
Abstract
The skin is the first line of defense to cutaneous microbes and viruses, and epidermal keratinocytes play a critical role in preventing infection by viruses and pathogens through activation of the type I interferon (IFN) response. Using RNAseq analysis, here we report that the conditional deletion of C/EBPβ transcription factor in mouse epidermis (CKOβ mice) resulted in the upregulation of IFNβ and numerous keratinocyte interferon-stimulated genes (ISGs). The expression of cytosolic pattern recognition receptors (cPRRs), that recognize viral RNA and DNA, were significantly increased, and enriched in the RNAseq data set. cPRRs stimulate a type I IFN response that can trigger cell death to eliminate infected cells. To determine if the observed increases in cPRRs had functional consequences, we transfected CKOβ primary keratinocytes with the pathogen and viral mimics poly(I:C) (dsRNA) or poly(dA:dT) (synthetic B-DNA) that directly activate PRRs. Transfected CKOβ primary keratinocytes displayed an amplified type I IFN response which was accompanied by increased activation of IRF3, enhanced ISG expression, enhanced activation of caspase-8, caspase-3 and increased apoptosis. Our results identify C/EBPβ as a critical repressor of the keratinocyte type I IFN response, and demonstrates that the loss of C/EBPβ primes keratinocytes to the activation of cytosolic PRRs by pathogen RNA and DNA to induce cell death mediated by caspase-8 and caspase-3.
Introduction
The epidermis, the outermost layer of the skin, is the first line of defense against pathogens, microbes, and environmental insults. 1 Not only is the epidermis a physical barrier, but epidermal keratinocytes sense infection and environmental insults that initiate and shape local immune responses. Four major subfamilies of pattern recognition receptors (PRRs) are expressed in keratinocytes consisting of Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)- Leucine Rich Repeats (LRR)-containing receptors (NLR), retinoic acid-inducible gene I (RIG-I) -like receptors (RLR), and C-type lectin receptors (CLRs).2–5 Upon sensing pathogen-associated molecular patterns (PAMPs) expressed by pathogens and viruses and host damage-associated molecular patterns (DAMPs), keratinocyte PRRs activate signaling pathways including the type I interferon (IFN) response.2,4,6 Activation of the keratinocyte type I IFN response results in production and secretion of IFNs that activate the heterodimeric transmembrane IFNα/β receptor (IFNAR) on the keratinocyte surface, leading to transcription of hundreds of IFN-stimulated genes (ISGs). The up-regulation of ISGs is widely recognized for its critical role in preventing infection by viruses and other pathogens. 6
The CCAAT/enhancer binding protein-β (C/EBPβ), a basic leucine zipper transcription factor, is expressed in a variety of cell types and regulates tissue-specific gene expression in a hormone, growth factor, cytokine and nutrient dependent manner.7,8 Epidermal keratinocytes are one of the most highly C/EBPβ expressing cell types.9,1°C/EBPβ has critical roles in fundamental cellular processes including differentiation, inflammation, survival, and energy metabolism.11–16 However, it is unknown if C/EBPβ regulates skin innate immunity in response to pathogen and viral mimics.
To model the actions of viruses and pathogens in keratinocytes, we utilized poly(I:C), a synthetic analog of dsRNA, and poly(dA:dT), a repetitive dsDNA synthetic analog of B-DNA.17,18 Outside of the cell, poly(I:C) is sensed by TLR3. dsRNA is a replication intermediate for RNA viruses, and when poly(I:C) is transfected into the cell it is sensed by cytosolic PRRs RIG-I and MDA-5. RIG-I and MDA-5 signal through the mitochondrial antiviral signaling protein MAVS, leading to the production of type I IFNs. When transfected into the cell, poly(dA:dT) is sensed by cytosolic DNA sensors including cGAS, AIM2, DAI (ZBP1), DDX41, and IFI16. 19 A key mediator of cytosolic DNA receptors signaling is the adaptor protein stimulator of interferon genes (STING) that triggers the production of type I IFNs.20,21 Poly(I:C) and poly(dA:dT) were used to directly activator cytosolic PRRs to determine the functional impact of the deletion of C/EBPβ on regulation of the skin innate immune type I IFN response and regulation of keratinocyte cell death.
Results
Conditional deletion of C/EBPβ in epidermis results enrichment of type I IFN pathways
The C/EBPβ transcription factor is highly expressed in keratinocytes of the epidermis. 9 To further investigate cellular processes in the skin regulated by C/EBPβ, we conducted RNAseq analysis on RNA isolated from epidermis of 4 K5Cre+/tg (Cre) and 3 K5Cre+/tg;C/ebpβflox/flox (CKOβ) mice. In this model, the keratin 5 (K5) promoter directs Cre recombinase expression to the epidermis to delete the floxed gene (44, 45). The deletion of C/EBPβ in the epidermis was confirmed by immunoblot analysis (Figure 1A). The conditional deletion of C/EBPβ in the epidermis had a significant impact on transcriptional responses. We identified a total of 2586 differentially expressed genes (1202 up-regulated, and 1384 down-regulated genes out of a data set of 20,400 genes) that were significantly altered in the epidermis of CKOβ mice compared to Cre mice (FDR p < 0.1) (Figure 1B and Supplemental Table 1). Overall, expression patterns in CKOβ mice were similar and distinct from expression patterns observed in Cre mice (Figure 1B). An annotated volcano plot analysis indicates C/ebpβ and Krtap19 as some of the most down regulated genes with H2-ea-ps and Nudt11 as the most upregulated genes in CKOβ epidermis (Figure 1C). Gene set enrichment analysis (GSEA) was performed using gene sets from within the Molecular Signature Database (MSigDB). Strikingly, GSEA revealed the IFN pathway was the most highly enriched pathway in the CKOβ epidermis (FDR ≤ 0.1) (Figure 1D). Using eXploring Genomic Relations (XGR) analysis using Reactome Ontology set from MSigDB and comparing Cre to CKOβ mice, we observed that ‘Interferon alpha/beta signaling’ was the most enriched pathway CKOβ mice (FDR =8.36E-8) (Figure 1E).

CKOβ epidermis displays significant alterations in gene expression and enrichment of type I interferon pathway. (A) Immunoblot analysis for C/EBPβ and β-actin in epidermal protein lysates from Cre and CKOβ mice. (B) Total RNA was isolated from the epidermis Cre and CKOβ mice and was subjected to RNA sequencing. Row-scaled heatmap showing total gene expression, with each column representing RNA isolated from a single mouse. Left DEG column indicates statistically up-regulated (top;green, 1202 genes) and down-regulated (bottom; purple, 1384 genes) and non-significant (middle; grey) genes in CKOβ mice compared to Cre control mice (FDR < 0.1). (C) Volcano Plot of RNAseq data in CKOβ mice (FDR < 0.1). (D) Results from gene set enrichment analysis with top 20 statistically significant (FDR < 0.1) positively enriched pathways displayed. (E) Reactome pathway enrichment results (top 10) in epidermis of CKOβ compared to Cre control.
Conditional deletion of C/EBPβ results in increased expression of IFNβ and subset of ISGs
Among 2586 differently expressed genes between CKOβ and Cre mice, we observed that 15 out of the top 75 (20%) most upregulated genes in CKOβ epidermis were interferon-stimulated genes (ISGs) (Supplementary Table 1). Further analysis of the MSigDB combined IFN ontologies showed a selective upregulation of ISGs as 24 out of 39 ISGs were significantly altered in CKOβ epidermis (FDR ≤ 0.1) (Figure 2A). Real-Time qPCR analysis revealed expression of Ifnβ was induced ∼ 30-fold in CKOβ epidermis compared to the control epidermis (Figure 2B). In addition to the upregulation of Ifnβ, TaqMAN Real-Time qPCR analysis confirmed the RNAseq findings that CKOβ mouse epidermis display significant increases in a subset of ISGs including cytosolic pattern recognition receptors (PRRs, including Dhx58 (LGP2), Zbp1, Ddx58 (RIG-I), and Ifi16), antiviral proteins (including Ifit1, Ifit2, Isg15 and Mx1) and regulators of IFN signaling (Stat1 and Stat2) (Figure 2C). Together, these results uncover a previously unknown function for C/EBPβ in regulating basal keratinocyte expression of Ifnβ and a subset of ISGs. Earlier studies in UVB-treated mouse epidermis revealed an increase in ISGs 22 ; the current results indicate that the increase in most ISGs is independent of UVB treatment and represents an intrinsic change in the innate immune system in keratinocytes.

Deletion of C/EBPβ enhances keratinocyte type I IFN system. (A) Heatmap of genes differentially expressed in combined Interferon α/β signaling pathway ontologies (enrichment FDR = 8.3E-8). Left DEG column indicates statistically up-regulated (top;green) and down-regulated (bottom; purple) and non-significant (middle; grey) genes in CKOβ mice compared to Cre control mice (FDR < 0.1). Each column represents RNA isolated from a single mouse. (B) TaqMAN Real-Time PCR analysis for Ifnβ expression in Cre and CKOβ mouse epidermis. (C) TaqMan Real-Time PCR analysis for ISG expression in Cre and CKOβ mouse epidermis. Data are expressed as the mean ± SD N ≥ 3 mice. * denotes p-value <0.05.
Loss of C/EBPβ results in an enhanced innate immune response to direct activators of cytosolic PRRs
We reasoned that increased basal expression of cytosolic PRRs in CKOβ mice may prime and enhance the keratinocyte innate immune response to viral or pathogen RNA or DNA. C/EBPβ deficient keratinocytes displayed significant upregulation of the RIG-I-like family of cytosolic RNA receptors, including RIG-I (Dhx58) and LGP2 (Ddx58) and upregulation of cytosolic DNA receptors, including Ifi16, and Zbp1 (Figure 2C). To determine the functional impact of the deletion of C/EBPβ on regulation of the skin innate immune type I IFN response, Cre and CKOβ primary keratinocytes were transfected with poly(I:C) and poly(dA:dT) to promote detection by cytosolic PRRs and minimize detection by cell surface TLRs.17–21 The activation of cytosolic PRRs leads to activation and translocation of transcription factors including IRF3 to the nucleus that subsequently induce transcription of interferons. 6 Compared to similarly treated control keratinocytes, poly(I:C) treated CKOβ keratinocytes displayed a ∼ 2-fold increase in phosphorylation of IRF3, despite no significant differences in total IRF3 protein levels (Figure 3A, quantification not shown). Immunoblot analysis for C/EBPβ confirmed deletion of C/EBPβ, and in the Cre keratinocytes we detected a doublet corresponding to LAP* 37 kDa, and LAP 35 kDa (Figure 3A). In addition to enhancing activation (phosphorylation) of IRF3, CKOβ primary keratinocytes transfected with poly(I:C) resulted in significant increases in Ifnβ transcript levels (Figure 3B), increases in secretion of IFNβ protein (Figure 3C) and increases in expression of numerous interferon stimulated genes (ISGs) compared to similarly treated control keratinocytes (Figure 3D). Poly(dA:dT) is recognized by cytosolic DNA sensors.5,17–21 Compared to similarly treated control keratinocytes, CKOβ primary keratinocytes transfected with poly(dA:dT) also displayed significant increases in expression levels of Ifnβ (Figure 3E), increases in secretion of IFNβ protein (Figure 3F), and increases in numerous ISGs compared to similarly treated control keratinocytes (Figure 3G). These results indicate that C/EBPβ regulates the type I IFN response and that the loss of C/EBPβ results in an enhanced innate immune response to direct activators of RNA and DNA sensing cytosolic PRRs.
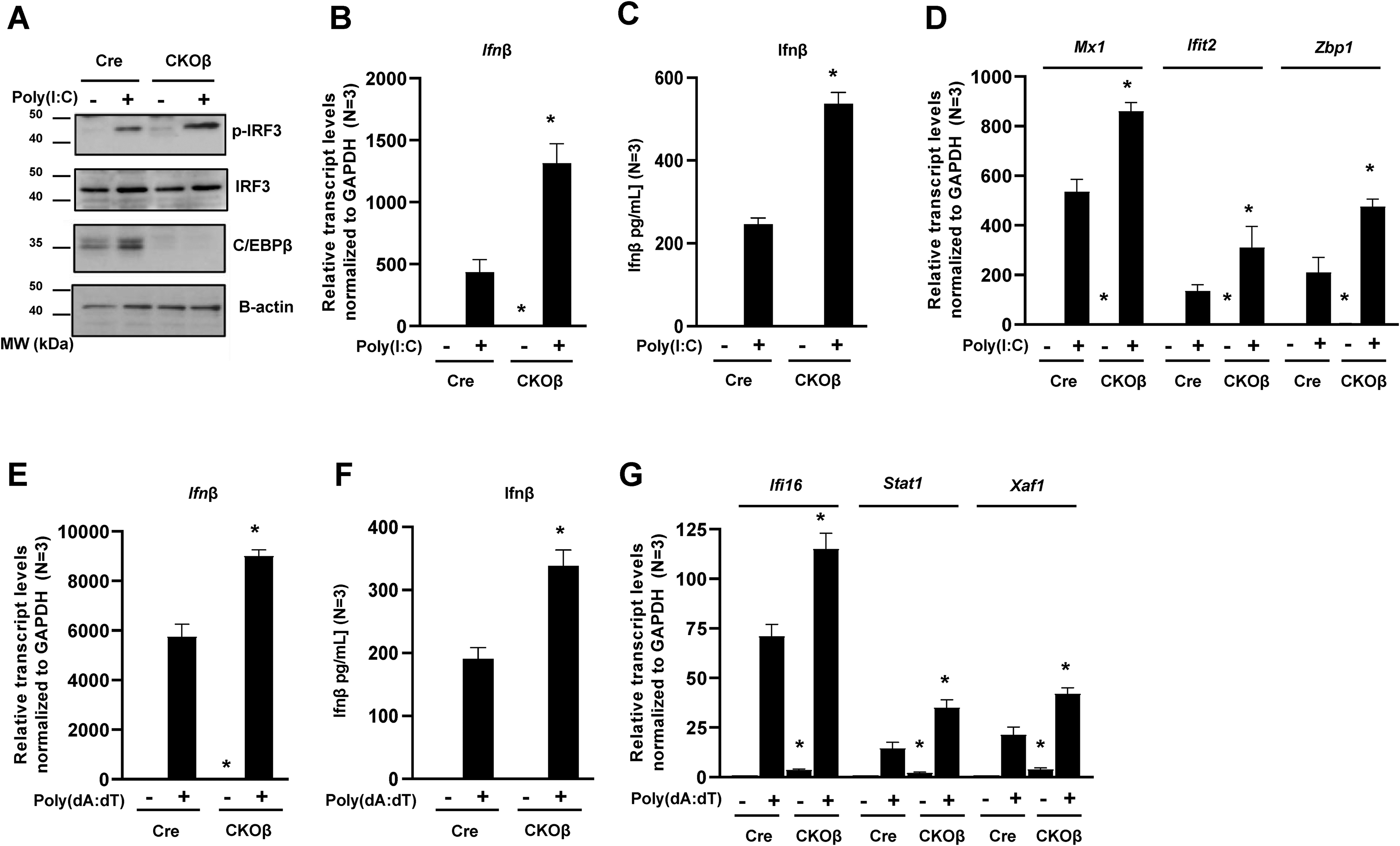
Deletion of C/EBPβ enhances the innate immune response to direct activators of cytosolic PRRs. (A) Cre and CKOβ primary mouse keratinocytes in culture were transfected with 800 ng/ml poly(I:C)-Lyovec. Cells were collected 8 h post transfection. Immunoblot analysis for C/EBPβ (the doublet is LAP* 37 kDa, and LAP 35 kDa), p-IRF3, IRF3, and β-actin in poly(I:C)-Lyovec treated cells. (B) Cre and CKOβ primary mouse keratinocytes were transfected with 800 ng/ml poly(I:C)-Lyovec. Cells were collected 18 h post transfection. TaqMAN Real-time PCR analysis for Ifnβ. (C) Cre and CKOβ primary mouse keratinocytes were transfected with 1600 ng/ml poly(I:C)-Lyovec. Growth media from cultured keratinocytes was collected 18 h post transfection. Secreted IFNβ protein levels were measured from cell supernatant by ELISA. (D) Cre and CKOβ primary mouse keratinocytes were transfected with 800 ng/ml poly(I:C)-Lyovec. Cells were collected 18 h post transfection. TaqMAN Real-Time PCR analysis for various ISGs. (E) Cre and CKOβ primary mouse keratinocytes were transfected with 2 μg/ml poly(dA:dT)-Lyovec. Cells were collected 18 h post transfection. TaqMAN Real-time PCR analysis for Ifnβ. (F) Cre and CKOβ primary mouse keratinocytes were transfected with 2 μg/ml poly(dA:dT)-Lyovec. Growth media from cultured keratinocytes was collected 18 h post transfection. Secreted IFNβ protein levels were measured from cell supernatant by ELISA. (G) Cre and CKOβ primary mouse keratinocytes were transfected with 2 μg/ml poly(dA:dT)-Lyovec. Cells were collected 18 h post transfection. TaqMAN Real-Time PCR analysis for various ISGs. Data are expressed as the mean ± SD N ≥ 3. * denotes p-value <0.05 compared to similarly treated control.
Deletion of C/EBPβ sensitizes keratinocytes to direct activators of cytosolic PRRs to induce caspase activation and cell death
C/EBPβ has been shown to regulate cell survival in response to DNA damage, toxicants or oncogenic stress.22–27 Inducing cell death and eliminating infected cells is an important component of immunity. The deletion of C/EBPβ and subsequent upregulation of the type I IFN response could sensitize keratinocytes to cell death. To test this we transfected Cre control and CKOβ primary keratinocytes with poly(I:C) or poly(dA:dT) to active cytosolic PRRs and the type I IFN response. Poly(I:C) and poly(dA:dT) transfected CKOβ primary keratinocytes display enhanced cell death as measured by Annexin V and PI staining of primary keratinocytes (Figure 4A). Treatment with poly(I:C) resulted in a ∼3-fold increase in cell death and poly(dA:dT) treatment resulted in a ∼2.5-fold increase in cell death compared to similarly treated controls (Figure 4A). Poly(I:C) and poly(dA:dT) transfected CKOβ primary keratinocytes displayed striking increases in cleavage (activation) of caspase-8 (Figure 4B-E). Caspase-8 is critical in numerous cell death pathways including extrinsic apoptosis and is activated by cleavage.28–30 Two active caspase-8 pools can be generated: a membrane associated pool of caspase-8 known as p43 and a cytosolic pool of casapase-8 known as p18. 31 CKOβ primary keratinocytes display increased levels of the p43 fragment and the p18 fragment of caspase-8 (Figure 4B-E). The enhanced activation of caspase-8 in CKOβ primary keratinocytes was coupled with increased cleavage of caspase-3 (Figure 4B-E). We observed the enhanced activation of caspase-8 and caspase-3 in CKOβ primary keratinocytes was dose-(Figure 4B and 4D) and time-dependent (Figure 4C and 4E). When primary keratinocytes were treated with poly(I:C) that was not transfected into the cell (“naked poly(I:C)”), CKOβ primary keratinocytes displayed enhanced activation of caspase-8 and caspase-3 compared to control keratinocytes (Figure 4F). However, this observed activation of caspases was diminished compared to the response when poly(I:C) was transfected into the cell (“Lyovec”, Figure 4F). These results indicate that delivery of pathogen and viral mimics to the cytoplasm enhances the cell death response and suggests that the overall priming of the basal type I IFN response in CKOβ primary keratinocytes results in an elevated cell death response to pathogen and viral mimics that stimulate TLRs.
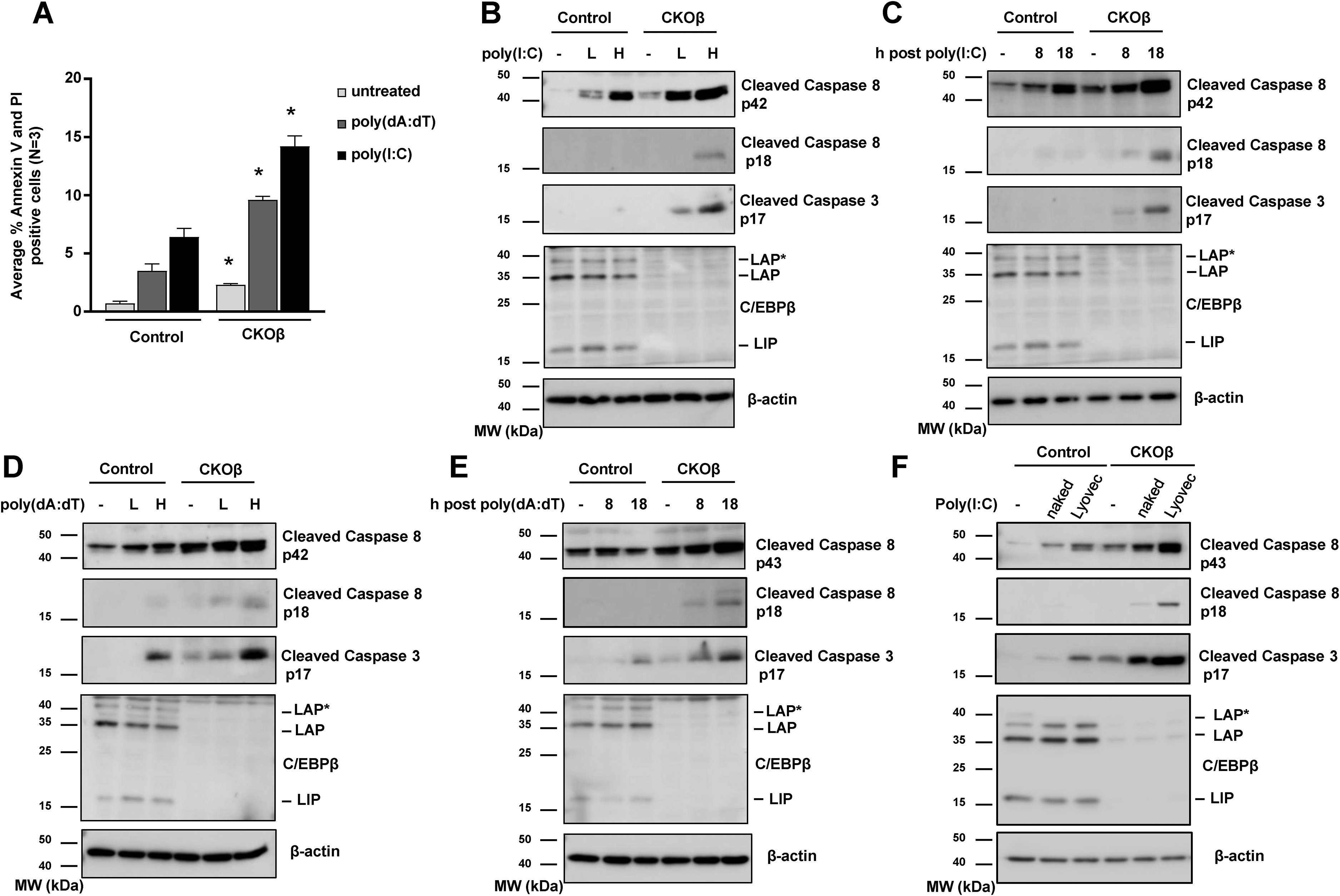
Deletion of C/EBPβ enhances activation of caspase-8 and apoptosis in response to direct activators of cytosolic PRRs. (A) Cre and CKOβ primary mouse keratinocytes were transfected with 1600 ng/ml poly(I:C)-Lyovec or 2 μg/ml poly(dA:dT)-Lyovec. 18 h post treatment cells were stained with annexin-V and PI and analyzed by FACS. Data are expressed as the mean + SD N > 3. * denotes p-value <0.05 compared to similarly treated control. (B) Cre and CKOβ primary mouse keratinocytes were transfected with 800 ng/ml (low dose, labeled as L) or 1600 ng/ml poly(I:C)-Lyovec (high dose, labeled as H) and collected 18 h post transfection. Immunoblot analysis for C/EBPβ, cleaved caspase-8, cleaved caspase-3 and β-actin. (C) Cre and CKOβ primary mouse keratinocytes were transfected with 1600 ng/ml poly(I:C)-Lyovec and collected at 8 h and 18 h post transfection. Immunoblot analysis for C/EBPβ, cleaved caspase-8, cleaved caspase-3 and β-actin. (D) Cre and CKOβ primary mouse keratinocytes were transfected with 1 μg/ml (low dose, labeled as L) and 2 μg/ml poly(dA:dT)-Lyovec (high dose, labeled as H) and collected at 18 h post transfection. Immunoblot analysis for C/EBPβ, cleaved caspase-8, cleaved caspase-3 and β-actin. (E) Cre and CKOβ primary mouse keratinocytes were transfected with 2 μg/ml poly(dA:dT)-Lyovec and collected at 8 h and 18 h post transfection. Immunoblot analysis for C/EBPβ, cleaved caspase-8, cleaved caspase-3 and β-actin. (F) Cre and CKOβ primary mouse keratinocytes were treated with1600 ng/ml poly(I:C) (labeled “naked”) or transfected with 1600 ng/ml poly(I:C)-Lyovec (labeled as “Lyovec”). Cells were collected 18 h post treatment. Immunoblot analysis for C/EBPβ, cleaved caspase-8, cleaved caspase-3 and β-actin.
C/EBPβ is highly expressed in human keratinocytes. To determine whether C/EBPβ could regulate survival of normal human epidermal keratinocytes (NHEKs) exposed to pathogen and viral mimics, we transfected C/EBPβ knockdown and control NHEKs with poly(I:C). Similar to mouse keratinocytes, the knockdown of C/EBPβ in NHEKs resulted in increased activation of caspase-8 and caspase-3 in poly(I:C) transfected NHEKs (Figure 5A). Taken together these results indicate that deletion of C/EBPβ sensitizes mouse and human epidermal keratinocytes to direct activators of cytosolic PRRs to induce cell death.

C/EBPβ-deficient normal human epidermal keratinocytes (NHEKs) display enhanced activation of caspase-8 and apoptosis in response to direct activators of cytosolic PRRs. (A) Normal human epidermal keratinocytes (NHEKs) were transfected with control or human C/EBPβ targeting siRNA. 48 h hours post transfection NHEKs were transfected with 800 ng/ml (low dose, labeled as L) or 1600 ng/ml poly(I:C)-Lyovec (high dose, labeled as H). Cells were collected at 8 h and 18 h post treatment and immunoblot analysis for human C/EBPβ (the doublet is LAP* 48 kDa, and LAP 46 kDa), cleaved caspase-8, cleaved caspase-3 and β-actin was conducted.
Discussion
We report that the conditional deletion of the C/EBPβ transcription factor in epidermal keratinocytes results in the upregulation of IFNβ and numerous ISGs in the epidermis, including cytosolic pattern recognition receptors (PRRs), antiviral proteins, and regulators of IFN signaling. We propose that enhanced basal expression of ISGs primes the type I IFN response in C/EBPβ deficient keratinocytes. Our results indicate that deletion of C/EBPβ results in an enhanced innate immune response to pathogen RNA or DNA as measured by enhanced expression of Ifnβ and multiple interrogated ISGs, and subsequent increased caspase mediated cell death. Earlier studies showed that deletion of C/EBPβ increased ISG expression in UVB-treated mouse epidermis and regressing skin tumors.22,24 However, these previous studies relied on DNA damage and; the current results indicate that the increase in most ISG is independent of UVB treatment and represents an intrinsic change in the innate immune system in keratinocytes.
C/EBPβ (also known as nuclear factor induced by IL-6 (NF-IL-6) has been shown to positively regulate numerous cell stress response pathways mediated by IL-6, TNFα, and IFNs.22,24,32,33 IFNα, β, and-γ have also been shown to induce C/EBPβ expression and enhance C/EBPβ's transcriptional activity in macrophages.34–36 Previous research indicating that C/EBPβ is a pro-inflammatory gene is in contrast with our reported findings that indicate that C/EBPβ is a suppressor of the keratinocyte type I IFN response. However, this previous work was conducted largely in macrophages, immune cells, and the liver whereas our work presented herein investigated the function of C/EBPβ in cultured primary mouse keratinocytes and in keratinocyte specific C/EBPβ knockout mice (CKOβ).32,33,37–41 C/EBPβ exhibits remarkable plasticity with respect to the range of transcriptional activities it participates in. 33 For example, depending on the tissue type, C/EBPβ has been shown to either stimulate or inhibit cellular proliferation.15,42–47 Similarly, the C/EBP family member C/EBPδ contributes to both pro-inflammation and anti-inflammation responses depending on the cell type and conditions.48,49 It is quite reasonable that C/EBPβ functions as a repressor or activator of stress response pathways, including the type I IFN response, in a cell specific manner.
The epidermis constitutes the outermost layer of the skin, and as such is the first line of defense to cutaneous pathogens, viruses and environmental DNA damage. Keratinocytes not only provide a physical barrier to infection and environmental insults through the formation of the stratum corneum but are also thought to function as sentinels of infection and damage that initiate and shape local immune responses. Epidermal keratinocytes are one of the most highly expressing C/EBPβ cell types.9,1°C/EBPβ can function as both a repressor and activator of gene transcription in part through regulation of chromatin marks and recruitment of the Mediator complex.50–53 C/EBPβ occupancy at the sites throughout the interferome could influence the active/repressive state of the promoter and modulate the transcriptional activities of ISG transcription factors to regulate the type I IFN system. It is also a distinct possibility that C/EBPβ functions to repress IFNβ itself as C/EBPβ in a heteromeric complex with C/EBPδ and CEBPζ can suppress transcription of IFNγ. 36 C/EBPβ can interact with the NfκB transcription factor, a critical regulator of IFNβ expression, to regulate cellular functions. 54 While beyond the scope of the functional consequences of loss of C/EBPβ on keratinocyte autonomous innate immunity described in the current study, future studies will focus on how C/EBPβ regulates the keratinocyte type I IFN response via protein-protein interactions or direct interactions with chromatin, specifically at gene promoters of the type I IFN pathway.
We report that deletion of C/EBPβ also results in enhanced activation of the IRF3 transcription factor, a known regulator of IFNβ transcription. The activation of cytosolic PRRs results in activation of the kinase TBK1 which phosphorylates IRF3 resulting in IRF3 activation and localization to the nucleus to induce IFNβ and ISG expression.4,6 We hypothesize that the increased activation of IRF3 is a result of a primed type I IFN response and increased activation of cytosolic PRRs due to the loss of C/EBPβ. We predict that multiple downstream targets of cPRR signaling including TBK1 will be affected in C/EBPβ deleted keratinocytes due to enhanced cPRR signaling. Further investigation on the role of cytosolic PRRs and TBK1 signaling in enhancing the type I IFN response and inducing cell death in C/EBPβ deleted keratinocytes is required.
Elimination of infected cells is a critical component of immunity. We show that treatment of C/EBPβ deleted keratinocytes with poly(I:C) or poly(dA:dT) resulted in enhanced activation of cPRRs and enhanced activation of caspase-8 and caspase-3 mediated cell death. Caspase-8 is the primary initiator caspase in cell extrinsic apoptosis and is typically initiated by extracellular stress signals including FasL, TNF and TNF-related apoptosis-inducing ligand (TRAIL).28,30,55 We did observe that the deletion of C/EBPβ in keratinocytes resulted in differences in key genes associated with the extrinsic apoptosis pathway, such as Tnfsf10 (TRAIL), Xaf1, and Cflar (c-FLIP) (data not shown).
In summary, we show that loss of C/EBPβ enhances the keratinocyte type I IFN response to regulate innate immunity and cell death and identify C/EBPβ as a critical regulator of the keratinocyte type I IFN response. Aberrant immune responses in the skin can be linked to diseases, as observed in inflammatory conditions such as atopic dermatitis, systemic lupus erythematosus (SLE) and psoriasis.56–58 Depending on the condition, increasing or decreasing C/EBPβ levels/activity to suppress or enhance an IFN-cell death response could yield positive therapeutic outcomes to restore proper regulation of the keratinocyte type I IFN response and cell death to treat cancer and autoimmune diseases.
Materials and methods
Mice
Conditional knockout mice; K5Cre+/tg;C/ebpβflox/flox (CKOβ) were obtained by crossing K5Cre (K5Cre+/tg) mice with C/ebpβflox/flox mice. 14 K5Cre mice were a gift from Angel Ramirez and Jose Jorcano. 59 All genotypes were backcrossed to SKH-1 hairless female mice (Charles River Labs) for at least five generations. 22 All aspects of animal care and experimentation described in this study were conducted according to the NIH guidelines and were approved by the NC State University Institutional Animal Care and Use Committee (IACUC).
Isolation of primary keratinocytes
Primary keratinocytes were isolated from newborn CKOβ and Cre SKH-1 mice (1–2 day old) by trypsin floatation. 60 Isolated epidermal cells were plated at 6 × 106 cells per 60 mm plate in Ca2+ free EMEM supplemented with 10% FBS and 4 ng/ml EGF for 12 h to enhance keratinocyte attachment. Cells were then gently washed with Mg2+ and Ca2+ free PBS twice to remove any remaining calcium and then cultured in Ca2 + free EMEM supplemented with 8% Chelex-treated FBS and 4 ng/ml EGF. Calcium chloride was then added to produce a final concentration of 0.05 mM Ca2+. After 4 days in culture, keratinocytes were treated and collected.
Cell treatments
Poly(dA:dT)-Lyovec (Invivogen), poly(I:C)HMW-Lyovec (Invivogen), and poly(I:C)HMW (Invivogen) were reconstituted following manufacturer's recommendations and cells were treated using the indicated concentrations for the indicated time.
Preparation of protein lysates and immunoblot analysis
Mice (8-10 week) were euthanized and dorsal skin was removed and subjected to 6 s heat shock in 60°C dH20 followed by 15 s in ice water. 22 Epidermis was quickly scraped from dermis and collected. Epidermal and cultured keratinocyte total protein lysates were prepared using RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM dithiothreitol, 1 mM sodium orthovanadate, 1 mM AEBSF, 1 x protease inhibitor cocktail (Roche) and 1x Halt phosphatase inhibitor cocktail (ThermoFisher) in PBS). SDS-PAGE and immunoblot analysis was conducted using the following antibodies against C/EBPβ (#3081 Cell Signaling), C/EBPβ (ab32358, Abcam), p-IRF3 (#29047, Cell Signaling), IRF3 (#4302 Cell Signaling), caspase-8 (#9746 Cell Signaling), cleaved caspase-8 (8592, Cell Signaling), cleaved caspase-3 (#9579 Cell Signaling) and β-actin (sc-8432 Santa Cruz). Antibodies were diluted in TBS containing 1% BSA and 0.1% Tween20. Membranes were imaged using an AI680RGB camera imager (Cytiva) and protein levels were quantified using ImageQuant TL 8.1 (Cytiva) and normalized to β-actin.
ELISA for Interferon-Beta
IFNβ secretion from Cre and CKOβ primary keratinocytes was measured in cell supernatants using Mouse IFN beta Quantikine enzyme-linked immunosorbent assay (ELISA) kit (MIFNB0, R&D Systems, Inc.). IFNβ concentrations were derived from the absorbance values measured at 450 nm with a correction at 540 nm using the Multiskan EX microplate spectrophotometer (ThermoFisher) and converted to concentration values based on the standards provided from each kit.
Flow cytometry
Cultured keratinocytes were washed with PBS and were detached from culture plates with trypsin and pelleted along with floating cells in the culture media. Pelleted cells were washed twice with ice-cold PBS and then resuspended in Annexin V Binding Buffer (BioLegend) at a concentration of 1 × 106 cells per ml. Five microliters of Annexin V APC (BioLegend) was added to 100 μl of the cell suspension, followed by the addition of 10 μl propidium iodide (PI) solution (BioLegend). Cells were incubated for 15 min at room temperature in the dark, followed by the addition of 400 μl Annexin V Binding Buffer. Cells were gated, and the median fluorescence intensities (MFIs) were determined with a flow cytometer (Accuri C6 Plus, BD Bioscience) and FlowJo software (BC Bioscience).
RNA and qRT-PCR
Total RNA was isolated from heat shock isolated mouse epidermis and from BALB/MK2 cells using QiaZOL lysis (Qiagen), followed by DNase I digestion. 22 cDNA was prepared from RNA by ImProm-II Reverse Transcription System (Promega), and gene expression was determined using the indicated TaqMAN Gene Expression Assays in combination with TaqMAN Fast Advance Master Mix (ThermoFisher). Data were analyzed using the comparative ΔΔCT method normalized to Gapdh.
RNA next-generation sequencing
Total RNA was extracted as described above from the epidermis of Cre and CKOβ mice (4 mice per genotype per treatment, 1:1 male female ratio). Total RNA samples were submitted to the North Carolina State University Genomic Sciences Laboratory for Illumina RNA library construction and sequencing. 24 In brief, quantified libraries were sequenced using Illumina's NextSeq 500 DNA sequencer, utilizing a 75 bp paired-end kit (Illumina), which gave around 200 million reads for the 8 samples (4 samples/group), which works out to ∼25 million reads/sample. The software package Real Time Analysis (RTA) was used to generate raw bcl, or base call files, which were then de-multiplexed by sample into fastq files for data submission.
RNA-Seq data analysis was conducted in consultation with the Bioinformatics Core of the NCSU Center of Human Health and the Environment. The quality of raw sequence data was assessed using FastQC and the first 12 poor-quality bases were trimmed based on the quality matrix from the FastQC application. The remaining good-quality reads were aligned to the mouse reference genome (mm38 version 87) using STAR aligner. 61 Per-gene counts of uniquely mapped reads for each replicate sample were calculated using the htseq-count script from the HTSeq python package. Genes with numerous aliases were removed and represented in the data as a single gene in the data. The count matrix was imported and normalized for sequence depth and distortion, and dispersion was estimated using DESeq2 Bioconductor package in the R statistical computing environment. 62 Differentially expressed genes were identified after applying multiple testing correction using the Benjamini–Hochberg procedure (false discovery rate (FDR) < 0.1). 63
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) software was used to show statistically significant differences between Cre and CKOβ datasets as previously described.24,64,65 Briefly, the genes from differential expression were ranked based on signed fold change − log10 p-value. The rank file contains genes with the strongest upregulation (top), strongest downregulation (bottom), and not changing are in the middle. The analysis was performed using Gsea Preranked application in the GSEA software using mouse C2 MSigDB gene set and default settings. Finally, the enriched pathways (FDR < 0.1) were plotted using Enrichment Map Visualization in the same software package and plotted the bar plot for the top 40 enriched pathways. For visualization of specific pathways, normalized counts from DESeq2 were row scaled (by gene), grouped by average linkage, and heatmaps generated. 65 Gene ontologies were combined from the curated C2.GSEA MSigDB database available at (http://software.broadinstitute.org/gsea/msigdb):
Supplemental Material
sj-xlsx-1-ini-10.1177_17534259231162192 - Supplemental material for C/EBPβ deficiency enhances the keratinocyte innate immune response to direct activators of cytosolic pattern recognition receptors
Supplemental material, sj-xlsx-1-ini-10.1177_17534259231162192 for C/EBPβ deficiency enhances the keratinocyte innate immune response to direct activators of cytosolic pattern recognition receptors by John S. House, Sophia Gray, Jennifer R. Owen, Dereje D. Jima, Robert C. Smart and Jonathan R. Hall in Innate Immunity
Footnotes
Abbreviations
Acknowledgments
This work was funded in part by a grant from the National Institute of Environmental Health Sciences (ES024471) awarded to R.C.S and J.R.H. and NC State University intramural startup funds awarded to J.R.H. Additional support for this research was provided by the National Institute of Environmental Health Sciences (P30ES025128) through the NC State University Center for Human Health and the Environment (CHHE) and its Bioinformatics and Genomics Cores. In addition, this research was supported in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. We would like to acknowledge Dr Jun Ninomiya-Tsuji for the use of the Accuri C6 Plus flow cytometer.
Author contributions
Conceptualization: JRH, RCS; Formal Analysis: JRH, RCS, JSH; Funding Acquisition: JRH, RCS; Investigation: JRH, JSH, SG, JRO; Writing - Original Draft Preparation: JRH; Writing - Review and Editing: JRH, JSH, RCS
Data availability
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Environmental Health Sciences, (grant number ES024471, P30ES025128)
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.