
Abstract
The purpose of this review is to evaluate the use of tadalafil as monotherapy and in combination regimens for the treatment of pulmonary arterial hypertension (PAH). A systematic English language search of the medical literature using PubMed was conducted between January 1960 and May 2012 using the search terms ‘tadalafil’, ‘therapy’, ‘pulmonary (arterial) hypertension’ and ‘combination therapy’. Special emphasis was given to controlled clinical trials and case studies relevant for the use of tadalafil in PAH. The search revealed 113 relevant publications, 31 of which were clinical trials, 52 were reviews and 12 were case reports. Of these, 12 were clinical studies in human patients with PAH who were treated with tadalafil alone, and seven were clinical studies in human patients with PAH who were treated with tadalafil in combination with other agents. Only clinical studies in human patients were included. Exclusion criteria were monotherapy other than using tadalafil and any combination therapy that excluded tadalafil as part of the treatment regimen. Overall, 1353 human subjects were studied; 896 were treated with tadalafil alone while 457 subjects were treated with tadalafil in coadministration. Tadalafil appears to be an effective and a safe treatment option for patients with PAH. It improves clinical status, exercise capacity, hemodynamic parameters, compliance issues and quality of life and reduces the occurrence of clinical worsening. Tadalafil in combination therapy seems to be additive and synergistic in relaxing pulmonary vascular muscle cells but more clinical trials on human subjects are warranted.
Introduction
Definition
Pulmonary hypertension (PH) is defined as an increase in mean pulmonary arterial pressure (mPAP) ≥25 mmHg at rest as measured by right-sided cardiac catheterization.
The normal value for mPAP at rest is 14 ± 3 mmHg. Mean PAH of 20 mmHg is considered an upper limit of normal but the significance of values between 21 and 24 mmHg are not clear [Badesch et al. 2009; Kovacs et al. 2009].
Classification of PH
The World Health Organization (WHO) has classified PH into five diagnostic groups [Chin and Rubin, 2008; Galiè et al. 2011, McLaughlin et al. 2009]:
Group 1: pulmonary arterial hypertension (PAH), which consists of (a) idiopathic PAH (IPAH), (b) familial PAH (FPAH) (due to mutations in the gene encoding for bone morphogenetic protein receptor type II or BMPR2, for activin-receptor-like-kinase-1 gene or ALK1, or for endoglin), (c) associated with connective tissue disorder (APAH) (congenital systemic to pulmonary shunts, portal hypertension, HIV infection, drugs and toxins) and others (thyroid disorders, glycogen storage diseases, Gaucher’s disease, hereditary hemorrhagic telangiectasia, hemoglobinopathies, chronic myeloproliferative disorders, splenectomy), (d) associated with significant venous or capillary involvement (pulmonary veno-occlusive disease [PVOD]), pulmonary capillary hemangiomatosis (PCH), and (e) persistent pulmonary hypertension of the newborn.
Group 2: pulmonary hypertension with left-sided heart disease (left-sided atrial or ventricular heart disease, left-sided valvular heart disease).
Group 3: pulmonary hypertension associated with lung diseases and/or hypoxemia (chronic obstructive pulmonary disease, interstitial lung disease, sleep-disordered breathing, alveolar hypoventilation disorders, chronic exposure to high altitude, developmental abnormalities).
Group 4: pulmonary hypertension due to chronic thrombotic and/or embolic disease (CTEPH) thromboembolic obstruction of proximal pulmonary arteries, thromboembolic obstruction of distal pulmonary arteries, nonthrombotic pulmonary embolism (tumor, parasites, foreign material).
Group 5: miscellaneous, including sarcoidosis, histiocytosis X, lymphangiomatosis, and compression of pulmonary vessels (adenopathy, tumor, fibrosing mediastinitis) [Arif and Poon, 2011; Chin and Rubin, 2008; Falk et al. 2010; Galiè et al. 2011; McLaughlin et al. 2009; Sidorenko et al. 2011].
PAH is a progressive and debilitating disease that is characterized by restriction of flow of blood through the pulmonary vasculature resulting in progressive increase in mPAP in the presence of normal pulmonary capillary wedge pressures (<15 mmHg). The condition eventually leads to right-sided heart failure and death. The mean survival period is 2.8 years after the establishment of the diagnosis without treatment [Arif and Poon, 2011; Frey and Lang, 2012; Katz, 2008; Liang et al. 2012; Naeije and Huez, 2007; Sanchez et al. 2010].
Pathogenesis of PAH
The pathogenesis of IPAH is not well understood. Contributing factors are an imbalance between vascular cell proliferation and apoptosis. During the early part of the disease process, there is dysfunction and/or destruction of the endothelial cells, which results in decreased synthesis of vasodilators (such as prostacyclin and nitric oxide) and overproduction of vasoconstrictors (such as endothelin-1 and thromboxane) [Arif and Poon, 2011]. The inflammation, reduced vasodilation and excess vasoconstriction lead to narrowing of the pulmonary vascular lumen, increase in pulmonary vascular resistance (PVR), thrombus formation and an increase in right ventricular afterload [Budhiraja et al. 2004; Humbert et al. 2004; Jing et al. 2011]. The insults that resulted to endothelial damage could be from viral infections, ingestion of certain drugs (e.g. dexfenfluramine), certain toxins (e.g. toxic rapeseed oil) or genetic loss of function mutation from BMPR2 [Humbert et al. 2004; Budhiraja et al. 2004]. Imbalance of inflammatory and repair mechanisms is one key mediator that leads to worsening of the disease [Affuso et al. 2010].
Epidemiology
The lowest estimates of the prevalence of PAH and IPAH are 15 cases and 5.9 cases per million adult population, respectively. The prevalence of PAH is between 15 and 52 cases per million population in Europe (according to registries from Scotland and other countries) [Humbert et al. 2006; Peacock et al. 2007], and the French registry has shown that 39.2% of patients had IPAH and 3.9% had family history of PAH [Peacock et al. 2007]. PAH can affect all ages but it is most frequently seen between 36 and 50 years of age. Women tend to be more affected than men [Chin and Rubin, 2008].
Clinical manifestation of PAH
Patients with PAH usually present with exertional dyspnea, dizziness and fatigue early in the disease process. As the condition worsens, signs and symptoms of right-sided heart failure may occur such as dyspnea at rest, exertional chest pain, palpitations, cyanosis, jugular vein distension, abdominal distension, pedal edema and syncope [Arif and Poon, 2011; Chin and Rubin, 2008]. On physical examination, there may be a left parasternal heave, an accentuated second heart sound, a pansystolic murmur, a diastolic murmur and/or third heart sound [Galiè et al. 2011]. The presence of some physical sign can provide clues to the underlying cause of PAH (e.g. the presence of telangiectasis, sclerodactyly, digital ulcers point towards scleroderma while the presence of inspiratory crackles on auscultation may point towards interstitial lung disease as a cause) [Galiè et al. 2011].
Diagnosis of PAH
The diagnosis of PAH requires a comprehensive evaluation including history and physical, chest X-ray, pulmonary function testing, echocardiography, often followed by connective tissue disease serology, vasodilatory testing for reversibility and tests to exclude thromboembolic disease as a contributing factor [Chin and Rubin, 2008; Sidorenko et al. 2011]. Once diagnostic evaluation has suggested PAH, right-sided heart catheterization is recommended to confirm the diagnosis and determine the severity. In addition, left-sided heart disease should be excluded as well as any correctable cardiac problems such as left-to-right heart shunting. Vasodilator testing can be performed during the diagnostic procedure. The use of echocardiography and magnetic resonance imaging (MRI) also can be used to determine the prognosis [Arif and Poon, 2011]. The New York Heart Association (NYHA) functional classification system is used to evaluate the functional status while the Medical Outcomes Study Short Form 36-item questionnaire (SF-36) is often used to monitor progress of the disease. The exercise and functional capacity can be assessed using cardiopulmonary exercise testing or 6-min walk distance (6MWD) tests [Arif and Poon, 2011]. A mPAP ≥25 mmHg at rest or >30 mmHg with exercise and/or a pulmonary capillary wedge pressure (PCWP) of ≤15 mmHg (without the presence of mitral stenosis) and a mean PVR of ≥2 or 3 Wood units confirm the diagnosis of PAH [Arif and Poon, 2011; Barst et al. 2011; Buckley et al. 2010; Chin and Rubin, 2008; Falk et al. 2010; Frey and Lang, 2012; Katz, 2008; Liang et al. 2012; Naeije and Huez, 2007; Sanchez et al. 2010].
Prostacyclic analogs: effects on endothelial cells
Prostacyclin (or PGI2) is released by the endothelial cells and exerts its function through a paracrine signal cascade that involves a G protein-coupled receptor, termed prostacyclin receptor (or IP), on endothelial cells and platelets. The endothelial prostacyclin receptor becomes activated when it binds to prostacyclin. This activation signals adenyl cyclase to produce cyclic adenosine monophosphate (cAMP) in the cytosol. cAMP then activates protein kinase A (PKA). The activated form of PKA continues to phosphorylate and thereby inhibit myosin light-chain kinase, leading to smooth muscle relaxation and vasodilation.
There is dysregulation of the prostacyclin metabolic pathways in patients with PAH as evidenced by decreased prostacyclin synthase expression in their pulmonary arteries and decrease prostacyclin metabolites in their urine [Galiè et al. 2011; McLaughlin et al. 2009]. Therapy with prostacyclic analogs causes vasodilatation of the pulmonary vasculature as well as improvements in the clinical signs and symptoms and in the hemodynamic parameters of patients with PAH.
Endothelin receptor antagonists
Endothelin-1 is a potent vasoconstrictor peptide with mitogenic properties, which is overexpressed in PAH [Affuso et al. 2010; McLaughlin et al. 2009]. Increased plasma levels of endothelin-1 correlate with the severity and prognosis of the PAH [McLaughlin et al. 2009]. Endothelin-1 binds to endothelin receptors A and B [ET(A) and ET(B)] to cause constriction of pulmonary vasculature. By blocking this interaction, endothelin receptor antagonists (ERAs) relax pulmonary arterial smooth muscle and decrease PVR.
Phosphodiesterase-5 inhibitors effect on endothelial cells
Phosphodiesterase-5 (PDE-5) inhibitors act through the nitric oxide (NO)-cyclic GMP pathway to increase cGMP, which is the final mediator in the NO-cyclic GMP pathway, and exert vasodilatory and antiproliferative effects on pulmonary vascular smooth muscles [Affuso et al. 2010; Klinger, 2011]. Sildenafil (Viagra™, Pfizer, USA; Revatio™, Pfizer, USA) was the first studied PDE-5 inhibitor and was approved in 1998 by the US Food and Drug Administration (FDA) for the treatment of erectile dysfunction. In 2005 it was approved for the treatment of PAH based on a large clinical trial [Galiè et al. 2005]. In this trial, 278 patients with PAH were randomly assigned to placebo or sildenafil (20 mg, 40 mg or 80 mg) for 12 weeks. The sildenafil treatment group demonstrated significant improvements in 6MWD, hemodynamic parameters (mPAP) and WHO functional class [Galiè et al. 2005].
Other approved PDE-5 inhibitors in the US are tadalafil (Cialis™, Eli Lilly, USA; Adcirca™, Eli Lilly, USA) and vardenafil (Levitra™, GlaxoSmithKline, USA; Staxyn™, GlaxoSmithKline, USA). These newer agents have potential advantages over sildenafil such as a faster onset and longer duration of action, higher selectivity for PDE-5, and increased absorption [Rosenkranz et al. 2007; Sharma, 2007].
The three PDE-5 inhibitors also differ in the pharmacokinetics of pulmonary vasorelaxation. Tadalafil and sildenafil are selective for pulmonary circulation, vardenafil has the most rapid effect and sildenafil has a greater effect on arterial oxygenation [Ghofrani et al. 2004]. Tadalafil has a longer half-life (t1/2 = 17.5 h) compared with sildenafil (t1/2 = 3-4h), and it is not associated with visual side effects (blurring of vision and blue/green color tinges) that might occur with sildenafil use [Falk et al. 2010; Klinger, 2011; Levin and White, 2011; Rosenzweig, 2010; Schwarz et al. 2007; Sharma, 2007]. Tadalafil has a greater affinity for PDE-5 when compared with other PDE-5 inhibitors [Wrishko et al. 2008].
Tadalafil monotherapy
Tadalafil is a selective long-acting PDE-5 inhibitor originally manufactured for the treatment of erectile dysfunction and was approved by the FDA in 2009 as a once-daily dose treatment for PAH [Affuso et al. 2010; Croxtall and Lyseng-Williamson, 2008; Falk et al. 2010; Frey and Lang, 2012; Galiè et al. 2009; Klinger, 2011; Rosenzweig, 2010]. In addition, to vasodilatory and antiproliferative properties, tadalafil has anti-inflammatory actions and antioxidant effects making it an effective agent for the treatment of hypobaric hypoxia-induced pulmonary hypertension [Rashid et al. 2012]. Tadalafil is mainly metabolized in the liver by cytochrome P450 3A4 (CYP3A4) and coadministration with other drugs that interfere with cytochrome P450 can reduce the half-life and hence the efficacy of the drug [Wrishko et al. 2008]. Tadalafil may cause transient decreases in blood pressure. Coadministration with nitrates should be avoided with all PDE-5 inhibitors (and vice versa), and in the case of tadalafil, should be avoided at least within 48 hours in order to avoid potential life-threatening hypotension.
Tadalafil was approved as a once-daily dose of 40 mg taken orally with or without food [Frey and Lang, 2012; Barst et al. 2011; Klinger, 2011; Rosenkranz et al. 2007; Levin and White, 2011; Rosenzweig, 2010]. It is convenient to use and is considered to be safe and effective with minimal side effects [Barst et al. 2011; Buckley et al. 2010; Levin and White, 2011; Rosenzweig, 2010]. It has shown to improve exercise capacity, hemodynamic parameters, time to clinical worsening (relative risk reduction = 68%, p = 0.038) and quality of life (QoL) in patients with PAH [Galiè et al. 2009]
The first case of the successful use of tadalafil for the treatment of a PAH patient was published in 2004 by Palmiera and colleagues [Palmiera et al. 2004]. The authors reported the use of 20 mg of oral tadalafil in a 72-year-old female patient with PAH who failed to respond to intravenous epoprostenol. The patient showed significant improvements in clinical status and hemodynamic parameters. Since then there have been series of case reports on the successful use of tadalafil to treat patients suffering from PAH refractory to other agents such as epoprostenol. In a case series, 12 PAH patients with prior sildenafil use demonstrated sustained improvement with the use of tadalafil monotherapy [Tay et al. 2008]. A study in 405 patients with PAH was the first placebo-controlled trial using tadalafil with a favorable safety profile [Galiè et al. 2009].
In a 16-week phase III (PHIRST) trial, 405 patients with PAH (idiopathic or associated), either on background therapy with bosentan or treatment-naïve, were randomized to placebo or 2.5, 10, 20 or 40 mg once daily of tadalafil. The study showed a significant increase in 6MWD (from 33 m at baseline to 44 m at week 16, p < 0.01 for treatment naïve patients receiving tadalafil 40 mg once daily). Tadalafil 40 mg once daily dose also delayed clinical worsening (p = 0.041), incidence of clinical worsening (68% relative risk reduction; p = 0.038), and QoL. There were no statistical significant changes in WHO functional class. In patients receiving bosentan 125 mg twice daily as a background therapy the 6MWD was insignificantly increased by only 23 m [Galiè et al. 2009]. Patients who completed the PHIRST trial could still continue with the extended study (PHIRST 2) [Galiè et al. 2009]. In the 52-week, double-blind, uncontrolled PHIRST 2 trial, the long-term safety and efficacy of tadalafil for treatment of PAH were evaluated. The safety profile and adverse events of tadalafil in PHIRST 2 and PHIRST trials were the same. The 6MWD achieved in PHIRST for the group of patients receiving tadalafil 20 mg and 40 mg in both PHIRST and PHIRST 2 [406 ± 67 m (n = 52) and 413 ± 81 m (n = 59) at PHIRST 2 recruitment, respectively] were maintained at the completion of PHIRST 2 [415 ± 80 m (n = 51) and 410 ± 78 m (n = 59), respectively]. Few patients on tadalafil 40 mg in PHIRST and PHIRST 2 experienced WHO functional class deterioration [6% (n = 5)] compared with patients randomized to tadalafil 20 mg [9% (n = 7)] in either trial. Both studies demonstrated that tadalafil is a safe and effective treatment option for patients with PAH and long-term treatment appears to be sustained [Oudiz et al. 2012].
In a 4-week study on the efficacy and safety of tadalafil compared with placebo in the treatment of PAH, tadalafil was associated with improvements in 6MWD (409.25 ± 40.25 m versus 319.37 ± 42.39 m, p < 0.0001), improvements in Borg Dyspnea Index (BDI; 4.62 ± 2.56 versus 6.37 ± 2.61, p = 0.021), reduction in pulmonary artery systolic pressure (PASP; 88.75 ± 23.26 mmHg versus 109.5 ± 23.78 mmHg, p < 0.0001), and an improvement in the WHO functional class [Bharani et al. 2007]. De Carvalho and colleagues reported a case of a patient with IPAH (NYHA class IV) who demonstrated improvement in functional capacity and hemodynamic after treatment with tadalafil [De Carvalho et al. 2006].
Tadalafil appears to have beneficial effects on PAH irrespective of the underlying etiology [Aggarwal et al. 2007]. A total of 13 PAH patients with various comorbidities demonstrated improvements in exercise tolerance (350.54 ± 255.06 s to 479.54 ± 195.00 s, p < 0.01) 4 weeks after addition of tadalafil to their baseline medication. Hemodynamics improved slightly but the differences were not significant (mPAP 63.5 ± 26.2 mmHg versus 62.2 ± 24.8 mmHg, mean total PVR 1858.6 ± 1138.9 dyne-sec.cm−5 versus 1737.3 ± 1017.2 dyne-sec.cm−5, mean pulmonary blood flow 3.26 ± 1.04 l/min versus 3.44 ± 1.26 l/min) [Aggarwal et al. 2007].
Tadalafil monotherapy has been shown to be a safe and effective treatment option for the pediatric population. A study to investigate the efficacy and the safety of tadalafil in 33 pediatric patients showed that 29 patients voluntarily switched from sildenafil to tadalafil because of its once-daily dosing. Furthermore, 14 of these patients demonstrated improvements in hemodynamics (mPAP 53.2 ± 18.3 mmHg versus 47.4 ± 13.7 mmHg, p < 0.05; PVR index 12.2 ± 7.0 versus 10.6 ± 7.2 dyne-sec.cm−5/m2, p < 0.05) [Takatsuki et al. 2012]. This study concluded that tadalafil is a safe treatment option for pediatric patients with PAH and might reduce the progression of the illness [Takatsuki et al. 2012].
Side effects of tadalafil
The most commonly reported side effects are headache, back pain and myalgias, which occur as a result of tadalafil affinity on PDE-5 [Arif and Poon, 2011; Galiè et al. 2009; Sharma, 2007]. Other possible side effects are flushing, allergic reactions, dyspepsia, migraine and rhinorrhea (or stuffy nose) and these are usually transient.
Tadalafil in combination therapy
Oral medications, which target the three main pathways involved in the pathophysiology of PAH, namely the prostacyclic–cAMP pathway, the endothelin pathway and the NO-cyclic GMP pathway, are currently available (Table 1). These oral medications are prostacyclic analogs, such as epoprostenol (Flolan™, GlaxoSmithKline, USA; Veletri™, Actelion, USA), treprostinil (Tyvaso™, United Therapeutics, USA and Catalent Pharm Solutions, USA, Remodulin™; Baxter Healthcare, USA), iloprost (Ventavis™; Actelion, USA) and beraprost (Berastolin; Taisho, Japan), endothelin receptor antagonists such as ambrisentan (Letairis™, Gilead Sciences, USA) and bosentan (Tracleer™, Actelion, Pharmaceuticals, USA), as well as PDE-5 inhibitors such as tadalafil (Cialis™, Eli Lilly, USA; Adcirca™, Eli Lilly, USA), sildenafil (Viagra™, Pfizer, USA; Revatio™, Pfizer, USA), and vardenafil (Levitra ™, GlaxoSmithKline, USA; Staxyn™, GlaxoSmithKline, USA) [Anderson and Nawarskas, 2010; Affuso et al. 2010; Buckley et al. 2010; Liang et al. 2012; Maki et al. 2011; Sidorenko et al. 2011].
FDA-approved agents for PAH.

A total of 11 medications had been approved by the FDA for the treatment of PAH.
PAH, pulmonary arterial hypertension; PDE-5, phosphodiestarase-5; ERA, endothelin receptor antagonist; BID, twice a day; TID, three times a day; QID, four times a day; IV, intravenous injection; SQ, subcutaneous injection.
Evidence-based guidelines recommend starting treating PAH patients with monotherapy [Anderson and Nawarskas, 2010; Barst et al. 2011]. The American College of Cardiology, the American Heart Association, and the European Society of Cardiology recommended starting IV epoprotenol as first-line treatment for patients with severe PAH (WHO functional class III and IV) and sildenafil or ambrisentan or bosentan as first-line treatment for patients with WHO functional class III [Maki et al. 2011]. Intravenous epoprostenol is highly efficacious but is expensive and also difficult for patients to use [Anderson and Nawarskas, 2010; Levin and White, 2011; Singh et al. 2006]. Combination therapy is recommended as second-line treatment for those patients who do not respond to the initial treatment [Maki et al. 2011]. Many patients respond to single-drug therapy [Tay et al. 2008] while others do not show significant improvements [Badesch et al. 2007; Barst et al. 2011; Galiè et al. 2004]. Some of the patients who improved initially with monotherapy do not sustain long-term success, which might lead to the use of combination therapy [Barst et al. 2011; Benza et al. 2007; Galiè et al. 2011; Liang et al. 2012; Maki et al. 2011; Sidorenko et al. 2011]. Combination treatment has been advocated also for patients with 6MWD of less than 380 m, signs of right-sided heart failure and persistent functional class III or IV symptoms refractory to treatment with one medication [Chin and Rubin, 2008; Hoeper et al. 2005]. The combination of these agents results in additive and synergistic relaxation of the endothelin-contracted pulmonary ring, pulmonary remodeling, and a delay of progression of the disease, as well as improvement of clinical outcome, functional capacity, hemodynamic parameters, and overall prognosis [Affuso et al. 2010; Benza et al. 2007; Buckley et al. 2010; Kiliçkesmez and Küçükoğlu, 2010; Liang et al. 2012; Maki et al. 2011]. Studies have demonstrated significant improvements in symptoms, hemodynamics, and the time to clinical worsening with combination therapy [Chin and Rubin, 2008; Hoeper et al. 2005].
Bendayan and colleagues combined tadalafil and prostacyclins in four patients with PAH and demonstrated that all symptoms and 6MWD (from 214 to 272 m) were improved after 3 months [Bendayan et al. 2008]. Faruqi and colleagues reported the use of tadalafil combined with sitaxentan in three patients with IPAH [Faruqi et al. 2010]. Sustained improvement in exercise capacity and hemodynamic parameters were noted and sustained without serious adverse effects. In another report, a 49-year-old patient with IPAH was treated with a combination of bosentan, tadalafil, and beraprost with clinical improvements and near-normal hemodynamics at 6 months of therapy [Maki et al. 2011].
The combination of two or more drugs might exert additive or synergistic effects [Barst et al. 2011; Liang et al. 2012]. Nowadays, the most widely used combinations are endothelin receptors blockers (bosentan) with PDE-5 inhibitors (tadalafil, sildenafil) [Sidorenko et al. 2011; Liang et al. 2012]. There is a possible pharmacokinetic interaction between the two agents since tadalafil is metabolized by CYP3A4, while bosentan induces CYP3A4 and cytochrome P450 2C9 (CYP2C9) [Wrishko et al. 2008]. A study demonstrated that after 10 days of coadministration, bosentan reduced tadalafil exposure by 41.5% with clinically irrelevant differences (<20%) in bosentan exposure [Wrishko et al. 2008]. Combination of tadalafil plus bosentan was well tolerated in a study of 405 patients with PAH [Barst et al. 2011]. At week 16, 6MWD increases were 44 m (95% confidence interval [CI] 20–69 m; n = 37) for tadalafil in treatment-naïve patients and 23 m (95% CI −2 to 48 m; n = 42) for tadalafil coadministered with bosentan. Adverse effects observed by the use of tadalafil in monotherapy and in coadministration with bosentan were identical [Barst et al. 2011].
The pharmacokinetic interaction between tadalafil and ambrisentan were evaluated in a crossover study of 26 healthy adults. In the presence of tadalafil, ambrisentan maximum plasma concentration was similar [105.0% (90% CI 95.9–115.0%)], and systemic exposure was reduced slightly [87.5% (90% CI 84.0–91.2%)], compared with ambrisentan alone. The maximum plasma concentration and systemic exposure of tadalafil [100.6% (94.4–107.1%) versus 100.2% (92.6–108.4%)] were the same in the presence and absence of ambrisentan. The side effect profile of the combination agents was similar to either of the drugs alone [Spence et al. 2009]. Tadalafil plus sitaxentan combination therapy has been shown to improve QoL, exercise performance, and outcome without serious adverse effects in three patients with severe IPAH [Affuso et al. 2010].
Clinical study data review
Tables 2–5 summarize 10 clinical studies and eight case reports involving human subjects. Tadalafil monotherapy appears to be safe and represents an effective treatment option for patient with PAH [Barst et al. 2011; Buckley et al. 2010; Levin and White, 2011; Rosenzweig, 2010]. Several studies demonstrated that tadalafil monotherapy improves exercise capacity, hemodynamic parameters, the time to clinical worsening, incidence of clinical worsening, and QoL [Affuso et al. 2006, 2010; Arif and Poon, 2011; Barst et al. 2011; Buckley et al. 2010; Croxtall and Lyseng-Williamson, 2010; Galiè et al. 2009; Levin and White, 2011; Pepke-Zaba et al. 2009; Sakuma and Shirato, 2008; Takatsuki et al. 2012]. It is a convenient and cost-effective treatment option for PAH.
Tadalafil as a monotherapy in the treatment of PAH (clinical studies).
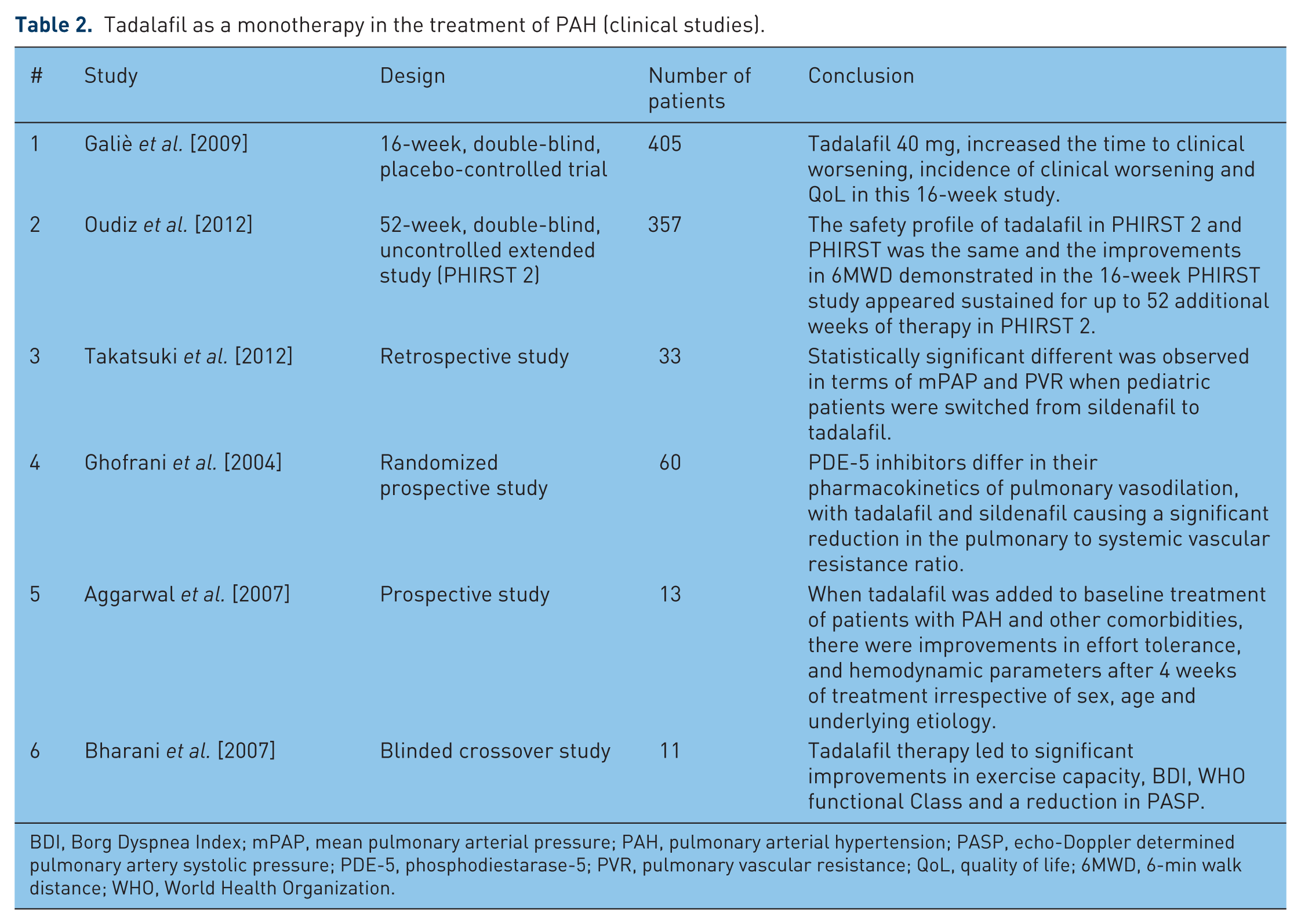
BDI, Borg Dyspnea Index; mPAP, mean pulmonary arterial pressure; PAH, pulmonary arterial hypertension; PASP, echo-Doppler determined pulmonary artery systolic pressure; PDE-5, phosphodiestarase-5; PVR, pulmonary vascular resistance; QoL, quality of life; 6MWD, 6-min walk distance; WHO, World Health Organization.
Tadalafil as a monotherapy in the treatment of PAH (case studies).

PAH, pulmonary arterial hypertension; PDE-5, phosphodiesterase-5.
Tadalafil in combination regimens for the treatment of PAH (clinical studies).

NYHA, New York Heart Association; PAH, pulmonary arterial hypertension; QoL, quality of life; 6MWD, 6-min walk distance.
Tadalafil in combination regimens for the treatment of PAH (case studies).

PAH, pulmonary arterial hypertension; QoL, quality of life.
Tadalafil in combination therapy demonstrated to exert additive and synergistic effects in relaxing pulmonary artery smooth muscles and in causing pulmonary vascular muscle remodeling, which eventually leads to slowing of the progression of the disease and to improvements in clinical outcome, functional capacity, hemodynamics, and prognosis [Barst et al. 2011; Benza et al. 2007; Liang et al. 2012; Levin and White, 2011; Maki et al. 2011; Kiliçkesmez and Küçükoğlu, 2010]. It has been advocated to start tadalafil coadministration in patients with 6MWD of less than 380 m, signs of right-sided heart failure, persistent functional class III or IV symptoms refractory to treatment with one medication, or those patients who did not respond to the first-line therapy [Chin and Rubin, 2008; Hoeper et al. 2005; Maki et al. 2011]. The availability of these different classes of medications specific for treatment of patients with PAH has improved the prognosis of this progressive and debilitating disease.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflicts of Interest statement
The authors declare no conflicts of interest in preparing this article.