
Abstract
Purpose. Hepatocellular carcinoma (HCC) is a common malignancy worldwide and has an annual occurrence of one million new cases. Novel therapeutic strategies of increased efficacy in the treatment of HCC-bearing patients would certainly be helpful. Hence, the authors explored the effect of combination treatment of roscovitine with chemotherapeutic drugs or quercetin (Qctn) in hepatoma cells, HepG2 and Hep3B. Methods. Cell viability was assessed by MTT assay, cell growth assay, and nuclear morphological changes by DAPI staining. The altered expression of signaling proteins and apoptotic molecules was established by Western blotting. Results. Roscovitine pretreatment considerably enhanced the drugs and Qctn-induced cell death in HepG2 and Hep3B cells. The exploratory studies revealed that augmented cell killing in HepG2 and Hep3B was mediated via Akt pathway and was independent of p53. pAkt was found to be significantly downregulated in combination treatment of roscovitine with carboplatin or Qctn. Corresponding to reduced expression of pAkt, the downstream molecules Bcl-2 and proactive forms of caspase 9 and caspase 3 were also downregulated indicating apoptosis. Conclusions. The present study reports for the first time, in hepatoma cells, the potentiation of carboplatin- and Qctn-induced cell death by the cell cycle inhibitor roscovitine. Roscovitine can thus be considered as a potential therapeutic target in combination with chemotherapeutic drugs or Qctn for treatment of HCC.
Introduction
Hepatocellular carcinoma (HCC), cancer of the liver, is worldwide the third most common cause of cancer associated deaths. 1 It is lethal in 75% of the cases. 2 Chemotherapy remains the first line of treatment for most of the tumor types, including HCC. Carboplatin and 5-fluorouracil (5-FU) are commonly used DNA-damaging chemotherapeutic drugs. 5-FU initiates apoptosis by targeting thymidylate synthase and by direct incorporation of its metabolites into DNA. 3 Carboplatin forms inter- and intrastrand cross-links and interstrand adducts with DNA by reacting with N 7 guanine residue. Alkylation at the N 7 position of guanine causes depurination leading to strand breakage. 4 However, HCC is resistant to most chemotherapeutic drugs. Presently, there is no effective treatment for HCC patients that is acquiescent to curative therapies. 5 Hence, alternate strategies that would render hepatoma cells susceptible to the action of chemotherapeutic drugs will certainly be advantageous in treating patients suffering from HCC.
Recently, Cdk inhibitors have gained attention as anticancer agents, and there have been reports highlighting the use of Cdk inhibitors as anticancer drugs. 6 Roscovitine is a small molecule that inhibits Cdks by directly competing for the ATP-binding site. It is particularly active against Cdk1 (Cdc2), Cdk2, and Cdk5 and induces G2-M arrest in cells. 7 Roscovitine has been reported to induce apoptosis in various tumor cells.8,9
Quercetin (Qctn), a phytoestrogen, exhibits antitumor, antiproliferative, and apoptosis promoting effects in wide range of cancer cells, including leukemia, 10 HCC, 11 and breast carcinoma. 12 It is also known to suppress the Multidrug resistance (MDR) by inhibiting the heat shock transcription factors in MDR-positive cells. 13 The growth inhibitory effect of Qctn on tumor cells arises because of its ability to obstruct the enzymatic processes that are involved in the regulation of cellular proliferation.14,15 Qctn is also reported to inhibit the phosphatidylinositol phosphate kinase activities in carcinoma cells, which causes reduction of second messengers IP3 concentration and consequently cell death.16,17 Thus, Qctn may be useful in the treatment of carcinomas with transformed signal transduction capacity. 18
In the present study, we report that in hepatoma cells HepG2 and Hep3B, Cdk inhibitor roscovitine significantly augments the pro-apoptotic action of DNA-damaging drugs, carboplatin and 5-FU and also that of Qctn. The enhanced cell killing occurs via Akt dependent pathway and is independent of p53 involvement.
Materials and Methods
Drugs and Chemicals
Carboplatin and 5-fluorouracil were purchased from Sigma (St Louis, MO) and dissolved in sterile water to prepare a stock of 25 mM. Qctn (Sigma, St Louis, MO) was dissolved in dimethyl sulfoxide to make a stock solution of 100 mM. Roscovitine (Calbiochem, San Diego, CA) was dissolved in dimethyl sulfoxide to prepare a stock solution of 50 mM. Stock solutions were diluted in culture medium immediately before use. Specific antibodies against p53, pAkt, basal Akt, pro-caspase 3, pro-caspase 9, caspase 3, caspase 9, Bcl-2, Bax, β-tubulin, and horse radish peroxidase–conjugated secondary antibodies were purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA). Roscovitine treatment was always given 1 hour prior to drugs or Qctn exposure and was present throughout the experiments.
Cells and Culture Conditions
Hepatoma cell lines Hep3B and HepG2 were obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained in our in-house cell repository. The cells were routinely cultured in DMEM containing 10% heat inactivated fetal bovine serum (HyClone Laboratories, Inc, Logan, UT), penicillin, and streptomycin, 100 U/mL (Invitrogen Life Technologies, Carlsbad, CA) at 37°C in the presence of 5% CO2.
Methylthiazole Tetrazolium (MTT) Cell Viability Assay
The MTT assay, which is based on conversion of yellow tetrazolium salt to purple formazan crystals by metabolically active cells, provides a quantitative determination of viable cells. Cells seeded at the density of 7500 per well in 96-well tissue culture plates were allowed to adhere for 24 hours at 37°C. They were then treated as indicated. Cell viability was assessed by MTT assay. A volume of 50 µL of MTT (1 mg/mL) was added to each well and incubated for 4 hours at 37°C. Formazan crystals were solubilized in 100 µL of isopropanol by incubating in shaking condition at room temperature for 5 minutes. Absorbance was taken at 570 nm. Absorbance given by untreated cells was taken as 100% cell survival.
Cell Growth Assay
HepG2 and Hep3B cells were plated at density of 5 × 105 cells in 60-mm dishes one day before treatment. The cells were treated with roscovitine, carboplatin, and Qctn alone or in combination as indicated. Thereafter, cells were counted once daily for 3 consecutive days. Floating cells were washed away, and only the living cells were detached from dishes by trypsinization and counted. Growth curves were constructed for each experimental group. Average and standard deviation at each time point were calculated based on 3 counts from an experiment. The experiment was repeated twice.
Morphological Changes
HepG2 and Hep3B cells were plated and treated with roscovitine, carboplatin, and Qctn alone or in combination as indicated for 48 hours. The cells were then photographed using DP30 camera (Olympus, Tokyo, Japan).
Immunofluorescence Confocal Microscopy
Cells were grown on collagen-coated coverslips and treated as indicated. They were washed with phosphate-buffered saline (pH 7.5) and fixed with 3.7% paraformaldehyde for 10 minutes at room temperature. Cells were permeabilized using 0.1% Triton X-100 and subsequently blocked with 5% bovine serum albumin for 1 hour at room temperature. Samples were mounted with DAPI (4′,6-diamidino-2-phenylindole)-containing mounting media and examined on a confocal microscope (LSM510, Carl Zeiss, Oberkochen, Germany). Images were subsequently processed by Adobe Photoshop software.
Cellular Protein Preparation and Immunoblotting
Following indicated treatments, cells were washed thrice with ice-cold phosphate-buffered saline and lysed in ice-cold lysis buffer (50 mM Tris–HCl, pH 7.5, with 120 mM NaCl, 10 mM sodium fluoride, 10 mM sodium pyrophosphate, 2 mM EDTA, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 1% NP-40, and protease inhibitor cocktail). Cellular lysates were clarified by centrifugation at 12 000 rpm for 30 minutes. Proteins (40 µg) resolved on 10% to 12% SDS-polyacrylamide gel was subsequently transferred onto nitrocellulose membrane (0.45 µm). The membranes were probed with respective primary antibodies followed by horse radish peroxidase–conjugated secondary antibodies. Immunoblots were detected by chemiluminescence reagent (Cell Signaling Technology, Beverly, MA). Whenever required, blots were stripped by incubating the membrane at 50°C for 15 minutes in stripping buffer (62.5 mM Tris–HCl, pH 6.8, 100 mM mercaptoethanol and 2% SDS) with intermittent shaking. Membranes were washed thoroughly with Tris-buffered saline and reprobed with desired antibodies. Otherwise, gels run in duplicates were probed for the desired proteins by Western blotting and then compiled together.
Statistics
Statistical comparisons were made using Student’s 2-tailed unpaired t test and value of P < .05 was considered statistically significant.
Results
Roscovitine Augments Cell Killing Activity of DNA-Damaging Agents 5-FU, Carboplatin, and Quercetin in HepG2 Cells
Development of effective therapies for advanced stages of HCC has been substantially lacking. We investigated the combinatorial effect of Cdk inhibitor roscovitine with chemotherapeutic drugs 5-FU, carboplatin, and with the flavonoid Qctn. Figure 1 depicts the combinatorial effects of roscovitine (10 µM) with 5-FU, carboplatin, or Qctn (50 µM). As can be seen from Figure 1A, roscovitine considerably enhances the cell death induced by carboplatin and 5-FU in comparison with cell killing observed when HepG2 cells were treated with either roscovitine or drugs alone at doses corresponding to 48IC50 value (calculated as described earlier in Sharma et al 19 ) of the drugs. Similar effects were observed when drugs exposure time was reduced to 24 hours at identical concentrations (Figure 1B). Roscovitine has been earlier reported to promote death induced by chemotherapeutic drugs in cancer cells.20,21 To explore the possibility that roscovitine could potentiate cell killing even at sublethal doses of the drugs, the experiment (48 hours) was performed with half IC50 values and interestingly, similar results were obtained. As depicted in Figure 1A, roscovitine significantly enhanced the cell death induced by 5-FU and carboplatin at sublethal doses. In combination therapeutics, apart from chemotherapeutic drugs, roscovitine has also been stated to enhance nutlin-induced apoptosis. 22 Similar to roscovitine, Qctn has also been reported to have synergistic effect with chemotherapeutic drugs on the induction of apoptosis 19 and with other molecules such as ribavirin 23 and genistein. 24 Hence, we explored the probability of synergistic effect of roscovitine with Qctn on Hep3B and HepG2 cells. Also, because Qctn mainly arrests the cells in the G1 and S phases of cell cycle 24 and roscovitine acts at the G2 phase of cell cycle 25 we hypothesized that the two might exert synergistic apoptotic effect in hepatoma cells. As can be seen in Figure 1C, an increase in cell death occurred in roscovitine and Qctn combination treatment in comparison with either of them alone. To further verify that roscovitine is indeed promoting death induced by carboplatin or Qctn and the effect observed by MTT assay is not merely cytostatic, we performed cell growth assay as described in experimental procedures. As can be observed from Figure 1D, all 3 agents (roscovitine, carboplatin, Qctn) had growth inhibitory effects on HepG2 cells whereas the combination treatments (roscovitine with carboplatin and roscovitine with Qctn) resulted in greater reduction in cell growth than either of the agent alone.

Roscovitine augments cells killing by DNA-damaging agents 5-FU, carboplatin, and Qctn in HepG2 cells
Roscovitine Augments Cell Killing Activity of DNA-Damaging Agents 5-FU, Carboplatin, and Quercetin in Hep3B Cells
To expand the study, we investigated the combination effects of roscovitine with chemotherapeutic drugs carboplatin, 5-FU, and Qctn in another hepatoma cell line, Hep3B. Similar results were obtained, which are illustrated in Figure 2. In Hep3B cells also, roscovitine (10 µM) significantly augmented death induced by carboplatin or 5-FU (at both IC50 for 24-hour and 48-hour treatment and half IC50 values for 48 hours; Figures 2A and 2B) and by Qctn (50 µM) as well (Figure 2C). Cell growth assay performed in Hep3B cells indicated that all 3 agents (roscovitine, carboplatin, Qctn) had inhibitory effects on Hep3B cells, though the combination treatments (roscovitine with carboplatin and roscovitine with Qctn) resulted in greater reduction in cell growth than either of 3 alone (Figure 2D).

Roscovitine augments cell killing by DNA-damaging agents 5-FU, carboplatin, and Qctn in Hep3B cells
Roscovitine in Combination With Carboplatin or Qctn Causes Significant Morphological Changes in HepG2 and Hep3B Cells
To further illustrate the effect of the combination treatment of roscovitine with either carboplatin or Qctn on cells, Figure 3 depicts the morphological changes following treatment with roscovitine, carboplatin, or Qctn alone or roscovitine in combination with carboplatin or Qctn, in both HepG2 (Figure 3A) and Hep3B (Figure 3B) cells. It can be clearly seen that combination treatments result in diminished cell density, increase in the number of floating cells (Figure 3), and enhanced apoptotic bodies/change in nuclear morphology observed by nuclear DAPI staining (Figure 4) indicative of increased cell death. As shown in Figure 4A (HepG2) and 4B (Hep3B), roscovitine in combination with carboplatin considerably disrupts the nuclear morphology in comparison with either of them alone. In combination with Qctn, the number of apoptotic bodies (marked by arrowhead) is considerably higher compared with independent treatments.

Roscovitine in combination with carboplatin or Qctn induces significant morphological changes in HepG2 and Hep3B cells

Roscovitine in combination with carboplatin or Qctn enhances cell death as depicted by apoptotic nuclei
Roscovitine Augments Pro-Apoptotic Action of Carboplatin and Qctn via Akt-Dependent Mechanism
In our quest to explore the mechanism by which roscovitine potentiates cell death, we checked for the involvement of p53 as it has been implicated in roscovitine, 26 carboplatin, 27 and also quercetin 28 induced cell death. Though roscovitine (Figures 5A and 5D), carboplatin (Figure 5A) and Qctn (Figure 5D) independently induced p53; the combination treatment of roscovitine with carboplatin or Qctn did not further increase p53 protein expression in HepG2 cells (Figure 5A and D). With reports documenting the downregulation of pAkt by roscovitine and involvement of Akt following Cdk5 knockdown29,30 we hypothesized Akt’s involvement in augmentation of death by roscovitine in HepG2 cells. As illustrated in Figures 5B and 5E, pAkt levels were significantly downregulated on combination treatment of roscovitine with carboplatin or Qctn respectively in comparison with either of them alone. To further investigate the involvement of downstream death executor molecules, the expression levels of Bcl-2, Bax, caspase 9, and caspase 3 were examined. Figures 5C and 5F clearly indicate that Bcl-2 is downregulated in combination treatments of roscovitine with carboplatin or Qctn without any significant change in expression of Bax causing lowering of Bcl-2:Bax ratio (indication of apoptosis) in combination treatments. The expression levels of proactive forms of both caspase 9 and caspase 3 were also reduced significantly in cells subjected to combination therapy and consequently their cleaved products were elevated in combination treatments. In contrast, in Hep3B cells, p53 being present in mutant form, 31 its levels were not altered when cells were treated with drugs alone or in combination (data not shown). Also, the expression levels of pAkt and the respective downstream molecules were investigated. The results were similar to that observed in HepG2 and are depicted in Figure 6. Figures 6A and 6B illustrate the result on combination treatment of roscovitine with carboplatin, and Figures 6C and 6D depict combination treatment of roscovitine with quercetin in Hep3B cells. It can be clearly seen that pAkt levels are diminished on combination treatments in comparison with either of them alone. In addition, the apoptotic effectors Bcl-2, pro-caspase 9 and pro-caspase 3 were downregulated in both combination treatments with no significant alteration in expression of Bax, and enhanced levels of cleaved products of caspase 9 and caspase 3.
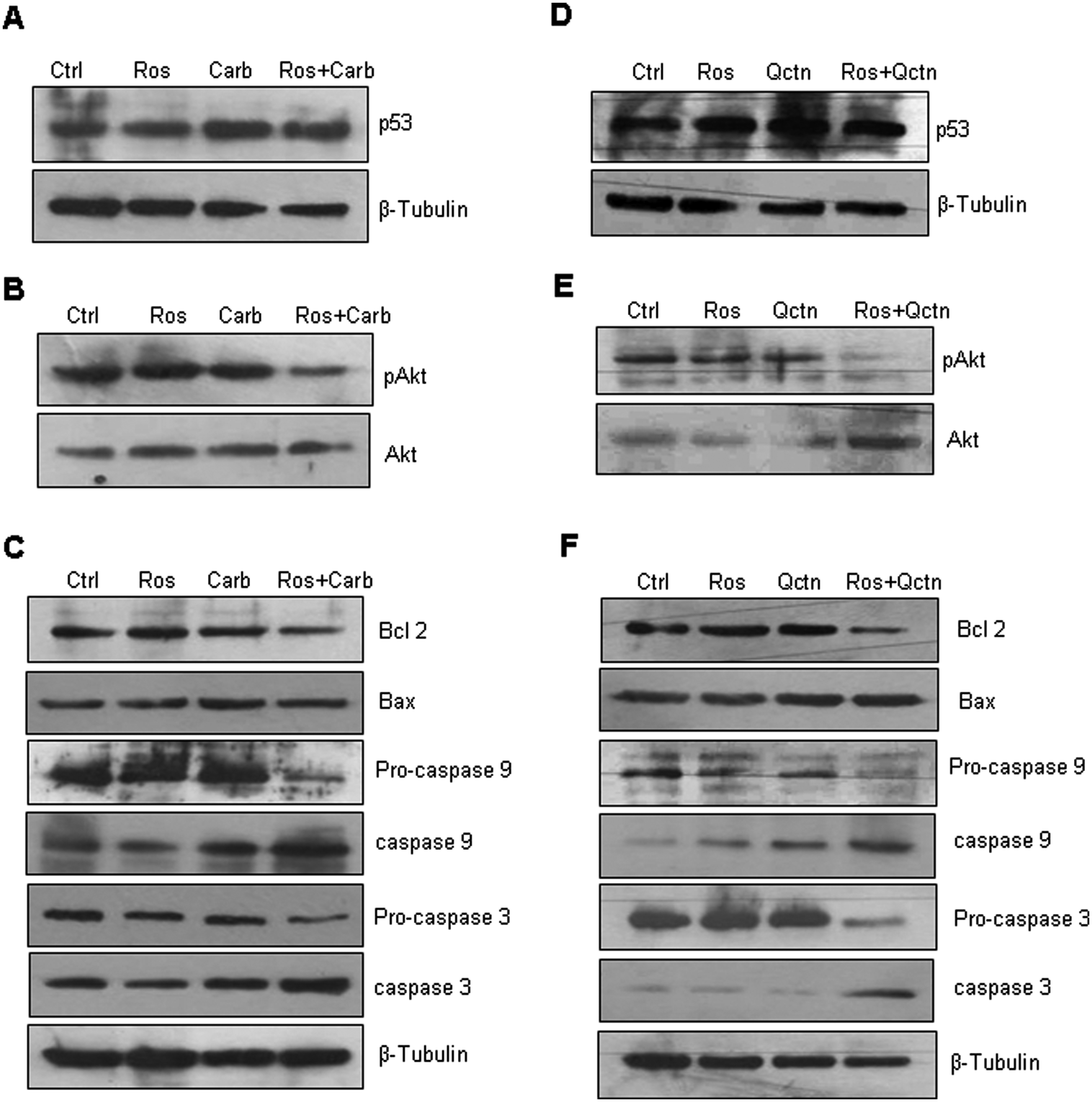
Roscovitine augments pro-apoptotic action of carboplatin via Akt-dependent mechanism in HepG2 cells

Roscovitine augments cell killing induced by carboplatin and by quercetin in Hep3B cells via Akt-dependent mechanism
Discussion
Cancer cells exhibit a typical cell cycle regulation to gain advantages for proliferation. A concept has now started to appear that cell cycle functions of Cdks can be exploited pharmacologically with Cdk inhibitors that have appropriate selectivity profiles.32,33 However, the functional redundancy of a number of Cdks may limit the effects of selective Cdk inhibitors. In vitro studies have shown that cell proliferation continues despite the lack of individual Cdks, probably because of the presence of other compensatory kinases. This implies that selective inhibitors of individual Cdks may potentially prove to be futile as single agent therapeutics. Hence, the conception of combinatorial therapeutics with other anticancer agents may be benefited by using Cdk inhibitors in cancer therapeutics. There are reports documenting the use of roscovitine in combination with either radiation or chemo therapeutics. Coley et al 34 reported the synergistic effect of roscovitine in combination with cisplatin for treatment of human uterine sarcoma. Hui et al 35 have reported the therapeutic efficacy of roscovitine in combination with ionizing radiation for human nasopharyngeal carcinoma, and an enhancement of radiation response by roscovitine in human breast carcinoma in vitro and in vivo was reported by Maggiorella et al. 36 An attempt was thus made to study the effect of combination treatment of roscovitine with chemotherapeutic drug carboplatin and with Qctn to improve their efficacy and therapeutic potential in hepatoma cells. Roscovitine was found to significantly enhance cell killing in HepG2 and Hep3B cells (Figures 1 and 2, respectively) when used in combination with either carboplatin or Qctn. To delineate the mechanism of enhanced cell killing, HepG2 cells were treated with carboplatin or Qctn alone or in combination with roscovitine and processed for detection of p53 and pAkt. Roscovitine is known to induce p53 in various cancer cell types. 37 Though an increase in p53 level was observed on roscovitine treatment, further increase in its expression was not observed in combination treatment with either carboplatin (Figure 5A) or Qctn (Figure 5D) in HepG2 cells. Following reports on involvement of pAkt pathway in roscovitine-mediated cell death, 38 we sought to verify any alteration in the levels of pAkt. Interestingly, pAkt level was considerably diminished when roscovitine treatment was combined with either carboplatin or Qctn exposure in both HepG2 (Figures 5B and 5E) and Hep3B cells (Figures 6A and 6C). The investigation of downstream molecules led to identification of involvement of Bcl-2, caspase 9, and caspase 3. Bcl-2 expression was observed to be downregulated with no alteration in Bax expression levels (Figures 5C, 5F, 6B, and 6D). Also, augmented cleavage was observed with enhanced levels of caspases 9 and 3, and concomitant decreases in pro-active forms of caspases 9 and 3 (Figures 5C, 5F, 6B, and 6D). To conclude, it can be stated that in hepatoma cells, roscovitine-mediated enhanced cell killing in combination with carboplatin or Qctn depends on pAkt and is independent of p53 involvement. Thus, roscovitine in combination with chemotherapy may prove to be an effective strategy to treat hepatocellular carcinoma in comparison with treatment with chemotherapeutic drugs alone.
Footnotes
Acknowledgements
We thank Dr G. C. Mishra, Director, National Centre for Cell Science (NCCS), for being supportive and giving all the encouragement to carry out this work. Support from other group members and all technical staff of NCCS is also duly acknowledged.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors thank the Department of Biotechnology, Government of India, for providing financial support. AS thanks the Council for Scientific and Industrial Research, New Delhi, India for a fellowship.