
Abstract
This review focuses on the advancements in the treatment of pulmonary hypertension (PH), especially after the Food and Drug Administration (FDA) approval of sotatercept and the advances in treatment recommendations after seven World Symposia on PH. PH, a complex and progressive condition defined hemodynamically by a mean pulmonary artery pressure >20 mmHg, encompasses multiple PH groups, each with distinct pathophysiological characteristics and treatment implications. Diagnosing PH can be challenging because symptoms like shortness of breath, fatigue, and chest pain are nonspecific. Contemporary treatment of pulmonary arterial hypertension aims to improve outcomes, symptoms, and overall quality of life, with a primary focus on preventing and treating right ventricular failure. Comprehensive risk stratification remains crucial, aiding in personalized therapy adjustments that improve patients’ outcomes. This review also touches upon the limited treatment options for other PH groups, like PH associated with left heart disease, parenchymal lung diseases, and chronic thromboembolic PH, underscoring the need for expanded therapeutic options. Despite advances, challenges remain: diagnostic delays, misdiagnosis, absence of head-to-head clinical trials, and the timing of introducing newer treatments such as sotatercept are discussed, emphasizing an integrated approach that transcends vasodilation to target underlying disease mechanisms. Future directions envision a comprehensive risk stratification incorporating right ventricular function and a mechanism-based treatment paradigm, encouraging a tailored therapeutic approach in PH management.
Plain language summary
Pulmonary hypertension is a condition characterized by high pressure in the vessels of the lung. During the last three decades, great progress has been made in the treatment of the disease. This review article describes the current treatment of pulmonary arterial hypertension, based on recommendations from the 7th World Symposium in Pulmonary Hypertension. It incorporates current knowledge on the use of a recently FDA approved medication, i.e. sotatercept, a medication with unique mechanism of action that directly acts on the disease process. It emphasizes the importance of risk stratification and early and aggressive treatment of the disease to improve outcomes. Furthermore it discusses the current treatment of different types of pulmonary hypertension, including pulmonary hypertension associated with left heart disease and parenchymal lung disease.
Introduction
Pulmonary hypertension (PH) is a hemodynamic condition (mean pulmonary artery pressure (mPAP) >20 mmHg) that can be seen in a large variety of diseases. PH is classified into five groups that reflect similarities in pathophysiology, clinical presentation, hemodynamic profile, and therapeutic implications. 1 The classification of PH includes five groups, with group 1, pulmonary arterial hypertension (PAH), notable for its severe prognosis with a median survival of 7 years, and a 3-year mortality rate of about 55% in high-risk patients.2,3 Other groups include PH associated with left heart disease (group 2), lung diseases and/or hypoxemia (group 3), pulmonary artery obstructions (group 4), and PH with unclear and/or multifactorial mechanisms (group 5). 1
Diagnosing PH can be challenging due to the nonspecific nature of its symptoms, such as shortness of breath, fatigue, and chest pain. 4 In fact, the time from symptom initiation to diagnosis of PAH with right heart catheterization (RHC) continues to be longer than 2 years. 5 The diagnosis and hemodynamic classification of PH requires RHC. PH is diagnosed in the presence of an mPAP >20 mmHg. The measurement of pulmonary artery wedge pressure (PAWP) 6 and the calculation of pulmonary vascular resistance (PVR), 7 allows a hemodynamic characterization as precapillary (PAWP ⩽15 mmHg and PVR >2 Wood Units (WU)), isolated post-capillary (ipcPH) (PAWP >15 mmHg, PVR ⩽2 WU), and combined pre- and post-capillary PH (cpcPH) (PAWP >15 mmHg, PVR >2 WU). 1 The most recent classification of PH also reintroduced exercise PH, which is characterized by an mPAP over cardiac output (CO) slope of >3 WU. 1
PAH (group 1 PH) is a progressive condition characterized by the narrowing of the pulmonary arteries, leading to an increase in the right ventricular afterload, eventually causing right heart failure and premature death. 8 The pathophysiology of PAH is multifactorial and yet not fully understood, involving complex interactions among genetic predispositions, environmental factors, and underlying diseases. 9
In this review, we will focus on the advancements in the treatment paradigm of PH, particularly after the Food and Drug Administration (FDA) approval of sotatercept and the updated recommendations of the 7th World Symposium on PH. 12
Treatment pathways in PAH
The key therapeutic pathways in PAH block endothelin, enhance nitric oxide and prostacyclin signaling and rebalance the activin/bone morphogenic protein signals.10 –12
The endothelin pathway is upregulated in PAH, which leads to pulmonary vasoconstriction through effects on endothelin A and B receptors. 13 Treatments include dual endothelin A and B receptor antagonists, bosentan and macitentan, as well as the selective endothelin A receptor antagonist, ambrisentan. The nitric oxide (NO)-cyclic guanine monophosphate (cGMP) signaling is reduced in PAH with lower levels of NO production and higher levels of destruction through phosphodiesterase-5 (PDE5). 14 Agents that boost cGMP include the PDE5 inhibitors, sildenafil and tadalafil, and the sGC stimulator riociguat, which enhances cGMP generation independently of NO.11,15,16
The prostacyclin (PGI2) signaling pathway is reduced in PAH, leading to lower cyclic adenosine monophosphate levels, which in turn cause vasoconstriction and pro-proliferative responses. 17 Therapeutic agents targeting this pathway include PGI2 analogues (epoprostenol, treprostinil, and iloprost) and a PGI2 receptor (IP) agonist (selexipag). 10 Recently, a medication (sotatercept) with a novel mechanism of action has received approval from the FDA for the treatment of PAH.18,19 Sotatercept is a fusion protein that acts as an activin signal inhibitor and ligand trap, which binds members of the transforming growth factor-β, such as activin and growth differentiating factors, particularly modulating the downstream pathway of the activin receptor type IIA /Smad2/3.19,20
Contemporary treatment of PAH
The goal of treatment in PAH is to improve outcomes, symptoms, and overall quality of life. 12 The key factor determining outcomes in PAH is the right ventricle’s (RV) ability to adapt to the increasing afterload resulting from the gradual narrowing of the pulmonary vasculature.4,8,21 Consequently, the primary focus of PAH management is to prevent and treat RV failure through a combination of interventions that would improve the coupling between the RV and the pulmonary artery. 22
Given the complexity in the management of PAH and poor prognosis if inadequately treated, experts emphasize the importance of promptly referring cases to centers specialized in the management of this life-threatening condition. 12 Centers specialized in PH can ensure an accurate diagnosis, risk stratification, and early initiation of effective treatments. The diagnosis of PAH requires, among other components, a reliable RHC showing precapillary PH. 1 Once the diagnosis of PAH is made, it is necessary to stratify the risk of dying at 1 year by using established risk score systems. 12 Based on careful risk stratification and assessment of RV function, the type and intensity of PAH treatment are selected.11,12 Once PAH treatment is initiated, a strict follow-up is essential to adjust PAH therapies with the ultimate goal of achieving a low-risk score and normal RV function.11,12
In addition to PAH-specific therapies, supportive treatment includes managing fluid balance by following a low-sodium diet and using diuretics at doses tailored to each patient’s volume status and renal function. Spironolactone, a mineralocorticoid receptor blocker, is frequently employed due to its role in modulating the renin-angiotensin-aldosterone system and its potential antifibrotic properties. Although randomized controlled trials are lacking, combining loop diuretics with mineralocorticoid receptor blockers is a common strategy in PAH management.23 –25 Supplemental oxygen should be considered after a thorough evaluation, with indications consistent with the American Thoracic Society (ATS) guidelines. 26
Medical conditions associated with the development of PAH should be adequately managed, including connective tissue diseases (i.e., scleroderma and systemic lupus erythematosus), portal hypertension, infections (i.e., HIV and schistosomiasis), and congenital heart diseases.11,27 Patients should also be screened for iron deficiency anemia and treated accordingly. 12 The use of anticoagulation therapy in idiopathic or heritable PAH remains controversial and should be considered on an individual basis, particularly avoiding it in groups at higher risk of bleeding, such as patients with scleroderma or porto-PH, or elderly individuals.28,29
Beyond the physical challenges, PAH imposes significant psychological stress on patients and caregivers. Facilitating access to resources such as social workers, mental health professionals, and support groups can greatly assist in coping with PAH. Women of childbearing age should be advised against pregnancy due to its risks. Alternative options like surrogacy or adoption should be discussed. In addition, supervised pulmonary rehabilitation is recommended for most patients with PAH, as it can improve quality of life and exercise capacity. 30 Other general supportive measures include immunization (i.e., severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), influenza, Streptococcus pneumoniae, and respiratory syncytial virus (RSV)), genetic and pre-transplant counseling.12,31,32 Overall, PAH treatment requires a holistic approach that goes beyond pharmacological therapies.
Targeted therapies in PAH
Current PAH treatments target one of four pathways. Phosphodiesterase-5 inhibitors (PDE5i) (sildenafil and tadalafil) and the soluble guanylyl cyclase stimulator (riociguat), enhance the nitric oxide-cyclic guanosine monophosphate (NO-cGMP) pathway and improve functional status, exercise tolerance, and pulmonary hemodynamic profile.33 –36 Endothelin receptor antagonists (ERAs), (bosentan, ambrisentan, and macitentan) block the effect of endothelin-1 and improve exercise capacity and pulmonary hemodynamics, while delaying disease progression.37 –39 Prostacyclin analogues (epoprostenol, treprostinil, and iloprost) improve exercise capacity and likely survival rates among PAH patients, particularly in those categorized as WHO functional class III or IV.40 –47 Selexipag, a PGI2 receptor agonist, delays disease progression and reduces the risk of hospitalization for PAH. 48 The activin signaling inhibitor (sotatercept) provides antiproliferative benefits 49 with a reduction in PVR, while increasing exercise capacity, improving WHO functional class, and reducing the risk of clinical worsening events, even in patients receiving triple background PAH therapy (PDE5i, ERA, and prostacyclin).50,51
Recent proceedings and consensus statements recommend a personalized PAH treatment approach, based on risk stratification at the initial visit and during follow-up, including a careful assessment of the response to PAH therapies11,12,31,52 (Figure 1). European guidelines recommend a three- and four-strata risk model, based on initial and follow-up assessments, respectively. 31 The Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) risk calculator (REVEAL 2.0 for initial and REVEAL 2.0 Lite for follow-up assessment) is also recommended at initial and follow-up visits.53,54

PAH treatment algorithm from the 7th World Symposium on PH.
At presentation, early and aggressive PAH treatment using dual combination therapy (PDE5i and ERA) is recommended for all patients, 55 with the addition of parenteral prostacyclin therapy for those at high-risk (REVEAL 2.0 score ⩾9 or high-risk per ESC/ERS guidelines). 12 In patients who do not achieve the low-risk stratum (REVEAL 2.0 Lite <6 or low-risk per ESC/ERS guidelines), activin signaling inhibitor therapy can be added. 12 Oral or inhaled prostacyclin analogues or PGI2 agonist therapy can be considered in patients at intermediate risk (REVEAL 2.0 Lite 7 or 8, or intermediate-low-risk by the ESC/ERS guidelines). Parenteral prostacyclin analogue therapy should be considered intermediate-high risk by ESC/ERS guidelines or high risk by REVEAL 2.0 Lite or ESC/ERS guidelines11,12,31 (Figure 2).
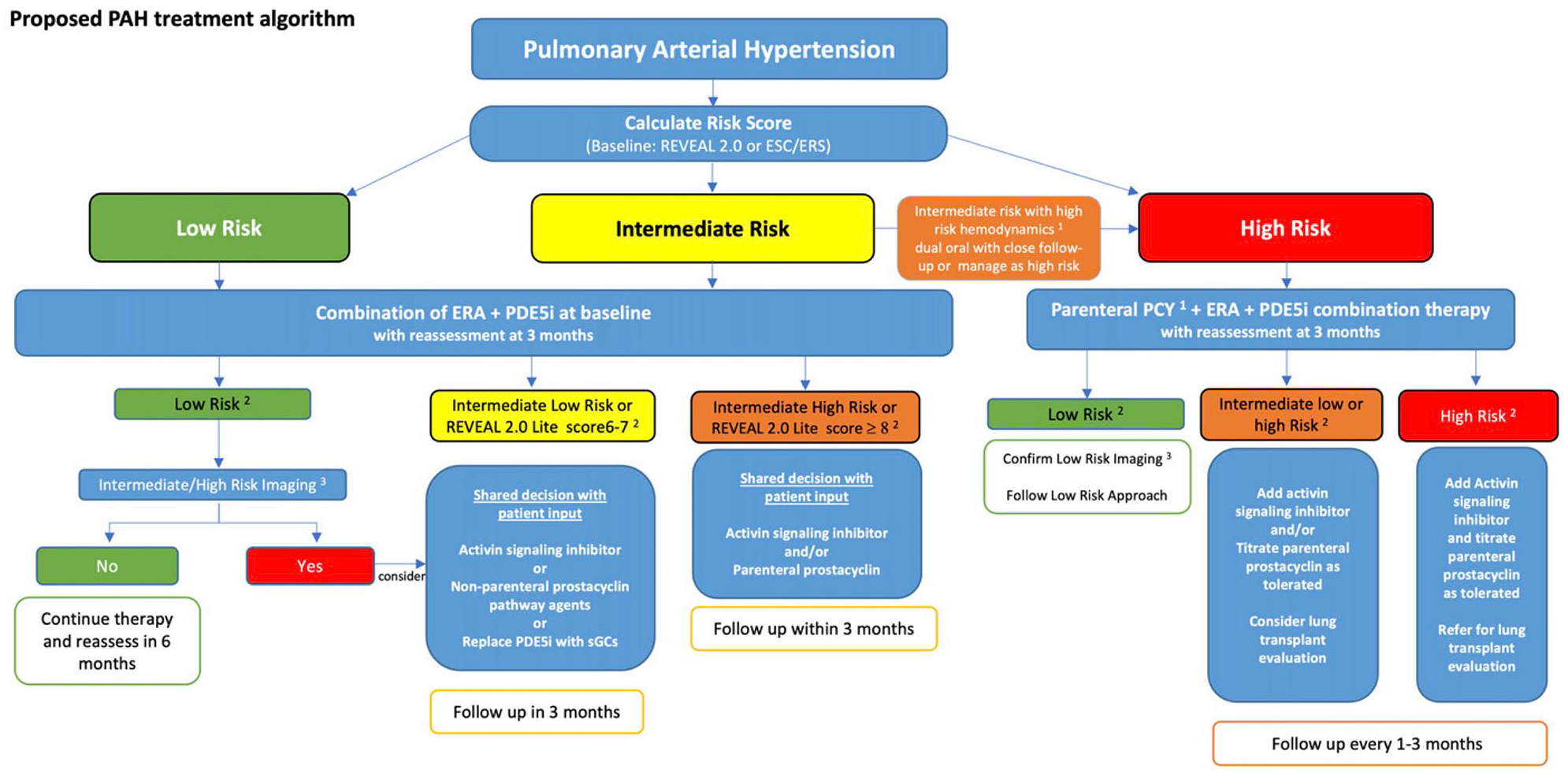
PAH treatment algorithm including expert opinion where evidence is lacking.
PAH treatment in special populations
PAH is a heterogeneous disease with multifactorial causes and diverse clinical presentations. 1 Its epidemiology is evolving, with an increasing age at the time of diagnosis56 –58 and burden of comorbidities. 59
Cardiac comorbidities in PAH
Many PAH patients, particularly women, present with concurrent cardiac conditions, often referred to as the “left heart phenotype.”60 –62 These conditions include systemic arterial hypertension, obesity (body mass index (BMI) ⩾ 30 kg/m2), atrial fibrillation, diabetes mellitus, and coronary artery disease. Such patients may exhibit echocardiographic features consistent with heart failure with preserved ejection fraction. Interestingly, a number of these patients may only show precapillary PH as they might be euvolemic at the time of RHC. 10 To identify underlying occult left ventricular diastolic dysfunction, these patients should undergo further evaluation with exercise or fluid challenge during RHC.63,64
Treatment with PAH-specific therapies should be considered if significant precapillary PH persists despite optimization of left-sided filling pressures and management of comorbid conditions. While ESC/ERS guidelines recommend starting PH monotherapy in these patients and adding a second agent based on response and tolerance, this conservative approach has been modified by the 7th World Symposium in PH, 12 based on the posthoc analysis of the AMBITION trial which showed beneficial effects of initial combination therapy with ERA and PDE5i were directionally the same, for patients with and without multiple risk factors for left ventricular diastolic dysfunction; however, those with risk factors experienced higher adverse effect rates with attenuated clinical response. 65 In addition, a similar analysis from the GRIPHON study evaluated the efficacy of selexipag in PAH patients with comorbidities, defined by BMI ⩾30 kg/m2, hypertension, diabetes, or coronary artery disease (CAD). Selexipag reduced morbidity and mortality across subgroups, regardless of the number or type of comorbidities. Furthermore, serious adverse events were comparable between treatment groups. 66
In the COMPERA registry, no survival differences were observed between PAH patients with comorbidities when adjusted for age and sex. Interestingly, most patients with comorbidities were treated with PDE5i as ERAs or prostacyclin analogues were poorly tolerated. 67
Pulmonary comorbidities in PAH
Patients with PAH often have pulmonary comorbidities, such as mild chronic obstructive pulmonary disease (COPD) or sleep apnea. Mild COPD or isolated nocturnal obstructive sleep apnea generally does not necessitate changes in the PAH treatment plan. 68 Sleep apnea alone rarely causes significant PH, though hypoventilation syndromes associated with daytime hypercapnia can contribute to the condition. 31
Severe parenchymal pulmonary disease falls under group 3 PH, but there is growing recognition of coexisting pulmonary conditions in PAH. A “lung phenotype” has been identified in PAH patients with significantly reduced diffusion capacity and minimal parenchymal abnormalities on computed tomography scans. 69 These patients are often males of older age with a smoking history, who have worse survival and poorer response to PAH therapies. Histopathological studies also support distinct differences between this “lung phenotype” and classic idiopathic PAH. 70
Treatment considerations for PAH patients with comorbidities
In clinical practice, managing patients with PAH and comorbidities requires an integral approach. It is crucial to optimize comorbid conditions, such as volume overload, diabetes, atrial fibrillation, and systemic arterial hypertension, as these patients are prone to PAH treatment discontinuation due to side effects and are less likely to achieve low-risk status. 11 Treatment with ERA may cause/worsen fluid retention that not always responds to diuretics. As PAH clinical trials often excluded patients with multiple comorbidities, the evidence for treatment efficacy and ideal treatment approach remains limited in this group of patients.
Monotherapy in PAH
There are specific scenarios in which monotherapy is appropriate in certain patients with PAH. According to the 7th World Symposium on PH, calcium channel blockers (CCB) monotherapy is recommended for patients with PAH who demonstrate vasoreactivity during RHC. 12 Long-term CCB responders show excellent outcomes. 71 Monotherapy may also be considered for older patients with multiple comorbidities, or those with pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis. In addition, monotherapy can be an option for PAH related to conditions like portal hypertension, congenital heart disease, or HIV, particularly when there are safety or tolerability concerns. 11
Treatment challenges in PAH
Despite significant advances in the treatment of PAH, there are still significant challenges that directly affect optimal patient care and outcomes. These challenges range from diagnostic delays, misdiagnosis, inadequate PAH treatment based on lack of a formal risk assessment, suboptimal use of background therapies, insufficient follow-up and assessments on treatment response and compliance, inadequate prevention and management of medication side effects, limited experience with more advanced diagnostic and therapeutic modalities, and late referral to lung transplantation.
An early and accurate diagnosis of PH remains a substantial hurdle, primarily due to the nonspecific nature of symptoms, the rarity of the condition, and the complexity of the diagnostic process, which requires specific tests and expertise.72 –75 Both late diagnosis and misdiagnosis are common, which create delays in treatment and inappropriate use of PAH therapies, respectively. 76
A significant challenge in PAH treatment selection is the absence of head-to-head clinical trials, 77 with current research focusing on new agents without direct comparison among available therapies. For example, for initial PAH treatment, has PDE5i combined with ERA a better alternative than PDE5i combined with oral prostacyclin analogues, or ERA combined with prostacyclin analogues? Is riociguat better than PDE5i in general, or only for those who have an “insufficient” PDE5i response? 78 Moreover, the incremental benefits of combining multiple therapies79,80 have not been adequately quantified, which might lead to under- or over-treatment. While combination therapy is generally considered more effective than monotherapy, more research is needed to determine the ideal combinations and sequence of medications, based on the patient’s characteristics and disease severity.11,12,31
Additionally, the adequate timing of the introduction of sotatercept is unclear. While promising results have been observed, its optimal placement in the PAH treatment algorithm remains unclear and is likely to be modified based on the results of newer studies such as HYPERION and ZENITH.51,81 The 7th World Symposium in PH suggests considering sotatercept for patients who do not reach a low-risk status after initial treatment with dual oral PAH therapy, including ERA and PDE5i.11,12,31 However, there is an emerging consensus that initiating sotartecept earlier might enhance its long-term benefits. The HYPERION trial (NCT04811092) specifically targets PAH patients diagnosed within the previous 12 months, who are treated with at least double PAH combination therapy and remain at intermediate- or high-risk. The ZENITH trial (NCT04896008) focused on high-risk patients with severe PAH on maximally tolerated double or triple combination therapy, who remained in WHO functional class III or IV. Importantly, this study was stopped earlier as the interim analysis showed overwhelming efficacy.
The potential effect of sotatercept in helping de-escalate other PAH therapies is under exploration, with no specific clinical trials currently addressing this possibility. Although further studies are needed, preliminary data on the SOTERIA study showed that few patients on parenteral prostacyclin analogues were able to lower the doses, transition to oral formulations, or discontinue altogether. 82 Furthermore, it remains unclear how to escalate treatment in PAH patients who remained at intermediate risk on dual oral combination therapy. Would these patients be considered for oral prostacyclin analogue, oral PGI2 agonist, inhaled prostacyclin analogue, or subcutaneous sotatercept? Similarly, for PAH patients that remain at intermediate-high risk, would sotatercept delivered subcutaneously every 3 weeks provide a better quality of life than parenteral prostacyclins that require continuous infusion, careful titration, and more involved management of side effects?
Economic factors and access to healthcare services also play a crucial role in the diagnosis and treatment of PAH. PAH therapies are often expensive and not universally available, limiting patient access to these life-saving interventions. Healthcare disparities, particularly in low- to middle-income countries, exacerbate these issues, with many patients unable to afford or access efficacious treatments.83,84
Treatment of other types of PH
PH associated with left heart disease (group 2 PH)
PH associated with left heart disease (PH-LHD) is the most common cause of PH and is associated with diseases of the left heart, such as systolic or diastolic heart failure and valvular disease, which lead to increased left ventricular filling pressures. This chronic vascular congestion results in the remodeling of pulmonary vessels, contributing to increased pulmonary pressures.84,85 It encompasses both ipcPH, where PH is solely due to diseases of the left heart, and cpcPH where additional pulmonary arterial remodeling has occurred.1,31
Establishing specific LHD subtypes, with accurate distinction between ipcPH and cpcPH are crucial steps for determining appropriate treatment strategies.86 –88 Treatment of PH-LHD focus primarily on optimizing the LHD, including the use of guideline-directed medical treatment. 89 In cases involving valvular heart disease, surgical interventions such as valve repair or replacement need to be considered. 88 Unfortunately, research using PAH-specific therapies in PH-LHD has yielded inconsistent results. 88
The VICTORIA trial demonstrated that the sGC stimulator vericiguat reduced the risk of death and hospitalization for chronic heart failure with reduced ejection fraction (HFrEF), but its specific impact on PH-LHD remains unclear. 90 The use of PAH-specific therapies in PH-LHD is generally not recommended outside of clinical trials, since PAH therapies may be harmful in this population.66,91 –94
PH associated with lung diseases and/or hypoxemia (group 3 PH)
The 7th World Symposium in PH emphasized the need to better phenotype patients with PH associated with parenchymal lung disease. 68 COPD, interstitial lung disease (ILD), and combined pulmonary fibrosis with emphysema are now recognized as separate categories within group 3 PH. These conditions cause PH by a combination of hypoxic vasoconstriction, loss of alveolar capillaries, and vascular remodeling. Importantly, the development of PH worsens prognosis and often exacerbates symptoms.95 –97
This refined categorization reflects the unique pathogenic mechanisms at play in each condition, allowing for a more precise and targeted approach to diagnosis and treatment. A multi-modal diagnostic approach, combining physiological assessments with high-quality radiographic imaging, is essential to accurately classify patients with group 3 PH. Treatment in PH-ILD is substantiated by data from a pivotal phase III randomized controlled trial, which demonstrated that inhaled treprostinil improved the 6-min walk distance, with a reduction in NT-pro brain natriuretic peptide and clinical worsening events compared to placebo. 98 However, a phase III randomized controlled trial of inhaled treprostil in patients with PH associated with COPD was halted early as there was no evidence of beneficial effect and a signal for harm. 99 This differential treatment response based on the type of parenchymal lung disease supports the need to carefully phenotype patients with group 3 PH.
In all group 3 PH patients, it is essential to manage the underlying lung disease and comorbid conditions, with consideration of early pulmonary rehabilitation and lung transplant referral.30,68,100 Other PAH medications have been studied in group 3 PH, but unfortunately, ambrisentan resulted in an increased risk of disease progression and respiratory hospitalizations, 101 and riociguat was associated with increased mortality in patients with idiopathic interstitial pneumonia. 94
PH associated with pulmonary artery obstructions (group 4)
Chronic thromboembolic pulmonary hypertension (CTEPH) is the most common type of PH in this group and is characterized by chronic thromboembolic disease of the pulmonary arteries, leading to PH and subsequently RV failure. This form of PH is unique because it can be potentially cured with surgical intervention.102,103
Treatment of CTEPH involves a multidisciplinary team tailored to offer the best possible treatment/s for each patient, that is, medical therapy, balloon pulmonary angioplasty (BPA), and pulmonary thromboendarterectomy (PTE). The multidisciplinary team encompasses PH experts, chest radiologists, interventionists with experience in performing BPA, and surgeons with expertise in PTE.102,104 PTE is the treatment of choice for eligible patients and can dramatically improve symptoms and pulmonary hemodynamics.31,105,106 BPA serves as a valuable alternative for patients who are not candidates for PTE or have residual PH following PTE.107,108 Riociguat is the only FDA-approved pulmonary vasodilatory therapy for CTEPH and can be used prior to BPA, when PTE or BPA are not feasible, or when residual PH persists after PTE or BPA.109,110 Other pulmonary vasodilator therapies have been explored in CTEPH, with variable results. 102 Lifelong anticoagulation is mandated for all CTEPH patients to prevent recurrent thromboembolic events.
Conclusion
Great progress has been made in the treatment of PAH during the last decades. Despite impressive advances, several challenges remain that directly affect the effectiveness of treatment. Future directions envision a comprehensive risk stratification incorporating right ventricular function and a mechanism-based treatment paradigm, encouraging a tailored therapeutic approach in PH management.