Superoxide anions produced by Streptococcus pyogenes group A-stimulated keratinocytes are responsible for cellular necrosis and bacterial growth inhibition
Free accessResearch articleFirst published online February, 2016
Superoxide anions produced by Streptococcus pyogenes group A-stimulated keratinocytes are responsible for cellular necrosis and bacterial growth inhibition
Gram-positive Streptococcus pyogenes (group A Streptococcus or GAS) is a major skin pathogen and interacts with keratinocytes in cutaneous tissues. GAS can cause diverse suppurative and inflammatory infections, such as cellulitis, a common acute bacterial dermo-hypodermitis with a high morbidity. Bacterial isolation yields from the lesions are low despite the strong local inflammation observed, raising numerous questions about the pathogenesis of the infection. Using an in vitro model of GAS-infected keratinocytes, we show that the major ROS produced is the superoxide anion (), and that its production is time- and dose-dependent. Using specific modulators of ROS production, we show that is mainly synthesized by the cytoplasmic NADPH oxidase. Superoxide anion production leads to keratinocyte necrosis but incomplete inhibition of GAS growth, suggesting that GAS may be partially resistant to the oxidative burst. In conclusion, GAS-stimulated keratinocytes are able to develop an innate immune response based on the production of ROS. This local immune response limits GAS development and induces keratinocyte cell death, resulting in the skin lesions observed in patients with cellulitis.
Infections of skin and soft tissues are characterized by microbial invasion of the epidermis, dermis and subcutaneous tissues generally inducing an immune response from the host. Various bacteria are able to trigger such infections, and they include the Gram-positive bacterium Streptococcus pyogenes (Group A Streptococcus or GAS), a major pathogen that interacts with keratinocytes in the epidermis. GAS are members of the pyogenic group of streptococci associated with infections in humans, and can be classified into more than 200 types based on the sequence of the 5′ variable region of the major surface M protein gene (emm types).1 GAS can cause numerous types of suppurative and inflammatory infections, ranging from mild superficial infections (impetigo) to more invasive and potentially life-threatening diseases (cellulitis, necrotizing fasciitis and toxic shock syndromes). Cellulitis is a common acute bacterial dermo-hypodermitis associated with high morbidity: its incidence has been estimated to be 10–100/100,000 of the population/yr; 20–50% of cases require hospitalization; and the estimated mortality is 0.5%.2 However, skin and blood specimen cultures are rarely positive for the causative bacterium, with reported isolation rates between 14% and 40% for skin cultures and between 2% and 3% for blood cultures.3 This low bacteriological yield contrasts with the often substantial local inflammation, raising numerous questions about the pathogenesis of cellulitis, which remains incompletely understood.
GAS strains may enter the skin through abrasions and other types of lesion allowing the bacteria to penetrate the epidermis and to adhere to keratinocytes via a receptor-mediated process.4 This invasion process involves the expression by GAS of many virulence factors facilitating the internalization of the bacteria into epithelial cells by a mechanism involving matrix and plasma proteins.5–7 The prominent M protein is important for the attachment of GAS to keratinocytes in skin infections, mediating the recognition of CD46 and α5β1-integrin.8–11 The fibronectin-binding protein F may mediate the attachment of GAS to Langerhans cells,12,13 and GAS plasminogen-binding proteins have been shown to interact with α1β1 and α5β1integrins.14 Once adhesion and colonization have been established, bacterial enzymes and exotoxins (streptolysin, erythrogenic toxin, DNase, streptokinase, proteinase, amylase and esterase) contribute to local inflammation and the dissemination of GAS.15
Keratinocytes are the major cell lineage in the epidermis, a multilayered stratified epithelium of the skin that is the essential first line of defense against microbial infection. It has repeatedly been shown that keratinocytes are more than a mere passive barrier to infection, and that they play an active role in the host innate immune response. Following stimulation of the PRR-related pathways by certain pathogens, keratinocytes produce various pro-inflammatory molecules that contribute to inducing the direct migration of various subpopulations of immune cells, which, in turn, amplify the local inflammatory response.16 Several surface molecules expressed by GAS are specifically recognized by cognate PRRs on the surface of host cells; the signaling adaptor MyD88 is involved in the subsequent cytokine production, although the PRRs specifically involved are not known. Indeed, GAS strains activate the p38 MAPK and NF-κB pathways via MyD88-dependent and MyD88-independent pathways without the involvement of TLR2, TLR4 or TLR9.17,18 Adherent streptococcal strains, such as S. pyogenes JRS4, induce the transcription of the IL-1α, IL-1β, IL-6 and IL-8 genes and the release of prostaglandin E2 (PGE2).19 The cytokine response is believed to be critical for the inflammatory response, but the production of ROS may also be involved, especially during the early phase of inflammation. Indeed, in other models of skin infection involving different pathogens, keratinocytes rapidly produce ROS, contributing to the destruction of the bacteria and to the production of pro-inflammatory cytokines.20,21
We report a study of the pro-inflammatory response of keratinocytes stimulated by cellulitis-related GAS strains in an in vitro model of infection. We investigated ROS production during the course of the immune response and the role of these molecules in both the inhibition of bacterial growth and keratinocyte cell death.
Materials and methods
Bacteria
Six S. pyogenes strains isolated from patients with cellulitis were obtained from the National Reference Center for Streptococci (https://www.cnr-strep.fr) (Table 1). They were classified on the basis of emm gene typing and included members of the three principal emm types encountered in French hospitals.22 Bacteria were grown in Todd Hewit broth (100 ml; Becton Dickinson), at 37℃, with shaking (250 rpm) and harvested by centrifugation at 7000 g for 10 min at 4℃. The exponential growth phase was identified experimentally as the point at which the OD620 reached 0.30 ± 0.03, corresponding to 3–4 h of culture. Stationary phase bacteria were obtained after 18 h. Bacterial pellets were washed in about 30 ml cold sterile PBS [1.5 mM KH2PO4, 2.7 mM Na2HPO4·7H2O, 0.15 M NaCl (pH 7.4)], centrifuged again and suspended in PBS or DMEM (Invitrogen, Cergy Pontoise, France). We used this suspension to prepare dilutions of 104–109 CFU/ml used immediately to stimulate keratinocytes at 37℃, such that the multiplicities of infection (MOI) with 0.1 ml of inoculum were 0.001–100 bacteria per cell. Bacteria inactivation was achieved by heating the bacterial suspension in PBS at 75℃ for 45 min and harvested by centrifugation at 7000 g for 10 min at 4℃.
Simplified number to identify GAS strains used in this study. bReference number of GAS strains used by the French National Reference Center for Streptoccoci. cThe genotype according to Protein M typing based on sequencing of the emm gene.
Cell culture and incubation assay
Immortalized human keratinocytes (HaCaT cell line) were cultured in DMEM with 0.1–10% (FCS; Invitrogen), 20 mM L-glutamine, 1 mM sodium pyruvate and antibiotic/antimycotic solution (10 U/ml penicillin, 10 mg/ml streptomycin, 0.25 mg/ml amphoterin), and primary human keratinocytes (NHEK cell line; Lonza, Walkersville, MD, USA) cells were cultured in the serum-free keratinocyte basal medium (KBM-2; Lonza), both at 37℃ under a humidified atmosphere containing 5% CO2 as previously described.23 Regular checks were carried out to ensure that the cell lines remained free from Mycoplasma infection. Cells at about 60–70% confluence were stimulated with GAS suspension, the concentration of which was adjusted to an appropriate value with DMEM supplemented with 0.1% FCS without antibiotics, for the desired period of time, at 37℃, under 5% CO2.
Spectrofluorimetric assessment of ROS production
HaCaT cells (2 × 104/well) were used to seed 96-well plates (Corning Costar, Brumath, France). The cells were incubated for 18 h, washed three times in PBS and incubated with 100 μl of a suspension of GAS (OD620 nm = 1.0) for various times between 15 min and 24 h. The cells were washed three times, 50 μl of 5 mM dihydroethidium [DHE, to assay superoxide anions (] or 5 mM H2-2′,7′-dichlorodihydrofluorescein diacetate (DCFDA; to assay H2O2) or 5 mM 4,5-diaminofluorescein diacetate (DAF2-DA; to assay NO) was added per well, and fluorescence intensity was recorded hourly, for 4 h. Fluorescent probes were purchased from Molecular Probes (Eugene, OR, USA). The fluorescence excitation/emission maxima were 495/515 nm for DAF2-DA, 480/610 nm for DHE and 507/525 nm for H2-DCFDA. At the end of the experiment, crystal violet assays were used to determine the number of adherent cells, as described below. The results of , H2O2 and NO assays were read by spectrofluorimetry in a Fusion spectrofluorimeter (PackardBell, Paris, France). The amount of ROS in each sample was calculated as follows: ROS rate (arbitrary units/min/106 cells) = [fluorescence intensity (arbitrary units) at T4 h – fluorescence intensity (arbitrary units) at T0/240 min/number of adherent cells as determined in the crystal violet assay]. Values are expressed in arbitrary units (AU).
Origin of GAS-induced superoxide anions and modulation of their levels
HaCaT cells (2 × 104/well) were used to seed 96-well plates and were incubated for 18 h in complete medium alone or in complete medium supplemented with the following: 2 mM diethyldithiocarbamate (or DDC or SOD inhibitor), 400 mM Cu(II)2(3,5-diisopropylsalicylate)4 (or CuDIPS a Cu/Zn SOD mimic), 100 mM Mn(III)tetrakis(4-benzoic acid (or MnTBAP a MnSOD mimic), 40 mM rotenone (inhibitor of mitochondrial complex I), 40 mM antimycin (inhibitor of mitochondrial complex III), 40 mM diphenyliodonium (inhibitor of NADPH oxidase) or 40 mM allopurinol (inhibitor of xanthine oxidase). The cells were then washed three times in PBS and incubated with 100 μl of a suspension of GAS (OD620 nm = 1.0) for various times between 15 min and 24 h. The cells were washed three times, 50 μl of 5 mM DHE was added to each well and fluorescence intensity was recorded hourly, for 4 h, as described above. At the end of the experiment, crystal violet assays were used to determine the number of adherent cells. The abundance of was calculated as described above.
Spectrofluorimetric analysis of cell death
Cell death was estimated spectrofluorimetrically with the fluorescent probe YO-PRO-1 (Molecular Probes) and a Fusion spectrofluorimeter (Packard Bell). HaCaT cells (2 × 104/well) were used to seed 96-well plates and were incubated for 18 h in complete medium alone or in complete medium supplemented with the following: 2 mM diethyldithiocarbamate, 40 μM rotenone, 40 μM antimycin, 40 μM diphenyliodonium, 40 μM allopurinol, 1600 μM reduced glutathione, 3200 μM N-acetylcysteine, 400 μM CuDIPS, 100 μM MnTBAP, 800 μM D,L-buthionine-[S,R]-sulfoximine, 400 μM aminotriazol or 20 U PEG-catalase. Cells were then washed three times in PBS and 100 μl of a suspension of GAS (OD620 nm = 0.5) in complete medium was added to each well and the incubation continued for 24 h. The cells were washed three times in PBS and incubated with 10 μM YO-PRO-1 for 30 min. Absorbance was read at an excitation wavelength of 480 nm and an emission wavelength of 525 nm for each sample, and cell death was evaluated by determining the fluorescence intensity (AU), as a measure of cell membrane disruption.
Flow cytometry assessment of cell death
HaCaT cells were incubated with or without GAS at an MOI of 0.01, 1.0 or 100, for 6 h at 37℃. The cells were washed twice with cold PBS, harvested by trypsin treatment, washed with PBS, incubated in 1 ml of PBS supplemented with both YOPRO-1 and propidium iodide for 15 min at 4℃ in the dark, and then analyzed by flow cytometry (FACScalibur; Becton Dickinson, Mountain View, CA, USA). Control experiments involved incubating cells with PBS alone and with YOPRO-1 and propidium iodide separately.
Inhibition of GAS growth by keratinocytes
HaCaT cells (2 × 104/well) were used to seed 96-well plates and were incubated for 18 h in complete medium. The cells were then washed three times in PBS and incubated with 50 μl of the GAS suspension (MOI = 1) in DMEM supplemented with 10% FCS without antibiotics at 37℃. After 3 h, 150 μl of liquid Todd Hewitt medium was added to each well and the plates were incubated for 18 h at 37℃. GAS growth was then evaluated by determining the OD620 of the culture supernatant. A control experiment was run in parallel, with GAS in DMEM supplemented with 10% FCS with no antibiotics. To assess the effect of production by the HaCaT cells on GAS growth, the HaCaT cells were pretreated for 18 h with 40 mM DPI, 2 mM DDC and 50 mM MnTBAP; the cells were then stimulated with GAS and the bacteria were cultured as described above.
Cell viability assays
Crystal violet staining was used to determine the numbers of adherent cells in 96-well plates. Briefly, after experimental incubations, the culture medium was discarded and the cells were incubated with 0.5% crystal violet solution (Sigma) for 30 min at room temperature (21℃). The cells were washed in PBS, 100% methanol was added and absorbance at 540 nm was measured spectrophotometrically.
The MTT [1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan] assay was used to assess cell viability in 96-well plates. The cells were incubated with 0.2% MTT in cell culture medium for 4 h at 37℃. The MTT solution was then discarded and DMSO was added to solubilize the MTT–formazan crystals formed in living cells. After thorough mixing, absorbance was measured at 540 nm.
Statistical analysis
The statistical significance of differences between experimental groups was analyzed by ANOVA. Values of P < 0.05 were considered significant.
Results
GAS-stimulated keratinocytes produce
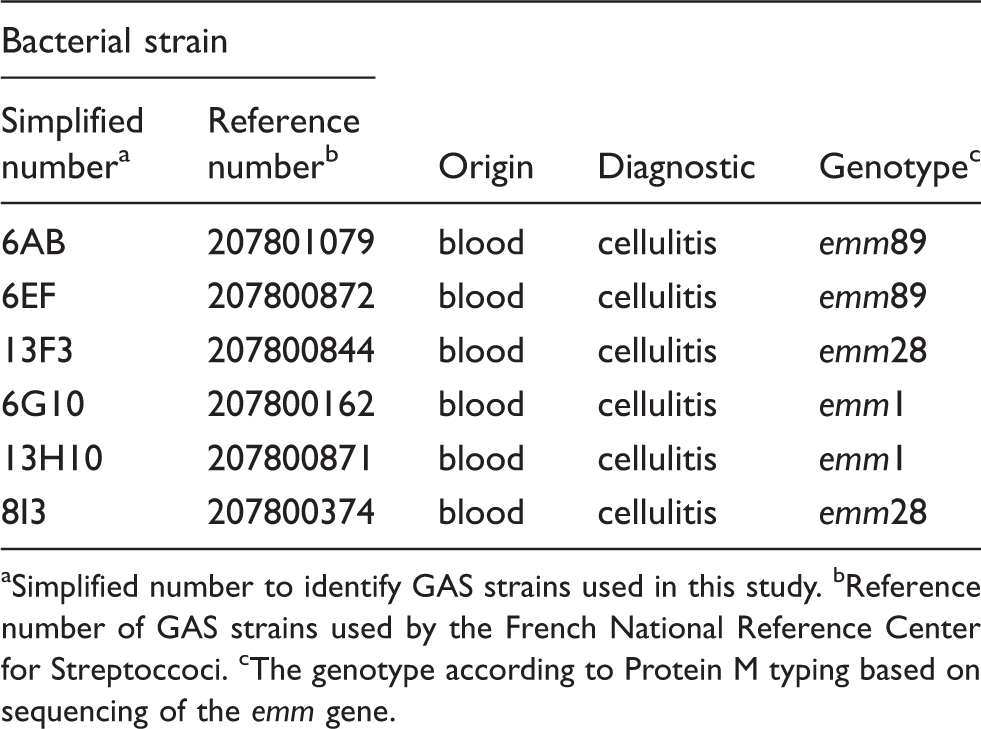
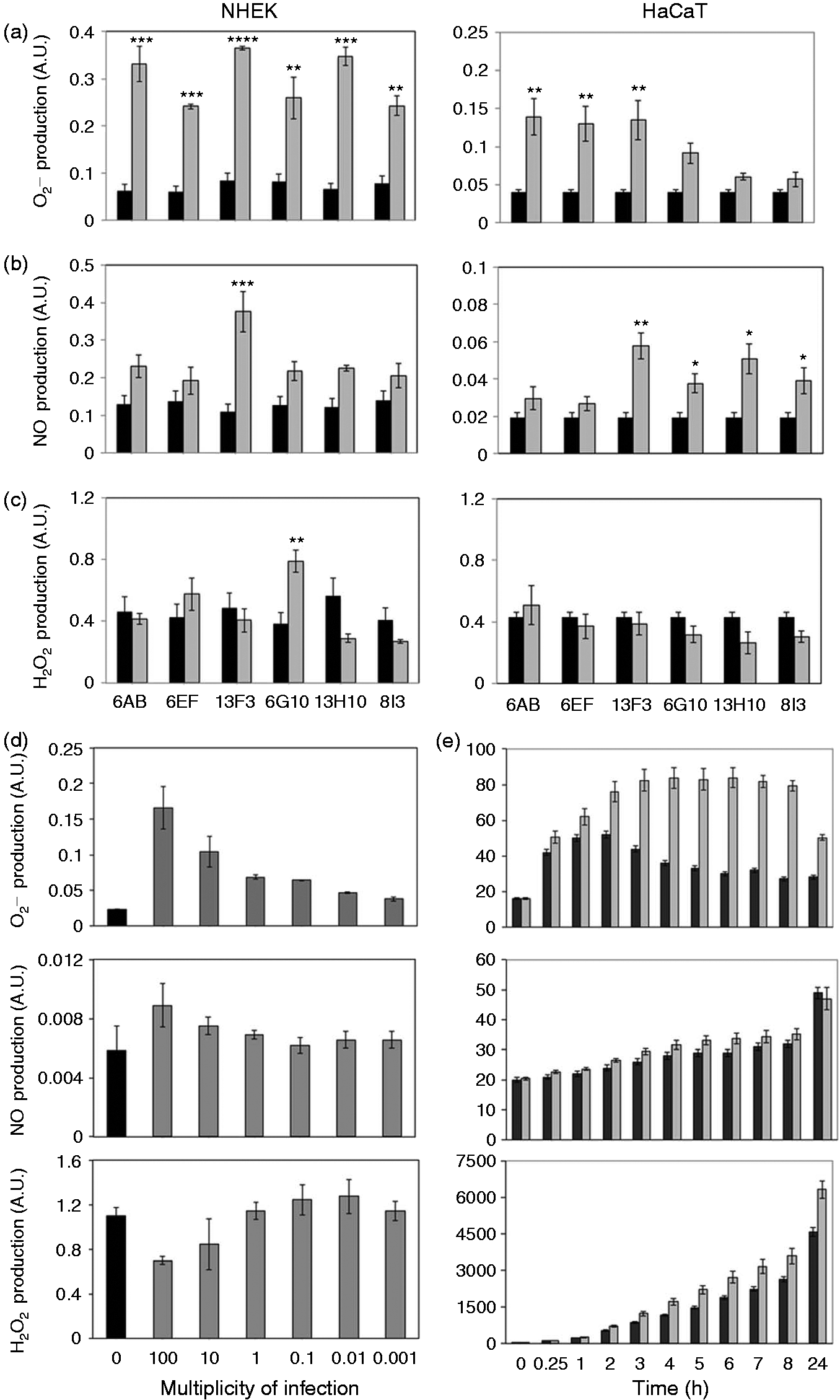
We assessed the ability of the six GAS strains isolated from patients with cellulitis to induce the production of the major ROS involved in oxidative stress. The production of and NO by keratinocytes was significantly stimulated in response to all six GAS strains tested (Figure 1a, b), but the production of H2O2 was not (Figure 1c). Indeed, five of the GAS strains tested did not induce the production of H2O2, the only exception being strain 6G10. ROS production was the same in cultured primary NHEKs and HaCaT cells which were used in subsequent experiments. The strain 6G10, displayed continuous reproducible ROS production and was used in further experiments to verify the specificity of the ROS production. The production of and, to a lesser extent, NO, by HaCaT keratinocytes was stimulated by the 6G10 strain in a dose-dependent manner, whereas H2O2 production was not. At the highest GAS concentration, and NO levels were 86% (P < 0.001) and 44% (P = 0.09) higher, respectively, than in the control (Figure 1d). levels increased significantly 1 h after GAS stimulation (P < 0.05), peaking 3 h after stimulation (P < 0.001). By contrast, we observed no time-dependent significant increase in NO production and H2O2 production increased very slowly, reaching a significant peak only after 5 h of stimulation (P < 0.05) (Figure 1e). However, heat-inactivated bacterial strains are not able to induce ROS production (Figure 2). Thus, stimulation with live GAS rapidly induced the production of large amounts of by keratinocytes.
Evaluation of ROS production by GAS-stimulated keratinocytes. NHEK and HaCaT cells were incubated for 6 h with (gray bar) or without (black bar) cellulitis-related GAS strains (Table 1) at an MOI of 10. (a) , (b) NO and (c) H2O2 were assayed by spectrofluorimetry, as described in the ‘Materials and methods’. (d) Dose dependence was determined by incubating HaCaT cells for 6 h with strain 6G10 at an MOI of 100, 10, 1, 0.1, 0.01 and 0.001. (e) HaCaT cells were incubated with strain 6G10 at an MOI of 10 and ROS production assayed at various times between 0.25 and 24 h. The values shown are means ± SD of three independent experiments. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Evaluation of ROS production by heat-inactivated (HI) GAS-stimulated keratinocytes. HaCaT cells were incubated for 6 h with live (light gray bar) or with HI (dark gray bar) or without (black bar) 6G10 GAS strains (Table 1) at an MOI of 10. (a) , (b) NO and (c) H2O2 were assayed by spectrofluorimetry, as described in the ‘Materials and methods’. The values shown are means ± SD of three independent experiments. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
The origin of the and the detoxification pathways in GAS-stimulated keratinocytes
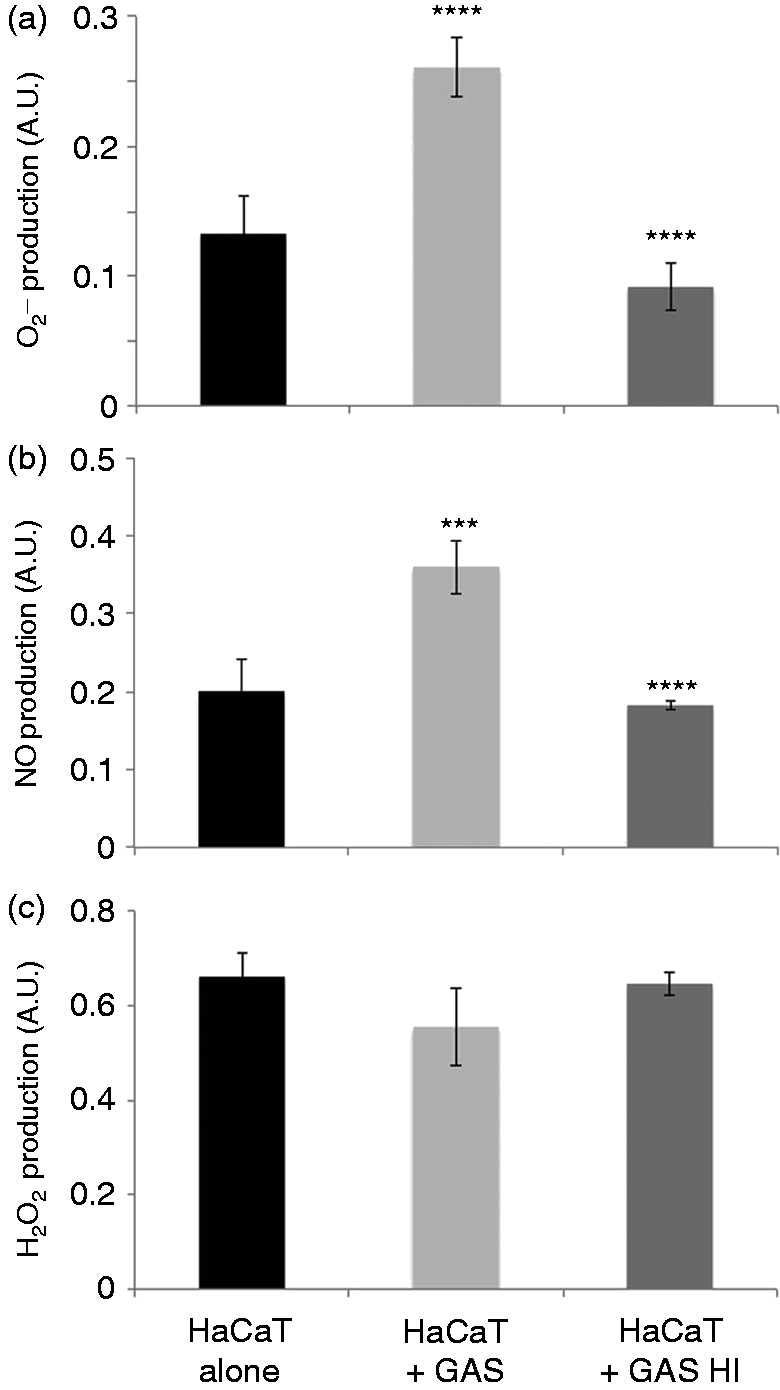
Superoxide anions may be produced by the cytosolic enzymes NADPH oxidase and xanthine oxidase, or by mitochondrial complex I or III of the respiratory chain. By using specific chemical inhibitors of the mitochondrial respiratory chain or cytoplasmic enzymes potentially responsible for production, we investigated the source of the produced. The prior treatment of keratinocytes with rotenone and antimycin, which inhibit complexes I and III of the mitochondrial respiratory chain, respectively, had no significant effect on the production of . By contrast, prior treatment with DPI, an NADPH oxidase inhibitor, significantly decreased production (P = 0.04). Incubation with allopurinol, a xanthine oxidase inhibitor, had no effect (Figure 3). The inhibition of superoxide dismutase (SOD) with a specific inhibitor, diethyldithiocarbamate (DDC), resulted in significantly higher levels of (P < 0.001). Moreover, the use of two SOD mimics, MnTBAP and CuDIPS, resulted in significantly lower levels of (P < 0.0001) (Figure 3), while the vehicles used to dissolve the ROS modulators are not (data not shown). Therefore, the produced by NADPH oxidase in GAS-stimulated keratinocytes is presumably converted into H2O2 by the enzyme SOD.
The superoxide anion detoxification pathway is involved in GAS-stimulated keratinocytes. was assayed after 6 h of incubation of HaCaT cells alone (black bar) or stimulated with GAS (dashed bar) or previously treated with specific modulators of enzymatic systems involved in ROS metabolism and then stimulated with GAS (strain 6G10, Table 1) at an MOI of 10 (gray bars). The concentrations used were as follows: 40 μM rotenone, 40 μM antimycin, 40 μM DPI, 40 μM allopurinol, 100 μM manganese [III] tetrakis (5,10,15,20)-benzoic acid porphyrin (MnTBAP), 400 μM ATZ, 0.8 mM BSO, 400 μM copper[II]diiosopropylsalicylate (CuDIPS) and 2 mM DDC. The values reported are means ± SD of three independent experiments. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
-induced cytotoxicity in GAS-stimulated keratinocytes
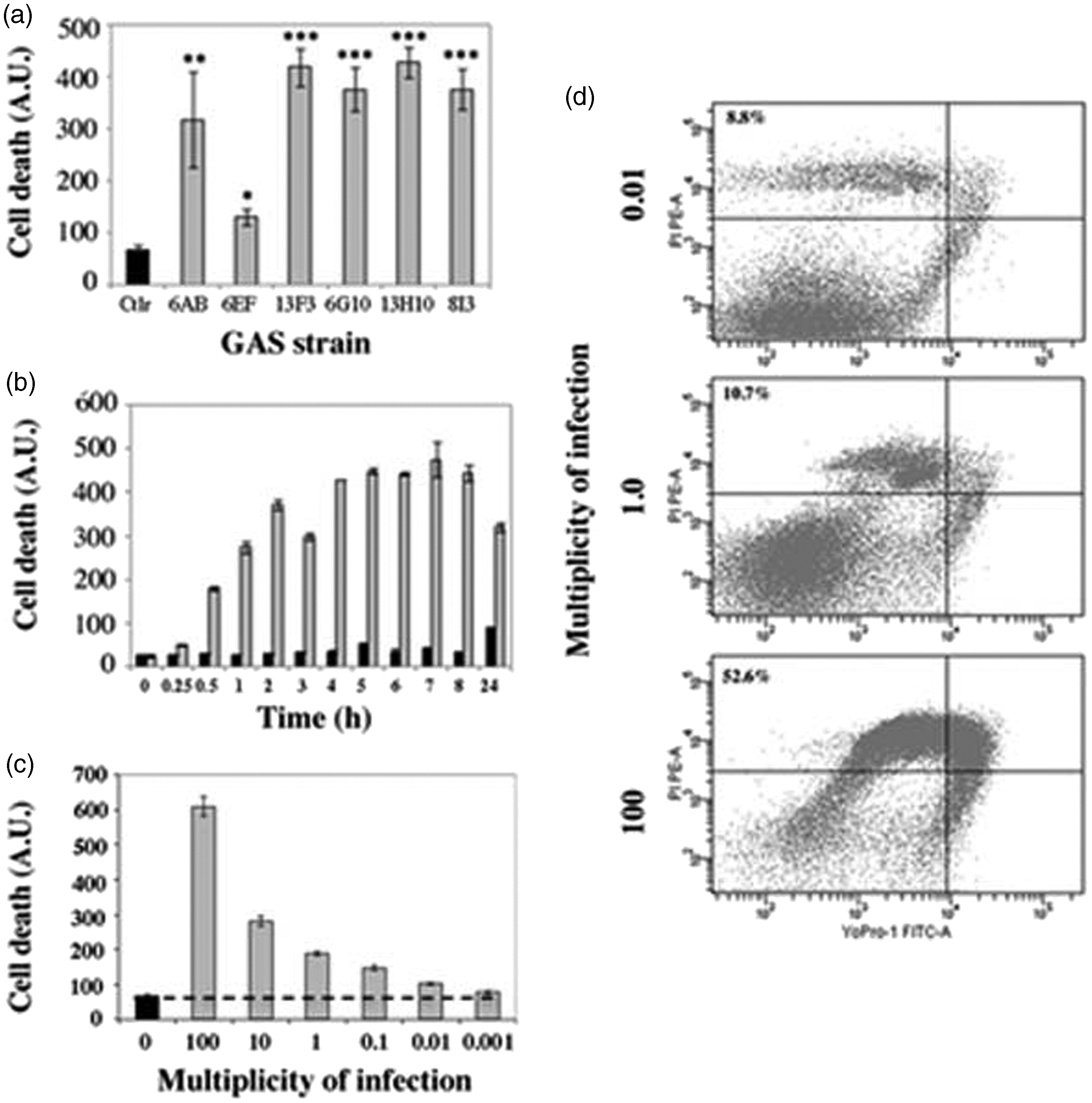
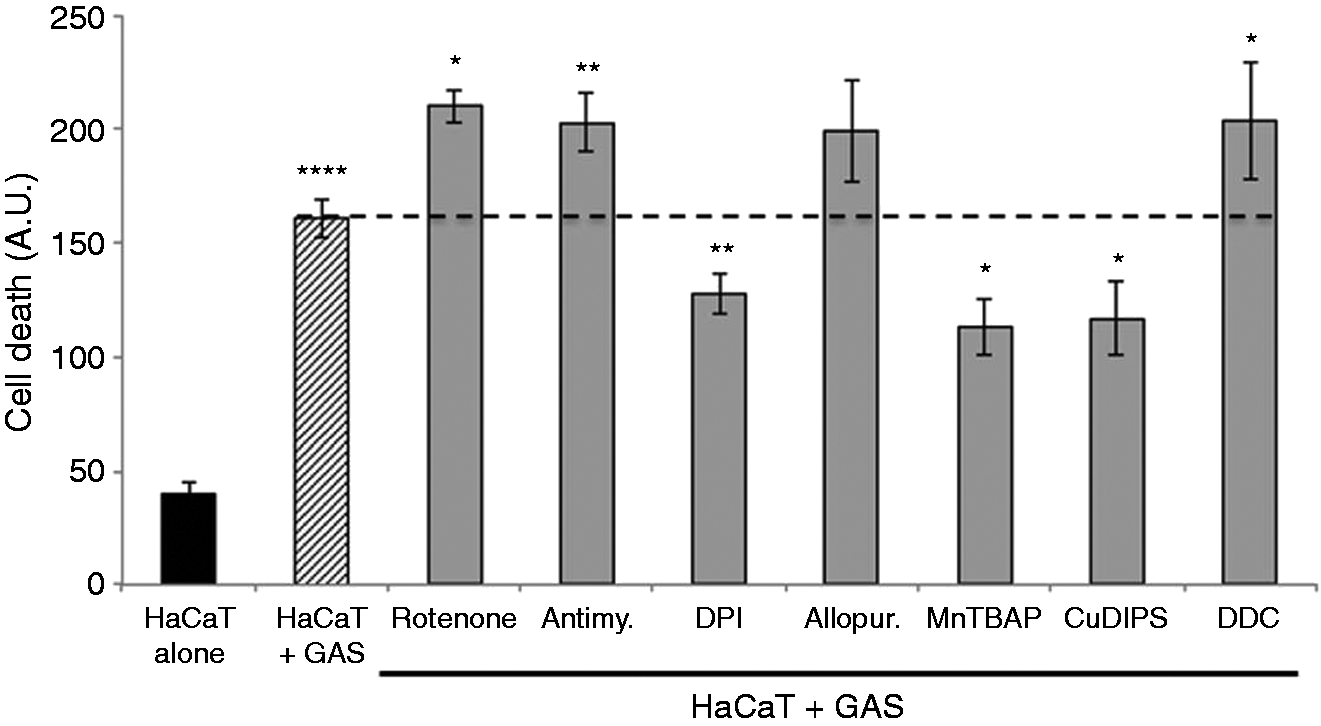
ROS, including , are highly toxic to cells so we investigated GAS-stimulated keratinocyte cell death and the role of in this phenomenon. We used YO-PRO-1 staining to evaluate the capacity of the six GAS strains to induce cell death. All but one (6EF) of the strains tested induced massive, significant (P < 0.001) levels of keratinocyte cell death (Figure 4a). GAS-induced death was detected within 30 min of infection, and was time- (Figure 4b) and dose-dependent (Figure 4c). We studied the nature of GAS-induced keratinocyte cell death, by a combination of YO-PRO-1 and propidium iodide staining, and analyzed keratinocytes by flow cytometry after stimulation with GAS strain 6G10. After 6 h of incubation, the cell death was mostly necrotic and cell death rates were dose-dependent (Figure 4d). We then investigated the role of in GAS-induced cell death, by treating the keratinocytes with specific modulators of enzymes involved in the production and detoxification of before incubation with GAS. Inhibition of production with DPI or the activation of detoxification by SOD with MnTBAP or CuDIPS all significantly decreased GAS-induced keratinocyte cell death (P < 0.001, P < 0.01 and P < 0.01, respectively). By contrast, DDC, rotenone and antimycin, all of which increase production, increased cell death rates (P < 0.01, P < 0.01 and P < 0.001, respectively) (Figure 5). These results suggest that the high rates of keratinocyte death caused by GAS are at least partly the consequence of inducing the production of by the cells themselves.
Cellulitis-related GAS strains induce keratinocyte death. HaCaT cells were incubated with GAS (gray bar) or left unstimulated (black bar). Cell death was assessed by spectrofluorimetry, with YO-PRO-1, as described in ‘Materials and methods’. (a) Cell death was evaluated in HaCaT cells after 24 h of incubation with six cellulitis-related GAS isolates (Table 1) at an MOI of 10. (b) Cell death kinetics of HaCaT cells incubated with GAS (6G10 strain, Table 1) at an MOI of 10 were followed for 24 h. (c) HaCaT cells were incubated with GAS (strain 6G10, Table 1) for 6 h at MOIs of 100, 10, 1, 0.1, 0.01 and 0.01 to test the dose dependence of the effect. (d) HaCaT cells were incubated for 6 h with GAS (strain 6G10, Table 1) at an MOI of 0.01, 1 or 100, and cell death was evaluated by flow cytometry after staining the samples with YO-PRO-1 and propidium iodide (PI), as described in the ‘Materials and methods’. The values shown are the means ± SD of two independent experiments. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
GAS induces cell death by stimulating production. HaCaT cells were incubated for 6 h with (dashed bar) or without (black bar) GAS (strain 6G10, Table 1) at an MOI of 10. We also tested HaCaT cells previously treated with the following specific modulators of enzymatic systems involved in ROS metabolism (gray bars): 40 μM rotenone, 40 μM antimycin, 40 μM DPI, 40 μM allopurinol, 100 μM manganese [III] tetrakis (5,10,15,20)-benzoic acid porphyrin (MnTBAP), 400 μM ATZ, 0.8 mM BSO, 400 μM copper[II]diiosopropylsalicylate (CuDIPS) and 2 mM DDC. The values reported are means ± SD of three independent experiments. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Role of in the defense mechanism of GAS-stimulated keratinocytes
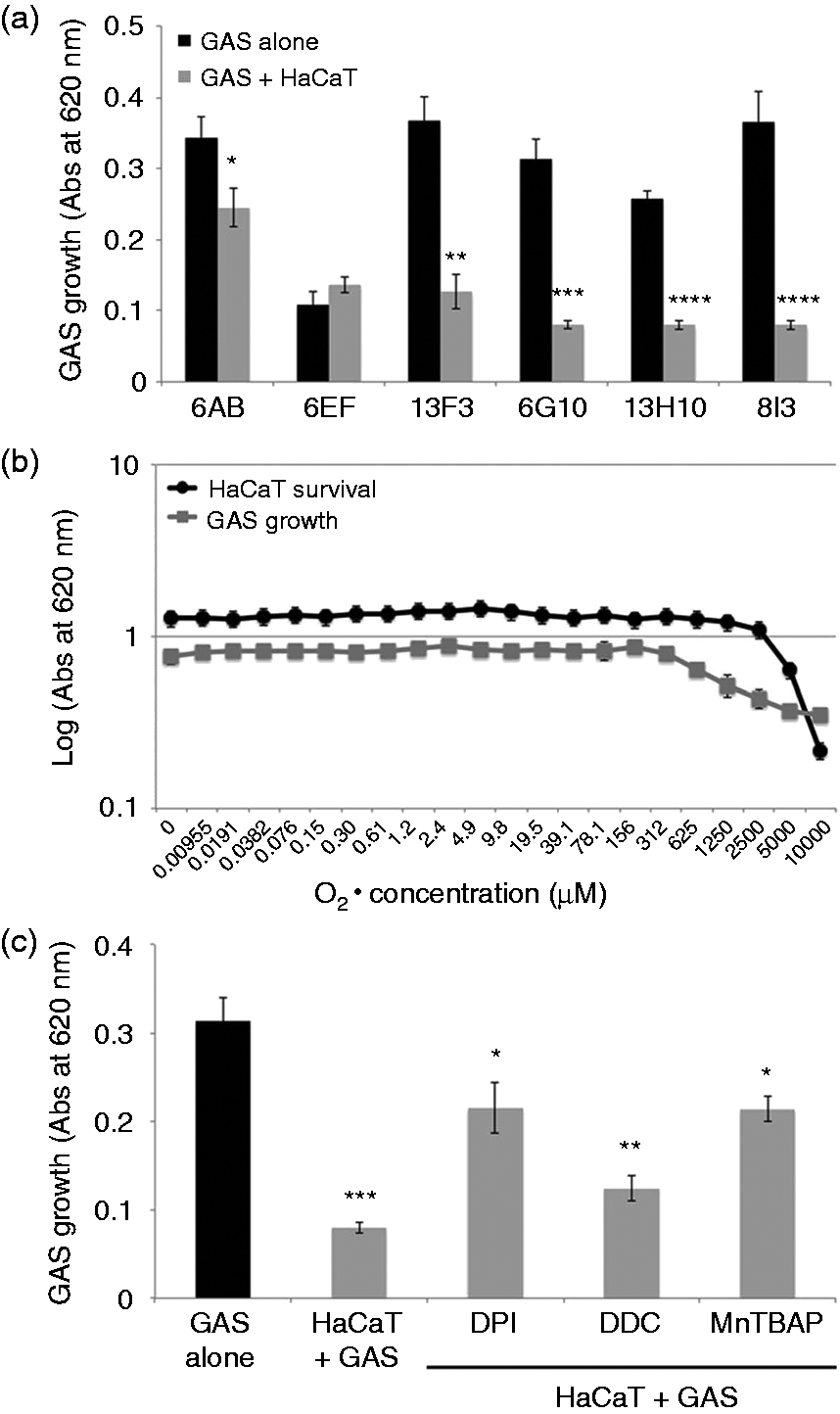
We measured the growth rates of the six GAS strains incubated with keratinocytes. All strains other than 6EF grew significantly more slowly after 18 h of incubation with keratinocytes in our experimental model (Figure 6a). However, when cultivating GAS strain in HaCaT conditioned media only the bacterial growth was not affected (data not shown). We then studied the effects of various concentrations of chemically generated on the survival of both keratinocytes and GAS. The 6G10 strain grew steadily until the concentration reached 156 μM, above which growth was dose-dependently inhibited; the HaCaT cells appeared to be more resistant to this concentration (Figure 6b). We then pretreated the keratinocytes with modulators of production: decreasing the amount of by blocking the NADPH oxidase (DPI) or activating SOD (MnTBAP) restored GAS growth, at least partly. Conversely, DDC, a SOD inhibitor that increases the amount of produced by keratinocytes, decreased GAS growth (Figure 6c).
GAS growth is inhibited by the produced by keratinocytes. (a) HaCaT cells were incubated for 4 h with six cellulitis-related GAS (Table 1) and (c) with GAS (strain 6G10) at an MOI of 1, in DMEM supplemented with 10% FCS, without antibiotics, at 37℃, under an atmosphere containing 5% CO2. We also carried out the same experiment with HaCaT cells previously treated with 40 μM DPI, 2 mM DDC and 50 μM MnTBAP. Control experiments were run in parallel with GAS in DMEM supplemented with 10% CFS without antibiotics and without keratinocytes (black bar). (b) HaCaT cells (dark line) and a GAS suspension (strain 6G10, OD620 nm of 0.5) (gray line) were incubated for 2 h under appropriate conditions with various concentrations of chemically generated superoxide anions, from 0.009 μM to 10 mM. The viability of HaCaT cells was determined by the MTT assay, as described in the ‘Materials and methods’. GAS growth was determined by adding liquid Todd Hewitt media to each well and incubating for 18 h at 37℃, then measuring optical density at 620 nm. The values shown are means ± SD of three independent experiments. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Discussion
GAS is a human pathogen, mostly transmitted by direct contact, which is amplified in overcrowded areas.24 Superficial impetigo is the most common manifestation of dermal GAS infection; these bacteria can spread into and/or penetrate the epidermis through epithelial abrasions and infect deeper tissues. Such secondary infection is characterized by damage to the epithelium, leading to the development of dermal and hypodermal invasion, which is a clinical entity (cellulitis) distinct from impetigo, which remains confined to the superficial layers of the skin.25 We studied GAS strains isolated from patients with cellulitis for their capacity to induce an oxidative burst in an in vitro model of infection of keratinocytes. ROS are small, short-lived molecular species, continuously produced in small amounts during the course of normal aerobic metabolism. Mitochondria is the typical site of ROS production: electrons leak from the electron transport chain and react with oxygen to form superoxide, and it has been estimated that 1–3% of oxygen reduced in cells may form superoxide in this manner. However, mitochondria are not the only site of ROS production in the cell. The enzymes NADPH oxidase and xanthine oxidase, located in cytoplasm, can also produce ROS. Several ROS have been described in cells, including and H2O2, which react strongly with cellular components.26 Incomplete reduction of O2 during respiration increases the level of and its abundance is determined by multiple enzyme systems, including SOD, which transforms into H2O2. Catalase, glutathione S-transferase and thioredoxin are then able to detoxify H2O2 into water. In this study, we show that GAS are also able to induce a massive production of ROS by both primary and immortalized keratinocyte cell lines. Using fluorescent probes, we demonstrate that all the GAS strains tested induced the production of by keratinocytes in a time- and dose-dependent manner, arguing for this production being specific. Moreover, we have shown that this ROS production needs live bacteria, suggesting an interaction between cellular receptors and surface components on the GAS strain that may have been altered, thus inhibiting the induction of ROS production. These results are in line with those describing that heat-inactivated GAS are not able to induce cell death as well as the production of pro-inflammatory molecules.27 None of the strains induced substantial H2O2 production, although three of the six strains induced NO production. The NO production in keratinocytes seems to be inconsistent; macrophages are able to produce NO when stimulated with GAS, suggesting that innate immune cells rather than keratinocytes are responsible for such production.28 Using specific chemical modulators able to activate or inhibit the production of , we confirmed that GAS induced the production of and we demonstrated that the production of was the result of NADPH oxidase activity. We showed that keratinocytes are able to produce ROS and this finding is consistent with previous work demonstrating that S. pyogenes is able to induce the production of ROS by HeLa cells and human leukocytes.29,30 Moreover, transcriptional studies with macrophages infected with GAS demonstrated up-regulation of the p47phox gene encoding a component of the NADPH oxidase (Ncf1), further evidence of activation of the oxidative stress response by GAS.31 In other in vitro inflammatory systems, keratinocytes are able to produce in response to UV stimulation, Propionibacterium acnes, Vitreoscilla filiformis and Mycobacterium ulcerans.20,21,32,33
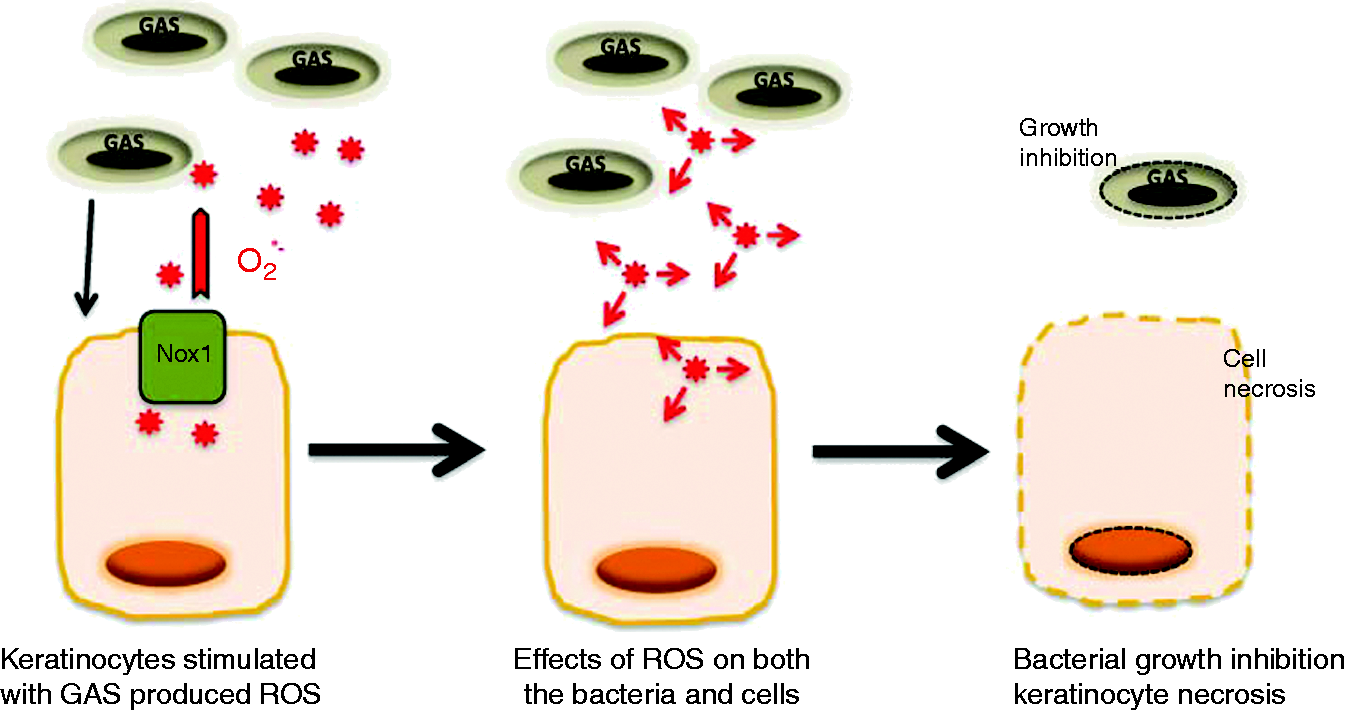
Superoxide anions are very toxic compounds and both bacteria and eukaryotic cells have molecular mechanisms to counteract their detrimental effects. However, production of ROS by cells infected with pathogens is often associated with high levels of cell death, and GAS induces apoptosis of infected human epithelial cells.1,34 All the GAS strains we tested here displayed cytotoxicity to keratinocytes, in a dose-dependent manner, within 30 min. At this early stage, the mechanism leading to the cell death seems to be mostly necrotic but not apoptotic. Moreover, modulation of production by specific modulators had a direct effect on the proportions of cells dying, implicating in the death of GAS-stimulated keratinocytes. These results are consistent with previous reports of ROS-induced necrotic cell death involving serine/threonine kinase receptor-interacting protein (RIP)1 and RIP3.35 Recently, it has also been shown that virulent GAS strains are able to modulate the PMN inflammatory response by reducing their apoptotic potential leading to a necrotic form of cell death.36 However, although the production of high levels of ROS is responsible of the death of keratinocytes, we show that this ROS production also contributes to eliminating the pathogen. This was demonstrated by the growth rates of GAS strains co-cultured with keratinocytes. In these experiments, keratinocytes were stimulated by GAS diluted in medium favorable for cell culture allowing the cells to react to the stimulation and produce ROS. Then, media favorable for the bacterial growth was added and the bacteria allowed to grow for a period of 18 h. In the presence of keratinocytes, GAS growth was significantly inhibited for five of the six strains tested. Indeed, the 6EF strain seems to present a low level of growth that was not affected by the co-culturing in presence of HaCaT cells. The use of specific inhibitors of production partly restored bacterial growth, suggesting that may be, at least in part, responsible for the inhibition of bacterial growth. However, the restoration of bacterial growth was only partial, suggesting that the keratinocytes may also produce other molecules to decrease GAS growth. Previously it has been shown that S. pyogenes was able to induce the production of β-defensin-2 (hBD-2),37 which may contribute to decrease the GAS growth. Further experiments will be necessary to evaluate it. Conversely, increasing the amount of did not completely abolish GAS growth, suggesting that there may be some mechanism conferring resistance to GAS against toxicity. GAS growth was inhibited only at high concentrations (625 μM) of chemically generated . Previously, we demonstrated that the lowest concentration able to abolish P. acnes growth in the same conditions was 9.8 μM, indicating that GAS is much less sensitive to .21 This observation is consistent with the demonstration of the regulation of GAS iron-binding protein (Dpr) gene expression under conditions of oxidative stress, protecting GAS against ROS.38 SOD expression in GAS strains is dependent on the superoxide anion concentration, which, in turn, directly interferes with the expression of the surface protein F; this protein binds fibronectin, promoting attachment to the host extracellular matrix and may contribute to protecting the bacteria against . However, it has been suggested that S. pyogenes may be more susceptible to oxidative stress than other species of the genus Streptococcus.39 This observation is interesting. Indeed, cellulitis is characterized by hyper-inflammation and necrosis associated with the absence of detection of the bacteria. GAS are able to activate innate immunity triggering the production of pro-inflammatory cytokines and chemokines that recruit macrophages and neutrophils. In addition to this production of pro-inflammatory cytokines, the adhesion of GAS to keratinocytes may also induce the production of ROS, which the cell is unable to detoxify, and consequently impedes bacterial growth as well as contributing to keratinocyte cell death (Figure 7). Further experiments will be necessary to elucidate in depth the molecular mechanisms involved in the keratinocyte and bacteria death.
Proposed role of ROS produced by keratinocytes upon stimulation by GAS. Keratinocytes are stimulated by GAS, triggering the production of through the NADPH oxidase pathway; this contributes to both keratinocyte necrosis and GAS growth inhibition.
Overall, our analysis of the oxidative burst response developed by keratinocytes in response to GAS stimulation suggests that innate immune response mediated by keratinocytes may contribute to the inhibition of GAS growth and the strong local inflammatory reaction. In this study, we show that six independent strains of GAS isolated from the blood of patients with cellulitis induce the production of by human primary keratinocyte cultures, and that this is associated with substantial cell necrosis.
Footnotes
Acknowledgements
This work was supported by the Association de Recherche en Virologie et Dermatologie.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
1.
JasirALuca-HarariBDarenbergJ. Clinical and microbiological characteristics of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol2009; 47: 1155–1165.
2.
DupuyA. Descriptive epidemiology and knowledge of erysipelas risk factors. Ann Dermatol Venereol2001; 128: 312–316.
3.
DenisFMartinCPloyMC. Erysipelas: microbiological and pathogenic data. Ann Dermatol Venereol2001; 128: 317–325.
4.
DarmstadtGLFleckmanPJonasM. Differentiation of cultured keratinocytes promotes adherence of Streptococcus pyogenes. J Clin Invest1998; 101: 128–136.
5.
LaPentaDRubensCChiE. Group A streptococci efficiently invade human respiratory epithelial cells. Proc Natl Acad Sci U S A1994; 91: 12115–12119.
6.
JadounJOzeriVBursteinE. Protein F1 is required for efficient entry of Streptococcus pyogenes into epithelial cells. J Infect Dis1998; 178: 147–158.
7.
WalkerMJMcArthurJDMcKayF. Is plasminogen deployed as a Streptococcus pyogenes virulence factor?Trends Microbiol2005; 13: 308–313.
8.
OkadaNLiszewskiMAtkinsonJ. Membrane cofactor protein (CD46) is a keratinocyte receptor for the M protein of the group A streptococcus. Proc Natl Acad Sci U S A1995; 92: 2489–2493.
9.
CueDRSouthernSOSouthernPJ. A nonpeptide integrin antagonist can inhibit epithelial cell ingestion of Streptococcus pyogenes by blocking formation of integrin α5β1-fibronectin-M1 protein complexes. Proc Natl Acad Sci U S A2000; 97: 2858–2863.
10.
RezcallahMSHodgesKGillDB. Engagement of CD46 and alpha5beta1 integrin by group A streptococci is required for efficient invasion of epithelial cells. Cell Microbiol2005; 7: 645–653.
11.
LövkvistLSjölinderHWehelieR. CD46 contributes to the severity of group A streptococcal infection. Infect Immun2008; 76: 3951–3958.
12.
HanskiECaparonM. Protein F, a fibronectin-binding protein, is an adhesin of the group A streptococcus Streptococcus pyogenes. Proc Natl Acad Sci U S A1992; 89: 6172–6176.
13.
OkadaNPentlandAPFalkP. M protein and protein F act as important determinants of cell-specific tropism of Streptococcus pyogenes in skin tissue. J Clin Invest1994; 94: 965–977.
14.
SiemensNPatengeNOttoJ. Streptococcus pyogenes M49 plasminogen/plasmin binding facilitate keratinocyte invasion via integrin-integrin-linked kinase (ILK) pathways and protects from macrophage killing. J Biol Chem2011; 286: 21612–21622.
15.
LukomskiSBurnsEHJrWydePR. Genetic inactivation of an extracellular cysteine protease (SpeB) expressed by Streptococcus pyogenes decreases resistance to phagocytosis and dissemination to organs. Infect Immun1998; 66: 771–776.
16.
KollischGKalaliBNVoelckerV. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunol2005; 114: 531–541.
GratzNSillerMSchaljoB. Group A streptococcus activates type I interferon production and MyD88-dependent signaling without involvement of TLR2, TLR4 and TLR9. J Biol Chem2008; 283: 19879–19887.
19.
WangBRuizNPentlandA. Keratinocyte pro-inflammatory responses to adherent and nonadherent group A streptococci. Infect Immun1997; 65: 2119–2126.
20.
MahéYFMartinRAubertL. Induction of the skin endogenous protective mitochondrial MnSOD by Vitreoscilla filiformis extract. Int J Cosmet Sci2006; 28: 277–287.
21.
GrangePAChéreauCRaingeaudJ. Production of superoxide anions by keratinocytes initiates P. acnes-induced inflammation of the skin. PloS Pathog2009; 5: e1000527–e1000527.
22.
LepoutreADoloyABidetP. Epidemiology of invasive Streptococcus pyogenes infections in France in 2007. J Clin Microbiol2011; 49: 4094–4100.
23.
BoukampPPetrussevskaRTBreitkreutzD. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol1988; 106: 761–771.
24.
RalphAPCarapetisJR. Group A streptococcal diseases and their global burden. Curr Top Microbiol Immunol2013; 368: 1–27.
25.
StulbergDLPenrodMABlatnyRA. Common bacterial skin infections. Am Fam Physician2002; 66: 119–124.
26.
FridovichI. Fundamental aspects of reactive oxygen species, or what's the matter with oxygen?Ann N Y Acad Sci1999; 893: 13–18.
27.
OkahashiNOkinagaTSakuraiA. Streptococcus sanguinis induces foam cell formation and cell death of macrophages in association with production of reactive oxygen species. FEMS Microbiol Lett2011; 323: 164–170.
28.
ZinkernagelASHruzPUchiyamaS. Importance of Toll-like receptor 9 in host defense against M1T1 group A Streptococcus infections. J Innate Immun2012; 4: 213–218.
29.
SaëtreTHoïbyEAKahlerH. Changed expression of leukocyte adhesion molecules and increased production of reactive oxygen species caused by Streptococcus pyogenes in human whole blood. APMIS2000; 108: 573–580.
30.
AikawaCNozawaTMaruyamaF. Reactive oxygen species induced by Streptococcus pyogenes invasion trigger apoptotic cell death in infected epithelial cells. Cell Microbiol2010; 12: 814–830.
31.
GoldmannOvon Köckritz-BlickwedeMHöltjeC. Transcriptome analysis of murine macrophages in response to infection with Streptococcus pyogenes reveals an unsual activation program. Infect Immun2007; 75: 4148–4157.
32.
GrimbaldestonMAGeczyCLTediaN. S100A8 induction in keratinocytes by ultraviolet A irradiation is dependent on reactive oxygen intermediate. J Invest Dermatol2003; 121: 1168–1174.
33.
LeeHMShinDMChoiDK. Innate immune responses to Mycobacterium ulcerans via toll-like receptors and dectin-1 in human keratinocytes. Cell Microbiol2009; 11: 678–692.
34.
TsaiPJLinYSKuoCF. Group A streptococcus induces apoptosis in human epithelial cells. Infect Immun1999; 67: 4334–4339.
35.
VanlangenakkerNVanden BergheTKryskoDV. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med2008; 8: 207–220.
36.
TsatsaronisJALyDPupovacA. Group A Streptococcus modulates host inflammation by manipulating polymorphonuclear leukocytes cell death responses. J Innate Immun2015; 7: 612–622.
37.
ChungWODaleBA. Innate immune response of oral and foreskin keratinocytes: utilization of different signaling pathways by various bacterial species. Infect Immun2004; 72: 352–358.
38.
TsouCCChiang-NiCLinYS. Oxidative stress and metal ions regulate a ferritin-like gene, dpr, in Streptococcus pyogenes. Int J Med Microbiol2010; 300: 259–264.
39.
GibsonCMCaparonMG. Insertional inactivation of Streptococcus pyogenes sod suggests that prtF is regulated in response to a superoxide signal. J Bacteriol1996; 178: 4688–4695.