
Abstract
Pulmonary arterial hypertension is a progressive and incurable disease. Over the past two decades, significant advances have been made in understanding and thus managing this disease. Multiple therapeutic options are currently available and optimizing the treatment of pulmonary arterial hypertension has become complex. Patients who meet the American College of Chest Physicians criteria for vasoresponsiveness can be safely and effectively treated with high-dose calcium channel blockers but require close follow up to assure durability of response. Patients with World Health Organization (WHO) functional class IV status and those with determinants of high risk for progression and death should be treated with an infused prostanoid agent without delay. These patients should also be referred early after stabilization for transplant evaluation. Patients with WHO functional class II status benefit from early initiation of oral therapies. Those with WHO functional class III status and lower determinants of risk for progression may receive treatment with one or more oral or inhaled agents, though many experts would advise early use of infused prostanoids for these patients as well.
Introduction
Pulmonary arterial hypertension (PAH) is a rare but progressive and fatal disease [McLaughlin et al. 2009]. It is estimated that PAH affects anywhere between 15 and 50 individuals/million in the USA and Europe. Management of PAH has advanced significantly in the past 15 years [Barst et al. 2009], but the mortality rate is still unacceptably high [Thenappan et al. 2010; Humbert et al. 2010] and it continues to be an incurable disease. Although there are currently nine drugs approved by the US Food and Drug Administration (FDA) for the treatment of PAH [Barst et al. 2009], they are not without side effects or complications [Augoustides et al. 2004; Ivy et al. 2009]. Patients with PAH span in age from newborns to older people, affecting both sexes, and all races [Frost et al. 2011]. With such diversity in the patient population and with the availability of different medication classes, routes of administration, and side effect profiles of the currently available PAH therapies, it becomes imperative to tailor therapy to the individual patient and to have a targeted approach to the treatment of PAH. This is especially true now with the advancement of the prognostication process for such patients [Benza et al. 2011a]. This article summarizes a practical targeted approach to the treatment of patients with PAH.
World Health Organization classification of pulmonary hypertension
During the most recent World Health Organization (WHO) World Symposium on Pulmonary Hypertension that was held in Dana Point in California in 2008, pulmonary hypertension (PH) classification was further refined to better reflect pathophysiologic pathways and treatment algorithms [Simonneau et al. 2009; Barst et al. 2009]. PH is still classified into five different categories, of which PAH (sometimes referred to as ‘precapillary’ PH in reference to its location of pathologic origin in the pulmonary arterial system) is one of these five groups (WHO group 1). It is the one that is most relevant to our discussion in this article because it is the only WHO group that has proven benefit from PAH-specific therapies (and so PAH-specific therapy is only FDA approved for patients classified as WHO group 1). The other four groups are PH owing to left heart disease (WHO group 2; sometimes referred to as ‘postcapillary’ PH or pulmonary venous hypertension), PH owing to lung disease or hypoxemia (WHO group 3), PH secondary to chronic thromboembolic disease (WHO group 4), and miscellaneous PH or PH with unclear or multifactorial mechanisms (WHO group 5) [Simonneau et al. 2009] (Table 1). It is important to note that PH is common, especially WHO group 2, but WHO group 1 PAH is much less frequently encountered.
World Health Organization (WHO) pulmonary hypertension classification.

Reproduced from [Simonneau et al. 2009] with permission from Elsevier.
Alk1, activin receptor-like kinase type 1; BMPR2, bone morphogenetic protein receptor type 2; COPD, chronic obstructive pulmonary disease; HIV, human immunodeficiency virus; ILD, interstitial lung disease; PAH, pulmonary arterial hypertension.
This classification is based on similar pathophysiologic mechanisms and treatment considerations [Simonneau et al. 2009]. Endothelial dysfunction and a proinflammatory milieu (local and systemic) are core features of WHO group 1 (PAH) histopathology [Morrell et al. 2009]. This happens in conjunction with in situ thrombosis or platelets activation, local abundance of vasoconstrictors (such as endothelin-1), and decreased presence of vasodilators and anti-inflammatory molecules (such as prostaglandin I2 and nitric oxide). Collagen and extracellular matrix deposition, chemotaxis, progenitor cells recruitment, and smooth muscle cells and fibroblasts hypertrophy and proliferation are key features of PAH pathophysiology as well [Hassoun et al. 2009; Barst, 2005]. Collectively, these pathophysiologic changes favor inflammation which leads to vascular wall thickening and ultimately the vasculopathy of PAH.
Pharmacotherapeutic pathways and key clinical trials
Among the different pathways and molecules implicated in the pathogenesis of PAH (WHO group 1), three pathways successfully stood out as viable targets to treat PAH: the nitric oxide pathway [targeted by phosphodiesterase type 5 (PDE-5) inhibitors], the endothelin-1 pathway [targeted by endothelin receptor antagonists (ERAs)], and the prostacyclin pathway (targeted by the prostanoid analogs) (Table 2) [McLaughlin and McGoon, 2006]. Nitric oxide, which is produced by endothelial cells, exerts its vasodilatory effects on the vascular smooth muscles via cyclic guanosine monophosphate (cGMP). PDE-5 inhibitors prevent the destruction of cGMP, thus potentiating the vasodilatory effects of nitric oxide. ERAs, however, block the endothelin receptors directly and prevent the vasoconstrictor and proinflammatory effects of endothelin-1. Prostanoids or prostaglandin I2 analogues stimulate adenylate cyclase which converts adenosine triphosphate to cyclic adenosine monophosphate and has antiproliferative downstream effects and induces vascular smooth muscle relaxation or vasodilation [McLaughlin and McGoon, 2006].
Pulmonary arterial hypertension-specific therapies approved by the US Food and Drug Administration (FDA).

Reproduced from [Fares et al. 2011] with permission from Bentham Science Publishers.
ERA, endothelin receptor antagonist; PDE5, phosphodiesterase-5.
Multiple randomized, controlled clinical trials (RCTs) have been performed looking at various therapeutic interventions in patients with PAH (WHO group 1). A summary of the key clinical trials that lead to FDA approval will be presented here. Prior to initiating any PAH-specific therapy, a right heart catheterization is mandatory in making the PAH diagnosis, and thus is required prior to PAH-specific therapy initiation. The first landmark RCT in PAH randomized 81 patients with idiopathic PAH (IPAH) with WHO functional class III or IV to standard care with or without epoprostenol infusion [Barst et al. 1996]. Of note is that this was the only trial that got FDA approval based on improved survival [Barst et al. 1996]. All other PAH-specific therapies were FDA approved based on improvements in hemodynamics, exercise capacity/6 min walk (6MW), and quality of life.
Seven years later, subcutaneously infused treprostinil in patients with idiopathic or associated PAH (n = 470) mostly with WHO functional class III also showed improvement in 6MW distance and hemodynamics [Simmoneau et al. 2002]. Inhaled iloprost (n = 203, idiopathic or associated PAH, or thromboembolic PH) and inhaled treprostinil (n = 235, idiopathic, heritable, or associated PAH) were also shown to be effective [Olschewski et al. 2002; McLaughlin et al. 2010].
The first ERA that was FDA approved was bosentan. This was based on two studies looking at 245 patients with idiopathic or associated PAH with WHO functional class III or IV and improvement in 6MW of 44 m in the study group compared with the placebo group [Rubin et al. 2002]. Ambrisentan was also subsequently approved based on two studies of a combined 394 patients with PAH with WHO functional class II or III [Galie et al. 2008a] with improvement in 6MW distance by about 50 m in the study group over a 12-week period. The two available PDE-5 inhibitors, sildenafil and tadalafil, both primarily tested in functional class II and III, were approved in 2005 and 2009, respectively. Sildenafil was tested in 278 patients with PAH (idiopathic or associated PAH WHO group 1) [Galie et al. 2005], and tadalafil was tested in 405 patients with PAH [Galie et al. 2009], both showing statistically significant improvement in 6MW distance achieved.
Baseline therapy (e.g. with diuretics and oxygen therapy) was continued in these RCTs and its value is not clear. There are no data suggesting improved survival with the use of diuretics, although they are an integral part of managing heart failure, be it right or left heart dysfunction. Oxygen has been proven to improve survival in patients with hypoxia who have chronic obstructive pulmonary disease. Designing an RCT to study the use of oxygen in patients with PAH may not be feasible or ethical. Anticoagulation in IPAH has been shown in observational studies to improve survival [Frank et al. 1997; Rich et al. 1992]. Although in situ thrombosis seems to be an integral part of the pathogenesis of PAH [Fuster et al. 1984], extrapolating such limited survival evidence from the above-mentioned populations to other PAH populations is controversial. Anticoagulating portopulmonary hypertension patients, for example, is often contraindicated in view of underlying coagulopathy. Patients with hereditary hemorrhagic telangiectasia and many patients with systemic sclerosis may have gastrointestinal arterio-venous malformations that place them at high risk of spontaneous bleeding in the setting of anticoagulation.
Prognostication and implications for therapeutic approach
Until 2001, the only drug available to treat PAH was epoprostenol (Flolan, GlaxoSmithKline Pharmaceuticals), and it was mostly used as a bridge to transplantation [Rich, 1995]. Since then, other therapies have evolved, and as a result the prognosis of patients with PAH has significantly improved from a historical median survival of less than 3 years to a 2-year survival of approximately 90% in the current era of PAH management [McLaughlin et al. 2002; McLaughlin, 2006; Oudiz et al. 2009; Thenappan et al. 2010]. More recent data show an approximate 80% 3-year survival rate [Oudiz et al. 2011]. Despite these advances, making individualized decisions on therapies continues to be a challenge.
The Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Disease Management (REVEAL), which was started in 2006 and ended enrollment in December 2009 after enrolling more than 3500 patients with WHO group 1 PAH [Badesch et al. 2011], is the largest PAH registry to date [Benza et al. 2010a]. Multiple independent prognostic factors have been identified (Table 3) [McLaughlin and McGoon, 2006], and an easy to use prognostic score calculator was developed and validated (Table 4) [Benza et al. 2011a]. Using this score can facilitate prognosis discussions with patients and thus assist in tailoring therapy that is patient specific. Although it has not been prospectively tested, such an objective score could theoretically assist in making informed decisions regarding timing of transplant evaluation and referral.
Risk determinants in pulmonary arterial hypertension.
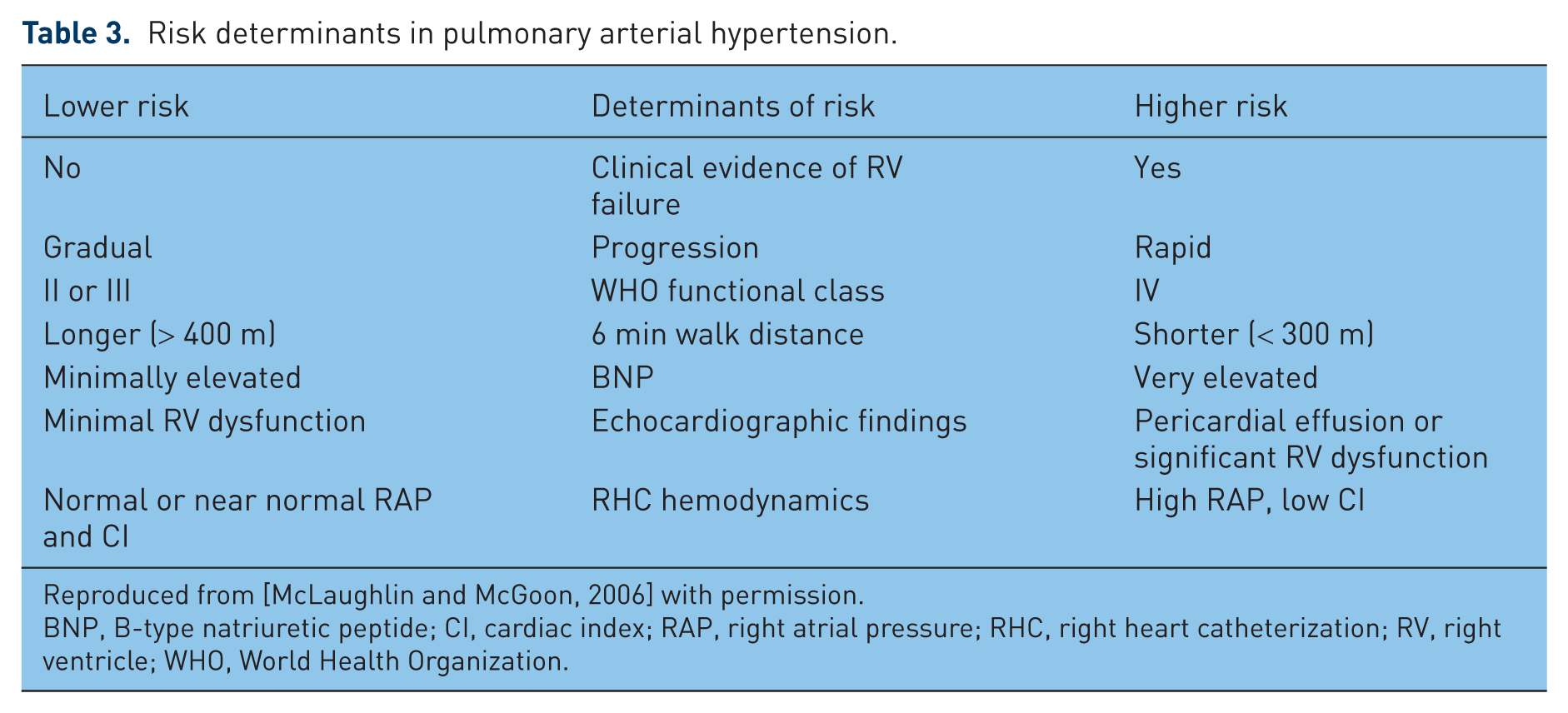
Reproduced from [McLaughlin and McGoon, 2006] with permission.
BNP, B-type natriuretic peptide; CI, cardiac index; RAP, right atrial pressure; RHC, right heart catheterization; RV, right ventricle; WHO, World Health Organization.
Targeted Approaches to the Treatment of Pulmonary Hypertension.

Reproduced from [Benza et al. 2011a] with permission.
Renal insufficiency was determined by each investigator, without a standardized definition. A glomerular filtration rate < 60 ml/h is reasonable to consider as renal insufficiency.
6MW, 6 min walk test; BNP, B–type natriuretic peptide; CTD, connective tissue disease; DLCO, diffusion capacity of the lung for carbon monoxide; NYHA, New York Heart Association; PAH, pulmonary arterial hypertension; PVR, pulmonary vascular resistance; RA, right atrial; REVEAL, US Registry to Evaluate Early and Long–term PAH Disease Management; WHO, World Health Organization.
For example, a young woman who has portopulmonary hypertension, WHO functional class III, walks 150 m on a 6MW test, with a pericardial effusion and a heart rate of 100/min, and thus a REVEAL risk score of more than 11 (out of 22 total potential points) would be treated very aggressively upfront since her predicted 1-year mortality rate would be in excess of 30%. This is in contrast to a young man with an HIV-associated PAH, WHO functional class II, who walks 450 m on a 6MW, without a pericardial effusion and with a B-type natriuretic peptide (BNP) of 100 pg/ml and right atrial pressure of 6 mmHg whose REVEAL risk score is less than 7. A more conservative approach may be elected for this patient since his predicted 1-year mortality rate would be less than 5%.
Patient with pulmonary hypertension and vasoresponsiveness
Unless contraindicated because of overt heart failure, vasoreactivity testing is mandatory for every patient with PAH during diagnostic right heart catheterization. The current American College of Chest Physicians (ACCP) definition of a vasoresponder is a drop in mean pulmonary artery pressure (mPAP) by at least 10 mmHg to an absolute level that is less than 40 mmHg without a simultaneous drop in cardiac output, during the administration of inhaled nitric oxide, intravenous adenosine, or intravenous epoprostenol. It is crucial to detect these vasoresponders because they have a much better prognosis than nonresponders when treated with high-dose calcium channel blocker (CCB) therapy [Sitbon et al. 2005]. Typically these patients will not require the more expensive PAH-specific therapies currently available, allowing them to avoid their inherent difficult-to-tolerate side effects. Earlier definitions used to define vasoresponders proved not to define true long-term CCB responders, leaving many patients in jeopardy of irreversible disease progression [Sitbon et al. 2005]. The key to CCB therapy is to use it only in patients with PAH who meet the above-stated ACCP definition and very close monitoring because some of these patients will not improve or even progress and thus will require more advanced therapies with time [Sitbon et al. 2005].
Unfortunately, less than 10% of patients with IPAH will demonstrate this degree of vasoreactivity [Sitbon et al. 2005]. The role of vasodilator testing in other associated forms of PAH is less clear [Montani et al. 2010]. Experience from the French Referral Center for Pulmonary Hypertension suggests that heritable and anorexigen forms of PAH may also have similar rates of vasoresponsiveness and may also have durable responses to long-term high-dose CCB therapy. However, PAH forms associated with HIV, congenital heart disease, pulmonary veno-occlusive disease, and pulmonary capillary hemangiomatosis do not typically demonstrate such vasoresponsiveness [Montani et al. 2010]. Portopulmonary-associated PAH rarely demonstrates such reactivity on vasodilator testing. Even if present, most experts consider high-dose CCB therapy contraindicated in this subset of patients because these agents increase the risk of worsening the portal hypertension [Rodriguez-Roisin et al. 2004].
Individualization of treatments according to risk stratification
Choice of therapy
The choice of the optimal therapy for a specific patient continues to be based as much on artistry as it is on pure science. However, reasonable approaches have already been published (Figure 1) [McLaughlin and McGoon, 2006; Fares et al. 2011], which can guide patient-specific therapies. High-risk patients as defined by the REVEAL calculator or other published determinants of risk (Tables 3 and 4) should be started on a prostanoid agent ideally delivered by infusion. The addition of either a PDE-5 inhibitor or an ERA is more controversial but data from the pulmonary arterial hypertension combination study of epoprostenol and sildenafil (PACES) trial [Simonneau et al. 2008] suggest that addition of high-dose sildenafil up to 80 mg three times daily to optimize intravenous epoprostenol improved exercise tolerance and decreased rates of clinical worsening compared with intravenous epoprostenol alone. Patients with PAH with determinants of lower risk (Table 3) can be initiated on oral agents with close monitoring and follow up. This would include patients in WHO functional classes II and III. There are currently no head-to-head data to suggest that one oral agent is superior to another as initial monotherapy. The availability of multiple validated indicators of disease severity and progression makes using such algorithms extremely helpful and potentially lifesaving. The early initiation of PAH-specific therapy in patients with functional class II disease is felt to result in slowed disease progression and improved long-term outcomes [Galie et al. 2008b].

Pulmonary arterial hypertension management algorithm. Modified with permission from [McLaughlin and McGoon, 2006].
Similar to goal-directed therapy that is the standard of care in patients with sepsis [Rivers et al. 2001], such an approach resonates for many PAH experts but lacks strong evidence supporting its routine implementation. Such an approach would guide therapy choice and titration based on set goals such as functional class, 6MW distance, hemodynamic thresholds, and quality of life assessments [Hoeper et al. 2005].
Reasonable goals to achieve in patients with PAH include WHO functional class I or II, 6MW distance of more than 380 m, a cardiac index of at least 2.2–2.5 l/min/m2, right atrial pressure of less than 10 mmHg, normal right ventricular size and function on echocardiography with a tricuspid annular plane systolic excursion (TAPSE) of more than 1.8 cm [Forfia et al. 2006], and a BNP level of less than 180 pg/ml [Sitbon et al. 2002; McLaughlin et al. 2002; Wensel et al. 2002]. Objective measures of quality of life and dyspnea scores should also be part of these goals. Therapy should ideally be escalated until the above goals are achieved. The key to the success of this approach is setting reasonable expectations in which the patient is directly involved, with close monitoring and frequent follow up. Normalizing a longstanding severely dilated and dysfunctional right ventricle might not be practical at some point since a significant proportion of this dysfunction might be due to irreversible fibrosis of the right ventricular myocardium. This underscores the importance of early and aggressive therapy to avoid irreversible remodeling. One such study that lends weight to such an approach is the COMPASS-3 trial in which sildenafil was added to bosentan in patients with PAH who failed to meet a goal of 6MW distance of 380 m on bosentan monotherapy [Benza et al. 2010b].
Despite improvement in survival rates of pregnant women with PAH in recent years [Kiely et al. 2010; Bedard et al. 2009], mortality rates in these patients continue to be unacceptably high. Thus pregnancy is a relative contraindication in women with PAH. This increased mortality is mostly related to increased cardiac output which is mediated by increased blood volume/stroke volume early on, decreased peripheral vascular resistance/decreased afterload, and increased heart rate later in pregnancy. These changes worsen PH because of inability to decrease pulmonary vascular resistance to accommodate the above physiologic changes in pregnancy in patients with pre-existing PH [Lane and Trow, 2011]. These elevations in the pulmonary artery pressures are augmented by the sudden increase in relative blood volume immediately postpartum due to diversion of blood away from the uterus.
This is further complicated by the fact that ERAs are contraindicated in pregnancy because of their potential teratogenicity (both ambrisentan and bosentan are category X in pregnancy). PDE-5 inhibitors and prostanoid analogues (except for iloprost), however, are pregnancy risk category B (i.e. there is no documented evidence of harm in humans, but teratogenic effects were not seen in animals). Inhaled iloprost, however, showed teratogenicity in some but not all animal studies and thus should be avoided in pregnant women. Based on this, PDE-5 inhibitors, epoprostenol, and treprostinil could be used with caution in pregnant women with PAH and then only if the benefit is perceived to outweigh the risks.
In view of this high mortality and the potential teratogenicity of some PAH medications (such as ERAs, vitamin K antagonists, and aldactone), two different kinds of contraception are recommended for patients with child-bearing potential. It is important to keep in mind that combined hormonal contraceptives increase the risk of thromboembolic disease on top of the already increased procoagulant state in pregnancy [Lane and Trow, 2011]. A barrier method is recommended to be one of the two contraceptive methods used. Counseling and detailed discussions about each contraceptive method in addition to discussing other options such as surrogate mothers should be addressed in detail with women of childbearing age. In the event of unplanned pregnancy or newly diagnosed moderate to severe PAH during pregnancy, therapeutic abortion should be one of the options provided to the patient.
Intense patient and family education are key to the successful management of patients with PAH. It is universally frightening to patients to receive a diagnosis of “PH” because of historically poor outcomes. Providing education, support, and motivation to the patients and their families, partners, and healthcare givers can define a successful management plan with the potential for improved outcomes.
Selection of therapy based on comorbidities
One potentially serious adverse effect of ERAs is hepatotoxicity [Humbert et al. 2007]. Despite this, there are anecdotal pieces of evidence, including small series, supporting the safety of ERAs in patients with liver disease (i.e. portopulmonary hypertension) [Cartin-Ceba et al. 2011; Hinterhuber et al. 2004]. A practical approach would be to use ERAs as a second-line therapy if PDE-5 inhibitors fail to be adequate in those patients who have not yet required prostanoid therapy. Alternatively, ERAs may be used as third-line agents in those whose condition has failed to respond to dual therapy with a PDE-5 inhibitor and an inhaled prostanoid and who are not accepting or intolerant of subcutaneous or intravenous prostanoid therapy. If ERA therapies are elected in portopulmonary hypertension, close monitoring of liver function tests (LFTs) is advised and the dose of these agents should be reduced or they should be stopped altogether if LFTs exceed three times the upper limit of normal. These agents are not currently FDA approved for this indication, and there is a theoretical advantage for ambrisentan compared with bosentan in this subpopulation in view of the lower liver toxicity seen with ambrisentan.
Patients who require nitrates for coronary heart disease should not be on PDE-5 inhibitors because of potentially excessive systemic hypotension. Both drugs increase cGMP and thus cause vasodilation via relaxation of vascular smooth muscle [Rybalkin et al. 2003]. Likewise, use of direct α receptor blocking agents used to treat benign prostatic hyperplasia such as tamsulosin, alfuzosin, terazosin, doxazosin, or silodosin with PDE-5 inhibitors is relatively contraindicated as systemic hypotension may also ensue.
Having three different classes of therapies with nine different FDA-approved medications for PAH treatment (Table 2) provides multiple options for combination therapy. Multiple trials have looked at the effects of these various combination therapies in PAH [Mukherjee and Howard, 2011]. The specifics of the combination trials are beyond the scope of this paper, but combination therapy is commonly used in real-world clinical practice as seen in the REVEAL registry, in which two out of five patients with PAH were on at least two PAH-specific therapies at the time of enrollment [Badesch et al. 2010]. There is growing evidence to support a combination therapy approach [Mukherjee and Howard, 2011], which in theory offers synergistic effects of drugs that act on different pathways.
Choice of the route of prostanoid therapy
Patients with more severe PAH that requires aggressive upfront therapy have a few options. The only available prostanoid option until recently was intravenous epoprostenol [Rich, 1995]. Since its approval in 1996, three other prostanoids have become available, and inhalational and subcutaneously infused routes have also become options. There are no head-to-head trials that provide evidence about the superiority or relative safety of these different options but each of them has its own advantages and disadvantages [Gomberg-Maitland and Olschewski, 2008].
The biggest advantage of intravenous prostanoids is the guaranteed bioavailability of the drug. Its main disadvantage, however, is the need for a permanent indwelling central venous access catheter (connected to a portable infusion pump system) with its associated potential complications, including infection and thrombosis. Although epoprostenol is the only PAH-specific therapy that has been shown to improve survival in patients with PAH [Barst et al. 1996], its main drawback is its very short half life [Vane and Botting, 1995]. This becomes critical because of adverse reactions that may occur in the event of inadvertent abrupt discontinuation of the infusion [Cuiper et al. 1996; Kingman et al. 2010], including rebound PH, arrhythmia, and even death. Treprostinil (Remodulin, United Therapeutics Corp.) is a prostanoid with a longer half life than epoprostenol, offering less risk in the event of pump failure or inadvertent interruption of infusion. The longer half life of treprostinil, while an advancement in PAH therapeutic safety, has encouraged patients to transiently discontinue their infusions (e.g. when they shower), resulting in more tubing changes and catheter manipulation, which some believe has contributed to a higher rate of catheter contamination and infections [Ivy et al. 2009]. The other factor that probably has contributed to a higher risk of infection with treprostinil infusion is its lower pH compared with increased alkalinity of the epoprostenol solution which has antibacterial effects [Rich et al. 2012; Zaccardelli et al. 2010]. The bioavailability of subcutaneously infused prostanoids also offers an advantage in eliminating the risk of bloodstream infection. Unfortunately up to 80% of patients treated with continuous subcutaneous treprostinil infusion endure pain at the site of the subcutaneous infusion site.
The availability of inhaled prostanoids was a potential breakthrough in PAH management since they do not require invasive catheters. However, the intermittent nature of exposure of the pulmonary vascular bed with peak and trough levels with this mode of delivery may make them less effective agents. In addition, pharmacokinetic data for treprostinil, for example given at the maximal FDA approved dose of 9 breaths four times a day, provides maximal plasma concentrations that are comparable to intravenous or subcutaneous treprostinil infused at 15 ng/kg/min [Voswinckel et al. 2006]. This is a relatively low dose but could be effective in a select few patients with PAH. In clinical practice, inhaled prostanoids do not seem to be as effective as intravenous or subcutaneous therapy [Opitz et al. 2005], but they remain an option for those who are not ready to commit to intravenous or subcutaneous therapy. These patients should be willing to take the inhalation four to nine times a day, depending on the specific inhaled prostanoid used [Olschewski et al. 2002; Benza et al. 2011b].
It can be challenging to anticipate patient compliance with each of the above modalities, and likewise a challenge to anticipate and manage complications inherent with each modality. At least part of the infectious complications can be related to patients’ (or their caregiver’s) strict adherence to sterile technique in the care of the central venous catheter. Patient dexterity is also important. For example, in patients with scleroderma and advanced sclerodactyly, handling vials and pumps can be a challenge. Providing an indwelling catheter or long-term intravenous access to a patient with a history of illicit drug use requires careful preconsideration. A patient’s tolerance to pain is a key factor in a patient who is being considered for subcutaneously infused prostanoids. Such pain is typically managed with local analgesics, anti-inflammatory medications or antihistamines [Mathier et al. 2010], and typically this pain lessens with time. These and other factors will influence the kind and route of the prostanoid therapy offered.
Conclusion
A lot of therapeutic advances have occurred in the past two decades in the field of PAH that have allowed improved exercise tolerance and improved outcomes for these patients. Patients with IPAH who are fortunate enough to meet ACCP criteria for vasodilator responsiveness should be treated with high-dose CCB therapy and generally enjoy excellent survival rates. Patients with IPAH without contraindications to anticoagulation should also be maintained on warfarin therapy. The remaining 90% of patients with IPAH and WHO group 1 patients with associated forms of PAH require individualized approaches to therapy. Patients with WHO functional class IV and those with determinants of high risk (Table 3) should be treated upfront ideally with an infused prostanoid. Addition of a PDE-5 inhibitor is controversial but there is some evidence to support this approach [Simonneau et al. 2008]. Addition of an ERA to an infused prostanoid is often done in clinical practice but double-blind, RCT data to support this approach are lacking. Patients with WHO functional class IV or those with determinants of high risk should be referred for transplant candidacy evaluation early after diagnosis and disease stabilization. Patients with WHO functional class II and III status have a variety of options as initial therapy (Figure 1) and individualization based on patient preferences, comorbidities, and drug–drug interactions is required.
No head-to-head studies of initial oral therapies have been conducted. After initial monotherapy has been started, close follow up is required with specific goals of therapy in mind. Specifically, decreasing WHO functional class to class I or II, improving 6MW distance to at least 380 m, improving right ventricular function and dilatation ideally with a TAPSE of greater than 1.8 cm, improving BNP levels to less than 180 pg/ml, improving cardiac index to more than 2.2 liters/min/m2, and lowering right atrial pressure to less than 10 mmHg are all laudable goals associated with improved survival. The concept of combination therapy to achieve these goals is often endorsed but further investigation of what agent to add and in what order is needed [Taichman, 2008]. Studies of upfront combination therapy are ongoing, such as AMBITION (ambrisentan and tadalafil) and COMPASS-2 (bosentan and sildenafil) trials. Patients not responding to aggressively escalated therapies should be referred to experienced centers for consideration of atrial septostomy or lung transplantation. Women with PAH should be counseled to avoid pregnancy and to use two forms of birth control if sexually active, given the high mortality rates associated with carrying a child to term.
Sadly, PAH continues to be an incurable disease. The relative rarity of this disease and the variety of primary etiologies that lead to its phenotype make finding unifying pathways and thus targets for therapy a huge challenge. However, in a relatively short period of time over the past two decades, multiple therapies have been developed to treat PAH and many more are in the pipeline [Girgis, 2010] offering hope for these patients.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Dr Terence K. Trow has served on advisory boards for Gilead Sciences, Actelion, United Therapeutics, and Bayer AG pharmaceuticals. He is also on Speaker’s Bureaus for Gilead Sciences, Actelion, and United Therapeutics pharmaceuticals. The University of North Carolina in Chapel Hill has received research funding from Gilead for a study on which Dr Wassim H. Fares is the Principal Investigator.