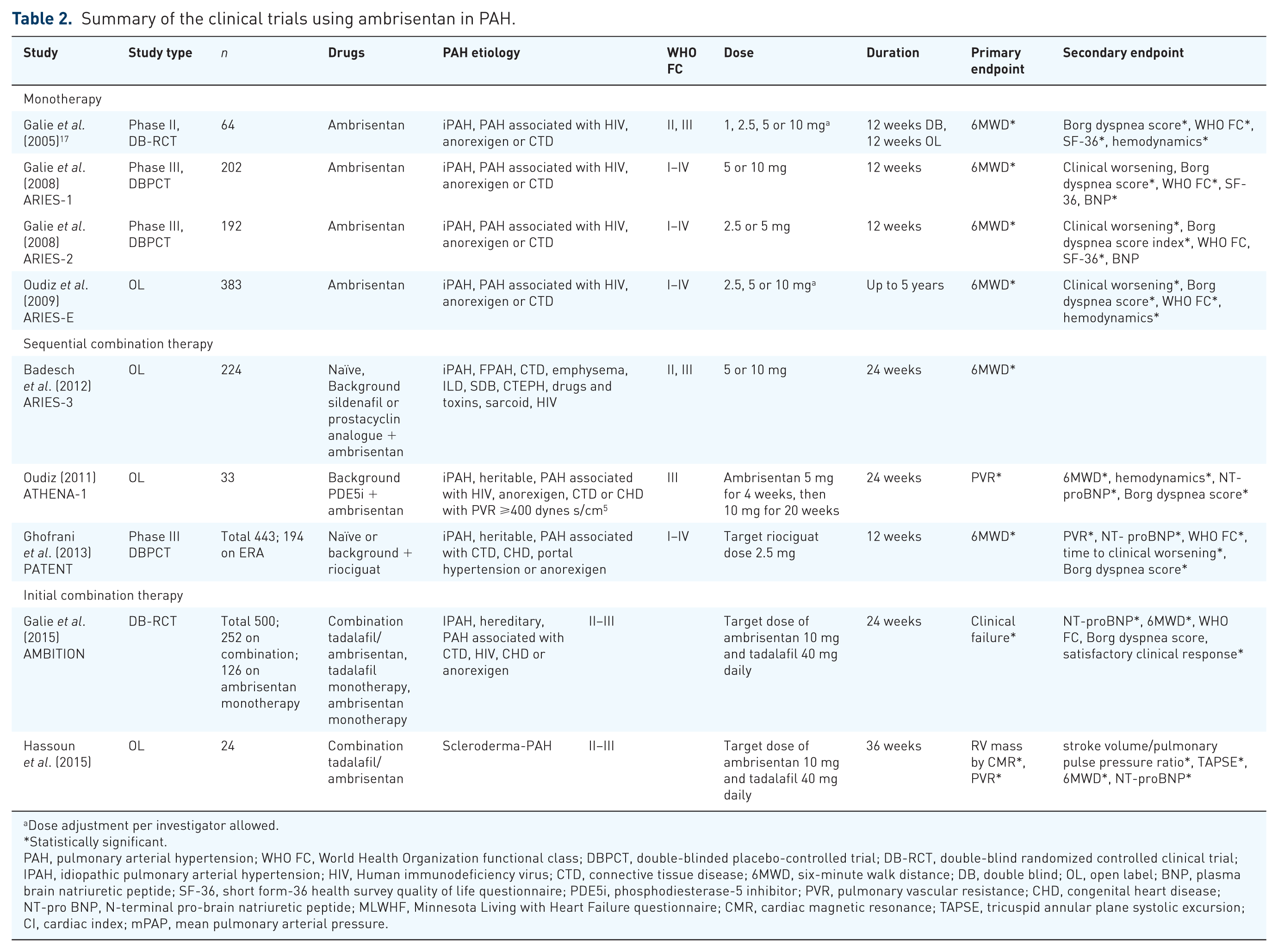
Dose adjustment per investigator allowed.
Statistically significant.
PAH, pulmonary arterial hypertension; WHO FC, World Health Organization functional class; DBPCT, double-blinded placebo-controlled trial; DB-RCT, double-blind randomized controlled clinical trial; IPAH, idiopathic pulmonary arterial hypertension; HIV, Human immunodeficiency virus; CTD, connective tissue disease; 6MWD, six-minute walk distance; DB, double blind; OL, open label; BNP, plasma brain natriuretic peptide; SF-36, short form-36 health survey quality of life questionnaire; PDE5i, phosphodiesterase-5 inhibitor; PVR, pulmonary vascular resistance; CHD, congenital heart disease; NT-pro BNP, N-terminal pro-brain natriuretic peptide; MLWHF, Minnesota Living with Heart Failure questionnaire; CMR, cardiac magnetic resonance; TAPSE, tricuspid annular plane systolic excursion; CI, cardiac index; mPAP, mean pulmonary arterial pressure.
In 2005, the results of a randomized, double-blind, multinational phase II clinical trial for the safety and efficacy of ambrisentan in 64 symptomatic Group 1 PAH patients due to iPAH or PAH associated with HIV, anorexigen or CTD. In the initial 12 weeks, patients were treated with fixed doses of ambrisentan (1, 2.5, 5 or 10 mg) followed by 12 weeks of open-label extension, which permitted dose adjustments based upon the investigator’s clinical assessment. 17 Exercise capacity was the primary efficacy endpoint measured by the change in 6-minute walk distance (6MWD) at 12 weeks after therapy compared with baseline. Secondary endpoints measured were change in Borg dyspnea score index, WHO FC and cardiopulmonary hemodynamics.
All four treatment groups experienced an increase in the 6MWD of 34–38 m, without a dose–response relationship. A total of 56 patients continued treatment in the optional open-label extension period, which is a smaller population that may have included a selection of the best responders to treatment, an often-recognized limitation in open-label extension trials. However, exercise capacity continued to improve at week 24 with an increase in 6MWD of +54.2 m. The patients with iPAH had a significant improvement in 6MWD at week 12 (+39.9 m) and 24 (+64.3 m) compared with other PAH etiologies. Improvements were also observed in the Borg dyspnea score index, quality of life and WHO FC as well as invasive cardiopulmonary hemodynamics [mean pulmonary arterial pressure (mPAP), pulmonary vascular resistance (PVR) and cardiac index (CI)] in patients with available repeat hemodynamics.
ARIES-1, 2 and E
The two concurrently run clinical trials ARIES-1 and 2, evaluated three different doses of the once daily oral ambrisentan (ARIES-1, 5 or 10 mg; ARIES-2, 2.5 or 5 mg) in 394 treatment-naïve patients diagnosed with iPAH, HIV, anorexigen and CTD-associated PAH. 18 This 12-week, multicenter, double-blind randomized placebo-controlled phase III trial met its primary endpoint of change in 6MWD. Both ARIES-1 and 2 demonstrated a combined improvement of 12-week 6MWD by +31 and +59 m respectively when compared with baseline. The secondary endpoints of Borg dyspnea score index and plasma brain natriuretic peptide (BNP) levels were also improved in both studies. The time to clinical worsening improved in ARIES-2, while WHO FC improved in ARIES-1.
In a single-center extension phase of ARIES-1, patients were followed for a range of 3–5.5 years in a small population of 12 patients. 19 After 1 year of ambrisentan monotherapy repeat hemodynamic studies demonstrated improved mPAP, Cardiac Output (CO) and PVR. At 1 and 2 years post therapy the baseline 6MWD improved from 350 m to 397 m (p < 0.01) and 393 m (p = 0.01). Follow-up cardiac magnetic resonance imaging (MRI) data demonstrated improved right ventricular ejection fraction from 29% to 46% at the 2-year mark (p = 0.02).
Patients who completed ARIES-1 and 2 were eligible to enroll in the open-label safety and efficacy extension study, ARIES-E (n = 383). 20 At 2 years of follow up, patients in the 5 and 10 mg ambrisentan group maintained 6MWD at +23 and +28 m, respectively. The WHO FC was maintained or improved at the 2-year mark in 79–89% of patients and 72% of patients remained free of clinical worsening events. A subset of 58 patients underwent right heart catheterization (RHC), which showed significant improvement in mPAP, PVR and CI from baseline. Patients treated with ambrisentan have demonstrated sustained hemodynamic and exercise capacity benefit on long-term follow up.19,21
ARTEMIS
Although the ARTEMIS trial evaluated the treatment of idiopathic pulmonary fibrosis (IPF) patients with ambrisentan, it included 48 RHC-documented patients with PAH. This phase III randomized, double-blind, placebo-controlled, multicenter event-driven clinical trial had a primary endpoint to evaluate the reduction in the rate of progression of IPF. 22 This study is notable in that use of an ERA may have been linked to clinical worsening of IPF. The study was terminated after enrollment of 492 patients as the interim analysis indicated a low likelihood for reaching efficacy endpoints. The ambrisentan-treated patients were more likely to have disease progression with 27.4% (n = 90) versus 17.2% (n = 28; p = 0.01) of placebo patients reaching this endpoint, with a hazard ratio of 1.74 [95% confidence interval (CI), 1.14–2.66]. Ambrisentan-treated patients had no worse lung function decline or death, but there was an increase in respiratory hospitalizations compared with placebo, 14.4% (n = 44) versus 5.5% (n = 9) respectively (p = 0.007). This study demonstrated that in patients with well-defined IPF, treatment with ambrisentan should be avoided.
POPH
There has been an interest in evaluating the effect of ambrisentan in other Group 1 disease such as POPH. Despite beneficial results, the lack of large-scale trials has not allowed for ambrisentan as an approved treatment for POPH.
In a small prospective single center, open-label observation study of 13 consecutive patients, with POPH was evaluated with ambrisentan monotherapy. 23 Patients were not permitted to be on other PAH-specific background therapy. Inclusion criteria were age >18 years; portal hypertension or chronic liver disease with mPAP >25 mmHg; PVR >240 dyn s cm−5 (3 Wood units (WU)); and Pulmonary artery wedge pressure (PAWP) < 15 mmHg or transpulmonary gradient > 12 mmHg. Patients were started on 5 mg ambrisentan for a month with a goal of 10 mg daily. A patient withdrew from the study due to side effects of periorbital bleeding, weight gain of 8 pounds and increased peripheral edema. Patients were followed for a median of 613 days [interquartile range (IQR) of 385–1011 days]. There were significant hemodynamic benefits noted at the time of repeat RHC. The mPAP dropped 29% from 58 mmHg (IQR range 37–63) to 41 mmHg (IQR range 27–48) (p = 0.004); PVR was reduced by 61% with an initial PVR of 445 dyn•s•cm-5 (IQR range 329–834) to 174 dyn•s•cm-5 (IQR range 121–361) (p = 0.008); there was no significant change in PAWP; cardiac output increased by 27%, 6 l/min (IQR range 4.7–7.7) to 8.6 l/min (IQR range 6.5–12) (p = 0.008). Additionally, the WHO functional class decreased by 50% from 3 (IQR range 2–3) to 1.5 (IQR range 1–2) and there was no significant increase in liver function tests (LFTs) over a 12-month evaluation period.
A small open-label study performed at four centers in Germany evaluated the effect of ambrisentan on exercise capacity in moderate-to-severe POPH. The median follow-up time was 16 months (IQR 12–21). The 6MWD was assessed at baseline, 6 and 12 months. Exercise capacity improved significantly from a baseline of 376 m (IQR 207–440) to 415 m (IQR 393–475) (p = 0.011) at 6 months and 413 m (IQR 362–473) (p = 0.005) at 12 months. There were significant improvements in WHO functional class and no significant changes in LFTs at the end of the study. The authors concluded that treatment of POPH with ambrisentan resulted in improved exercise capacity and clinical symptoms without relevant safety concerns.
Sequential combination therapy
Therapies for PAH are mainly aimed at the dysregulation of three key intracellular pathways, implicated in the pathogenesis of PAH: the endothelin, nitric oxide and prostacyclin pathways. 3 The concept of combination therapy for PAH had emerged from the heart failure, oncology and HIV literature that targeting multiple distinct pathways may be beneficial in PAH. Historically, a single agent was initiated and subsequent therapeutic response was assessed at a later date; additional medications are added when a patient fails to improve on current therapy. It is not clear however if the individual patient has predominant dysregulation of one pathway over another. The choice of therapy often strikes a balance among anticipated side effects, comorbid conditions and desired outcomes. 3 There have been mixed outcomes in clinical trials investigating sequential combination therapy, which may be in part related to drug–drug interaction, study design or small study size.24–28 The following trials highlight sequential combination therapy with ambrisentan as one of the therapeutic agents.
ARIES-3
ARIES-3 included 224 patients in a long-term, open-label, multicenter, single arm study. It evaluated a more diverse patient population that including Group 1 PAH, (iPAH and familial PAH); Group 3 PH (emphysema, interstitial lung disease and sleep-disordered breathing); Group 4 (chronic thromboembolic PH); and Group 5 (sarcoidosis) for the evaluation of safety and efficacy with ambrisentan. 29 Hemodynamic inclusion criteria were mPAP > 25 mmHg, PVR > 3 WU and PAWP < 15 mmHg; patients with Group 3 PH had stricter hemodynamic criteria with mPAP > 35 mmHg and PVR > 3.5 WU. Patients were not permitted use of other ERAs at the time of enrollment but could be on background therapy with prostacyclin analogues or sildenafil. Over half of the study patients were on background therapy, which included 41% on sildenafil and 5% on prostacyclin analogue therapy; 7% were on both therapies. The primary endpoint was improvement in 6MWD at 24 weeks. Patients in all groups, except for Group 3 PH, had improvements in 6MWD. The overall improvement in exercise capacity was +21 m. Patients on ambrisentan monotherapy had a +32 m improvement in 6MWD. The addition of ambrisentan to patients on background sildenafil monotherapy experienced an increase in 6MWD by +25 m; those on background prostanoid therapy ± sildenafil had a +46 m improvement in 6MWD. 29
ATHENA-1
In an open-label efficacy and safety trial, Oudiz and colleagues studied the addition of ambrisentan to 33 PAH patients on a background phosphodiesterase-5 inhibitor (PDE5i) with a suboptimal therapeutic response. 30 The primary endpoint of change in PVR was reached at 24 weeks of therapy with a decrease of –249 dyn•s•cm−5 (95% CI, −338 to −160) from a mean 761 ± 307 dyn•s•cm−5 (p < 0.0001). Other hemodynamic parameters improved, including mPAP, which decreased −5.4 mmHg (95% CI, −8.3 to −2.5) (p = 0.0007) and CI increased +0.58 l/min/m2 (95% CI, 0.25–0.90) (p = 0.0011). The 6MWD also improved +18 m (95% CI, 0.5–36) (p = 0.0437). Additionally, the plasma N-terminal pro-brain natriuretic peptide (NT-proBNP) decreased by 31% from baseline (p = 0.0216). At 24 weeks, ambrisentan was well tolerated and provided clinically relevant improvements in exercise capacity and hemodynamic parameters (mPAP, CI and PVR).
PATENT
Riociguat, a soluble guanylate cyclase stimulator, was approved for the treatment of PAH in 2013 after a randomized, multicenter, placebo-controlled trial evaluated its use in PAH (idiopathic, familial, CTD-associated, congenital heart disease, portal hypertension or anorexigen). 31 An initial 12-week trial (PATENT-1) included escalating doses of riociguat up to 2.5 mg three times daily in 443 patients. All patients completing this initial phase, were eligible for an extension trial (PATENT-2). A total of 44% of patients (n = 194) were receiving background ERA (bosentan or ambrisentan) and 6% were receiving prostanoid therapy. The study met its primary endpoint of improvement in 6MWD both in treatment-naïve and in patients with background therapy.
Zhuang et al. 2014
The addition of tadalafil to ambrisentan was investigated in 124 hemodynamically-confirmed Group 1 PAH Chinese patients aged 18–70 years in a 16-week double-blind study. Patients were considered stable on ambrisentan for at least 4 months and were treated with either tadalafil 40mg or placebo. Use of prostanoids or PDE5i precluded enrollment. The majority of patients enrolled were WHO functional class II, III and were diagnosed with severe pulmonary hypertension (PH). Although the study demonstrated no significant difference in hemodynamic parameters between those on tadalafil versus placebo there were significant improvements in 6MWD compared with placebo. At 16 weeks the addition of tadalafil to ambrisentan improved median 6MWD by 54.4 m compared with baseline and compared with the 18.3 m improvement in the placebo group. Patients in the tadalafil group experienced a significant delay in clinical worsening than the placebo group during the 16-week study. Combination was well tolerated and there was not a significant signal of increased edema, unlike the significant edema that was noted in the AMBITION trial.32,33
Initial combination therapy
Combination therapy had long been a mainstay of therapy for PAH; however most clinical trials performed employed a sequential, add-on therapy to existing PAH medications.28,31,34,35 The concept of initial combination therapy stemmed from the recognition that early aggressive therapy in Group 1 PAH improved outcomes. 36 Initial combination therapy primarily targets more than one pathway simultaneously with rapid and early up-titration of medications for an enhanced therapeutic effect. Part of the success of the combination of ambrisentan and tadalafil may be due to the lack of significant drug–drug interactions, unlike other ERA and PDE5i combinations.26,37 To date, ambrisentan is the only ERA with an indication for initial combination therapy to reduce the risk of disease progression and hospitalization for worsening PAH and improve exercise ability.
AMBITION
The Ambrisentan and Tadalafil in Patients with PAH (AMBITION) trial evaluated two simultaneous treatment strategies in Group 1 PAH comparing upfront combination therapy of ambrisentan and tadalafil with ambrisentan or tadalafil monotherapy in drug-naïve PAH patients. 33 Patients were randomly assigned in a 2:1:1 ratio to a target dose of ambrisentan 10 mg and tadalafil 40 mg daily. This event-driven, double-blind, multicenter study included WHO FC II and III, Group 1 PAH patients. Revised inclusion criteria were 6MWD ⩾ 125 m to ⩽500 m at screening and cardiopulmonary hemodynamics of mPAP ⩾ 25 mmHg, PVR ⩾ 300 dyn•sec•cm−5, Pulmonary Capillary Wedge Pressure (PCWP) or Left Ventricular End Diastolic Pressure (LVEDP) ⩽ 12 mmHg if PVR ⩾ 300 to ⩽500 dyn•sec•cm−5, or PCWP or LVEDP ⩽ 15 mmHg if PVR ⩾500 dyn•sec•cm−5. A total of 500 patients met the amended entry criteria. Overall, 18% of the combination therapy group (n = 46) reached the primary composite endpoint of time to clinical failure, defined as death, first occurrence of hospitalization for worsening PAH, disease progression or unsatisfactory long-term clinical response (defined as any decrease from baseline 6MWD at two consecutive visits separated by at least 14 days and WHO FC III symptoms assessed at two visits separated by 6 months) compared with 34% of the ambrisentan (n = 43) and 28% of the tadalafil (n = 34) monotherapy groups and 31% of the pooled monotherapy group (n = 77).
The combination therapy group demonstrated a 50% reduction in the risk of first event of clinical failure compared with the monotherapy groups. A decrease in hospitalization was the primary driver of the composite endpoint. Of the secondary endpoints, the combination therapy group at 24 weeks had a larger change in median 6MWD from baseline and percent change in NT-pro BNP levels compared with the other groups. Combination therapy also demonstrated a higher satisfactory clinical response at week 24 compared with pooled monotherapy and tadalafil groups, but not to ambrisentan monotherapy. The most common adverse events included peripheral edema, headache, diarrhea and dizziness. The rate of study drug discontinuation was not impacted by treatment assignment and the most common reasons included peripheral edema and dyspnea.
The differences in outcomes and therapeutic response to PAH-specific therapy in the connective tissue disease PAH populations compared with iPAH have been noted. 38 A post hoc analysis of the AMBITION study safety and efficacy data for CTD-PAH (n = 187) and scleroderma-associated PAH (SSc-PAH; n = 118; 63% of the connective tissue disease population) demonstrated that initial combination therapy reduced the risk of clinical failure compared with pooled monotherapy. 39 The hazard ratios were similarly reduced for CTD-PAH [0.43 (95% CI, 0.24–0.77)] and SSc-PAH patients [0.43 (0.22–0.89)]. Overall, 19% of the CTD-PAH (n = 20/103) treated with initial combination therapy reached the primary endpoint versus 36% (n = 30/84) of the pooled monotherapy patients. Overall, 21%, n = 15/71) of SSc-PAH patients treated with combination therapy experienced the primary endpoint as compared with 40% (n = 19/47) of the pooled monotherapy group. Patients with connective tissue disease-related PAH, including SSc may benefit from initial combination therapy with ambrisentan and tadalafil.
Although not powered as a mortality study, a post hoc analysis of the modified intention-to-treat population utilized two approaches to assess mortality in the AMBITION study. 40 A pre-specified analysis performed at the end of the study did not demonstrate a difference in deaths in the initial combination therapy (29/302, 10%) group versus (41/303, 14%) the pooled monotherapy group (hazard ratio 0.67; 95% CI, 0.42–1.08; stratified log rant p = 0.10). Utilizing a novel endpoint of 7 days after the end of randomization showed that the combination therapy group had fewer deaths (3/302, 1%) compared with pooled monotherapy (13/303, 4%; hazard ratio 0.21; 95% CI, 0.06–0.73). A firm conclusion regarding mortality cannot be drawn from this post hoc analysis. The survival advantage for the initial combination of ambrisentan and tadalafil over pooled monotherapy in newly diagnosed PAH patients warrants further evaluation.
Initial combination in scleroderma-PAH
Hassoun and colleagues performed a trial similar to AMBITION, which evaluated the upfront combination of ambrisentan and tadalafil in WHO FC II and III PAH patients. 41 This prospective, multicenter, 36-week open-label trial of 24 treatment-naïve patients with SSc-PAH demonstrated improvements in the co-primary endpoints of Right ventricle (RV) mass reduction by cardiac MRI and decrease in PVR. There were improvements in pulmonary artery compliance (median stroke volume/pulmonary pulse pressure ratio) measured by RHC and in mean tricuspid annular plane systolic excursion (TAPSE) at 36 weeks. Exercise capacity, as measured by 6MWD and serum biomarker NT-proBNP, also demonstrated significant improvements.
Comparison of ambrisentan with other ERAs
There are no ‘head-to-head’ clinical trials comparing the three US FDA-approved ERAs, bosentan, ambrisentan and macitentan. There are pharmacodynamics differences, drug–drug interactions and side effects that should be considered when prescribing these medications to patients. There are currently no clinical data to establish the benefit of one ERA over the other.
Although not intended as a direct comparison of ERAs, the 2015 European Society of Cardiology and European Respiratory Society updated guidelines on the management and treatment of PAH. 41 Within the guidelines, ambrisentan and bosentan monotherapy received a class/level of 1A (recommended/is indicated with data derived from multiple randomized clinical trials or meta-analyses) for WHO FC II, III patients and IIbC (conflicting evidence or divergence of opinion about the usefulness or efficacy of the treatment arrived at by consensus opinion or small studies) for WHO FC IV Group 1 PAH patients. Macitentan in comparison was given a class/level of 1B for WHO FC II, III patients (recommended/is indicated with data derived from a single randomized clinical trial) and IIbC (conflicting evidence or divergence of opinion about the usefulness or efficacy of the treatment arrived at by consensus opinion or small studies) for WHO FC IV Group 1 PAH patients.
Additionally, the upfront combination of ambrisentan and tadalafil received a class/level of 1B (recommended/is indicated with data derived from a single randomized clinical trial) for WHO FC II, III patients and a IIbC (conflicting evidence or divergence of opinion about the usefulness or efficacy of the treatment arrived at by consensus opinion or small studies) for WHO FC IV Group 1 PAH patients. For sequential drug combination therapy, ambrisentan added to sildenafil was recommended as a IIbC for WHO FC II–IV based upon the ARIES-3 results. 42
Post hoc analyses
PAH severity
Data from ARIES-1, 2 and E were utilized to evaluate outcomes at 12 weeks for 329 patients grouped by WHO FC. Both groups of WHO FC I–II and III–IV patients treated with ambrisentan (5 or 10 mg) had +48.4 m and +46.8 m improvement respectively in 6MWD compared with placebo; however patients with more advanced disease represented by a BNP level > 130 pg/ml had greater placebo-corrected improvement of +72 m in 6MWD. 43 This study also showed that while most patients on ambrisentan sustained the 6MWD improvement for around +40 m at 2 years, there was more clinical worsening and lower survival observed in patients with WHO FC III/IV compared with WHO FC I/II.
Follow-up cardiopulmonary hemodynamics
The hemodynamic effects of ambrisentan in patients who had a RHC at ⩾3 months enrolled in ARIES-E was analyzed retrospectively. 44 A total of 58 patients had a RHC at a median time of 60 months after beginning ambrisentan monotherapy. There were significant improvements in mPAP, PVR and CI with 5 and 10 mg therapy groups when compared with baseline values after approximately 2 years of follow up. The RHC cohort showed less improvement in 6MWD when compared with the 315 follow-up patients without available RHC data. This study showed a statistically significant correlation between improvement in 6MWD and decreases in mPAP and PVR.
Effectiveness of spironolactone and ambrisentan for PAH
Aldosterone levels are increased in patients with PAH and decreased pulmonary artery endothelial nitric oxide levels through inactivation of an ETB receptor. The coupling of therapies such as ETA receptor inhibitor (ambrisentan) with aldosterone antagonist (spironolactone) may reduce pulmonary artery vasoconstriction. A post hoc analysis of patients from the ARIES 1 and 2 trials on 10 mg ambrisentan with concurrent use of spironolactone demonstrated a trend toward improved 6MWD and reduced BNP compared with ambrisentan alone. 45 The retrospective nature of the study may have impacted clinical significance; this was a small, underpowered study with discordance in severity between the ambrisentan ± spironolactone groups. The concurrent use of spironolactone and ambrisentan warrants additional study.
Side effects
Edema
Edema is a well-documented phenomenon related to ERA use in PAH due to systemic vasodilation. ARIES-1 and 2 demonstrated that 17% of ambrisentan-treated patients (n = 261) developed edema compared with 11% in placebo patients (n = 132). 18 In the AMBITION trial, peripheral edema was the most common adverse event. It was observed at a higher rate in the ambrisentan plus tadalafil combination group (45%) compared with ambrisentan (33%) and tadalafil (28%) monotherapy. 33
In a post hoc analysis of the ARIES-1 and 2 clinical trials, Shapiro and colleagues evaluated the effect of edema on patients treated with ambrisentan. In the subgroup analysis, all forms of edema were considered, including peripheral edema, pitting edema, gravitational edema, localized edema, anasarca, fluid retention, and fluid overload. This led to an increase in the percentage of patients with edema, which occurred in 14.4% and 23% of patients for placebo and ambrisentan respectively. The edema was considered to be mild-to-moderate in severity. Patients that experienced edema while on ambrisentan were older and weighed more when compared with those without edema. Age > 65 years portended increased risk for developing edema while on ambrisentan. Patients without edema had a +38.9 m improvement in 6MWD compared with those with edema that had a +19.4 m improvement (p < 0.001). Treatment with ambrisentan in the presence of edema appears to ameliorate the beneficial response in exercise capacity in patients with Group 1 PAH. 46
Hepatotoxicity
Elevation in serum aminotransferase concentrations has been described with older ERAs, such as bosentan. 47 Upon approval for ambrisentan the US FDA mandated a ‘black box’ warning regarding hepatotoxicity and a risk minimalization and action plan required patients to enroll in a LEAP program (Letairis Education and Action Plan) where monthly LFTs were monitored. Clinically, the incidence of hepatic injury of ambrisentan is less than that of bosentan. The ARIES-1 and 2 study had no patients with serum aminotransferase >3-times the upper limit of normal. 18 In ARIES-E, a low risk of liver enzyme abnormalities was seen with long-term use of ambrisentan, with a 2% incidence in a 2-year follow-up period. 20
Post-marketing analysis of 10,927 patients exposed to ambrisentan and 9893 patient years studied, revealed a low risk of hepatotoxicity. The US FDA removed the black box warning in 2011 and no longer requires monthly LFT testing. 48 The LEAP program is still available as a service to clinicians. It has been postulated that the different hepatic effects are related to the underlying chemical structures of the ERA. Ambrisentan is a propanoic acid derivative and bosentan and sitaxsentan are sulfonamides. 49 In a sandwich-cultured human hepatocytes study of ERAs (ambrisentan, darusentan, bosentan and sitaxsentan), it was demonstrated that ambrisentan had little or no human hepatic transporter inhibition, compared with the other ERAs analyzed.50,51 This finding likely accounts for significantly less hepatic injury.
Anemia
A decrease in hemoglobin is observed with ERAs, likely in the setting of systemic vasodilation and hemodilution. Hemoglobin concentration decreased by a mean of 0.84 g/dl with the use of ambrisentan in the ARIES-1 and 2 trial. 18 In the AMBITION trial, anemia was seen more frequently in ambrisentan plus tadalafil combination (15%) than with ambrisentan (6%) or tadalafil (12%) monotherapy. 33
Teratogenicity
All ERAs are teratogenic and should not be used during pregnancy. Bosentan, unlike ambrisentan, may decrease the efficacy of oral contraceptives. 52 It is recommended that patients who are treated with ERAs employ two forms of contraception.
Conclusion
Ambrisentan is an ERA approved for the treatment of idiopathic, heritable PAH and CTD-PAH. It has shown to be effective in improving exercise capacity, cardiac hemodynamics, WHO FC and quality of life in patients with PAH. Ambrisentan displays an acceptable safety profile when used as monotherapy and in combination therapy with other vasodilators, such as a PDE5i.
The pivotal AMBITION trial has changed the conceptual approach to treating Group 1 pulmonary hypertension, with a shift from monotherapy and sequential add-on therapy to initial combination therapy. This combination therapy has been proven beneficial in the AMBITION trial and will be tested in other clinical trials such as the OPTIMA [ClinicalTrials.gov identifier: NCT02968901] and TRITON [ClinicalTrials.gov identifier: NCT02558231] trials. Aggressive early therapy however may need to be assessed in light of its contribution to increasing drug costs on healthcare systems. Additionally, in a rare disease population there has been increasing difficulty in enrolling patients due to the number of available clinical trials. As a result, there is an interest in the application of pulmonary vasodilators to non-Group 1 PH patients such as Group 3 related to interstitial lung disease [ClinicalTrials.gov identifier: NCT02630316] or exercise PAH [ClinicalTrials.gov identifier: NCT01338636].
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.