
Abstract
The authors provide the first in vitro and in vivo evidence that perturbations in mitogen-activated protein kinase (MAPK) signal-transduction pathways are involved in the pathophysiology of traumatic brain injury. In primary rat cortical cultures, mechanical trauma induced a rapid and selective phosphorylation of the extracellular signal-regulated kinase (ERK) and p38 kinase, whereas there was no detectable change in the c-jun N-terminal kinase (JNK) pathway. Treatment with PD98059, which inhibits MAPK/ERK 1/2, the upstream activator of ERK, significantly increased cell survival in vitro. The p38 kinase and JNK inhibitor SB203580 had no protective effect. Similar results were obtained in vivo using a controlled cortical impact model of traumatic injury in mouse brain. Rapid and selective upregulation occurred in ERK and p38 pathways with no detectable changes in JNK. Confocal immunohistochemistry showed that phospho-ERK colocalized with the neuronal nuclei marker but not the astrocytic marker glial fibrillary acidic protein. Inhibition of the ERK pathway with PD98059 resulted in a significant reduction of cortical lesion volumes 7 days after trauma. The p38 kinase and JNK inhibitor SB203580 had no detectable beneficial effect. These data indicate that critical perturbations in MAPK pathways mediate cerebral damage after acute injury, and further suggest that ERK is a novel therapeutic target in traumatic brain injury.
Traumatic brain injury (TBI) is the leading cause of death and disability in young adults, affecting approximately 2 million patients per year in the United States alone (Kraus et al., 1996). No effective treatment exists. A major conceptual advance in the management of TBI was the realization that tissue damage after trauma comprised both primary and secondary mechanisms of cell death (McIntosh et al., 1998). After the initial injury resulting from direct mechanical impact, secondary processes of cytotoxicity can continue to evolve over hours or days. The underlying processes that mediate secondary cell death after brain trauma remain to fully defined, but clearly include overlapping mechanisms of excitotoxicity (Smith and McIntosh, 1996), oxidative stress (Lewen et al., 2000), and apoptosis (Raghupathi et al., 2000).
Despite promising preclinical data, pharmacologic blockade of excitotoxic and oxidative injury have yielded mostly disappointing results in various clinical trials for TBI (Morris et al., 1999; Young et al., 1996). Although there are likely to be multiple reasons for these difficulties with clinical trials, a possibility remains that excitotoxicity, oxidative damage, and apoptotic mechanisms represent downstream effectors of cell death. It is conceivable that examination of upstream pathways in the pathophysiology of acute brain injury may lead to the identification of novel therapeutic targets.
Accumulating data now suggest that many downstream mechanisms of cell death may broadly involve perturbations in upstream cell-signaling pathways (Guyton et al., 1996; Mielke and Herdegen, 2000; Stanciu et al., 2000; Xia et al., 1995). Mitogen-activated protein kinase (MAPK) is an evolutionarily conserved mechanism of transducing external stress and injury to internal cellular responses that balance cell survival versus cell death (Chang and Karin, 2001; Robinson and Cobb, 1997). Hence, MAPKs are good candidates for mediating upstream signals that amplify brain cell death after injury. Indeed, recent studies have shown that MAPK is implicated in the progression of acute brain damage after ischemia (Alessandrini et al., 1999; Hu et al., 2000; Namura et al., 2001; Sugino et al., 2000). In this report, we show that the extracellular signal-regulated kinase (ERK) and p38 MAPK pathways are upregulated after TBI, and that pharmacologic inhibition of the ERK pathway significantly improves cell survival in both in vitro and in vivo models of trauma. These data show for the first time the involvement of MAPK pathways in the pathophysiology of TBI, and further suggest that the ERK pathway may present a novel therapeutic target.
MATERIALS AND METHODS
All experiments were performed following an institutionally approved protocol in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
In vitro mechanical trauma model
Standard methods were used to prepare mixed neuron and astrocyte cortical cocultures (Wang et al., 1999, 2001). Briefly, cortical tissue from rat embryonic day-17 rats were isolated and dissociated, and individual cells (2 × 103 cells/mm2) were seeded onto previously prepared confluent 15-to-18-days in vitro astrocyte cultures using a supplement of high-glucose Dulbecco modified Eagle medium containing 10% horse serum (Life Technologies, Gaithersburg, MD, U.S.A.). After 10 to 12 days, cocultures were subjected to a standard mechanical injury model of trauma (Katano et al., 1999). The culture medium was replaced with 0.2% G5 supplement (Life Technologies) 12 to 14 hours before mechanical injury, and control cultures were maintained in the same medium. A previously described standard model of mechanical trauma (Katano et al., 1999; Lau and Yu, 2001; Morrison et al., 1998; Shah et al., 1997; Tecoma et al., 1989) was induced by using a sterile 21-gauge needle to draw parallel scratches across the circular wells of culture plates (12 scratches in six-well plates and eight scratches in 24-well plates, respectively). For the pharmacologic inhibitor studies, cultures were pretreated with micromolar concentrations of PD98059 (New England Biolabs, Beverly, MA, U.S.A.) or SB203580 (Calbiochem, San Diego, CA, U.S.A.) (20-mmol/L stock solution in dimethyl sulfoxide) for 1 hour before injury. The dose ranges studied here were selected based on previously reported efficacies and specificities for these two inhibitors (Garrington and Johnson, 1999). To assess cell survival, the activity of lactate dehydrogenase was measured 24 hours after mechanical injury (Cytotoxicity Detection Kit; Boehringer Mannheim, Indianapolis, IN, U.S.A.). A culture maintained in high-glucose Dulbecco modified Eagle medium containing 10% horse serum was used as the baseline (100%) survival control.
In vivo controlled cortical impact model
Adult male C57BL/6 mice (22–25 g, Charles River, Wilmington, MA, U.S.A.) were used. Methods to induce controlled cortical impact brain injury have been previously described (Smith et al., 1995). Briefly, mice were anesthetized with halothane (1%–1.2%) under spontaneous respiration in an air–oxygen mixture in a stereotactic head holder. Rectal temperature was maintained at 37.5°C with a thermostat-controlled heating pad. A 5-mm craniotomy was performed over the right parietal cortex between the bregma and lambda, a 3-mm flat-tipped impactor was placed on the dural surface at a 20-degree angle, and injury was induced using 4-m/s impact velocity, 1-mm impact depth, and 150-millisecond impact dwell time. For the inhibitor studies, 2 μL 200-μmol/L PD98059 (n = 7) and 2 μL 100-μmol/L SB203580 (n = 7) were stereotactically administered into the right lateral ventricle 30 minutes before trauma. Controls received equal volumes of 0.4% dimethyl sulfoxide (n = 7). Intraventricular administration was used because the PD98059 compound does not reliably penetrate the blood–brain barrier (Garrington and Johnson, 1999). Inhibitor doses were chosen based on previously determined dose ranges used in ischemic studies of mouse brain (Alessandrini et al., 1999). Behavioral deficits were assessed at 1, 2, and 7 days after TBI using the standard rotarod test (Hamm et al., 1994; Wang et al., 2000). Rotarod scores were measured as the latency or time successfully spent running on the rotating rod (35 revolutions/min), and expressed as a percentage of preinjury baselines to reduce interanimal variability. At 7 days after injury, mice were deeply anesthetized with sodium pentobarbital, and transcardially perfused with phosphate-buffered saline (PBS) and 4% paraformaldehyde. Using standard procedures (Wang et al., 2000), the brains were rapidly removed and sectioned with a freezing microtome into 25-μm-thick coronal slices. Every twentieth section was mounted onto a glass slide and stained with 0.1% cresyl violet. Histologic lesion areas were quantified with a standard computer-assisted image analysis program, and lesion areas were integrated to obtain total lesion volumes. Lesion delineation of the necrotic cavitation after trauma was performed blindly using criteria established in previous studies in this mouse model (Wang et al., 2000).
Western immunoblot analysis
For the in vitro cell culture studies, the medium was removed and plates were washed twice with chilled PBS. The cells were quickly scraped and collected by centrifugation, then stored at −80°C until assayed. For the in vivo models, damaged cortex (approximately 30–40 mg) from the traumatized hemisphere was rapidly excised and frozen in liquid nitrogen. Standard methods were used to perform immunoblot analysis (Asahi et al., 2000; Wang et al., 2000). Three major MAPK pathways were assessed: ERK, p38 kinase (p38), and c-jun N-terminal kinase (JNK). Primary antibodies against phosphospecific ERK 1/2, phospho-stress–activated protein kinase and phospo-JNK, total stress-activated protein kinase and JNK, and phospho-activating transcription factor (ATF)-2 (Thr 71) were obtained from New England Biolabs. Antiphospho-p38 was obtained from Promega (Madison, WI, U.S.A.), and antibodies against total p38 (c-20) and total ERK2 (c-14) were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA, U.S.A.). Western blots were quantified using standard densitometry techniques (Asahi et al., 2001).
In-gel c-jun N-terminal kinase activity assay
The JNK kinase assays were performed using stress-activated protein kinase and JNK kinase assay kit (Upstate Biotechnology, Lake Placid, NY, U.S.A.). Each protein sample (200 μg) was incubated with 5 μg antipan-JNK/stress-activated protein kinase 1 antibody (Upstate Biotechnology) overnight at 4°C. Immunocomplexes were collected with 20 μL protein A-agarose beads (Life Technologies), and the pellet was washed three times with PBS and once with kinase buffer (20-mmol/L MOPS, pH 7.2; 25-mmol/L b-glycerol phosphate; 5-mmol/L ethyleneglycoltetracetic acid; 1-mmol/L sodium or-thovanadate; 1-mmol/L chlorophenothane). After the addition of 5 μCi [r-32 P]ATP (NEN, Boston, MA, U.S.A.), 50-μmol/L ATP, and 1 μg c-jun (1–169)-GST (Upstate Biotechnology) in 30 μL kinase buffer, the contents were incubated at 30°C for 30 minutes. Samples were then subjected to autoradiography by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Stress-activated protein kinase 1a and c-jun N-terminal kinase 2 fusion proteins served as positive controls.
Immunohistochemistry
Immunohistochemistry was performed on free-floating coronal sections to evaluate cell specificity. Mice were anesthetized deeply with an overdose of intraperitoneal sodium pentobarbital (100 mg/kg) and then transcardially perfused with ice-cold PBS, followed by 4% paraformaldehyde in 0.1-mol/L PBS (pH 7.4). The brains were removed quickly and stored in the same fresh buffer containing 20% sucrose. The brains were cut into 40-μm thick coronal sections on a freezing microtome. After they were washed three times in PBS containing 0.1% Triton X-100 (PBS-T, pH 7.4; Sigma, St. Louis, MO, U.S.A.), the sections were incubated with 10% normal goat serum (Sigma) in Tris-base saline/Tween 20 for 1 hour. Then sections were incubated with primary antibody against phospho-ERK 1/2 (1:100, New England Biolabs), neuronal nuclei marker (NeuN, 1:100; Chemicon, Temecula, CA, U.S.A.), and glial fibrillary acidic protein (1:100, Zymed, South San Francisco, CA, U.S.A.) at 4°C overnight. After washing with PBS-T, they were incubated with goat antirabbit immunoglobulin G against phospho-ERK conjugated with fluorescein isothiocyanate (1:100, Santa Cruz), goat antimouse immunoglobulin G against NeuN conjugated with cy3 (1:100, Zymed), and goat antirat immunoglobulin G against glial fibrillary acidic protein conjugated with rhodamine (1:100, Santa Cruz). All fluorescent-stained sections were subsequently analyzed on a Leica DMRB/Bio-Rad 1024 confocal microscope with krypton–argon laser.
Statistical analysis
Data were expressed as mean ± SD. Analysis of variance followed by Tukey-Kramer tests were used for multigroup comparisons. P values less than 0.05 were considered significant.
RESULTS
We used in vitro and in vivo systems to assess ERK, p38, and JNK, three major MAPK pathways. Our first series of experiments were conducted using a standard needle scratch model of mechanical trauma in primary cortical cocultures of neurons and astrocytes derived from rat brain. Western blots showed that ERK and p38 were rapidly phosphorylated after trauma, indicating the upregulation of these two pathways (Figs. 1A and 1B). These changes occurred within minutes but were transient, and returned to baseline by 1 to 3 hours. No change in the phosphorylation of JNK was observed within the limits of our detection system (Figs. 1A and 1B). However, the antiphospho-JNK antibody showed a cross-reactivity against p42 phospho-ERK; this was detected as the emergence of a new band close to the level of phospho-JNK on the Western blot (Fig. 1A). We further performed an in-gel kinase activity assay using c-jun as a substrate to check the specificity of our findings. This assay confirmed that there was no detectable upregulation in the JNK pathway in our in vitro model of mechanical trauma (Fig. 1A).
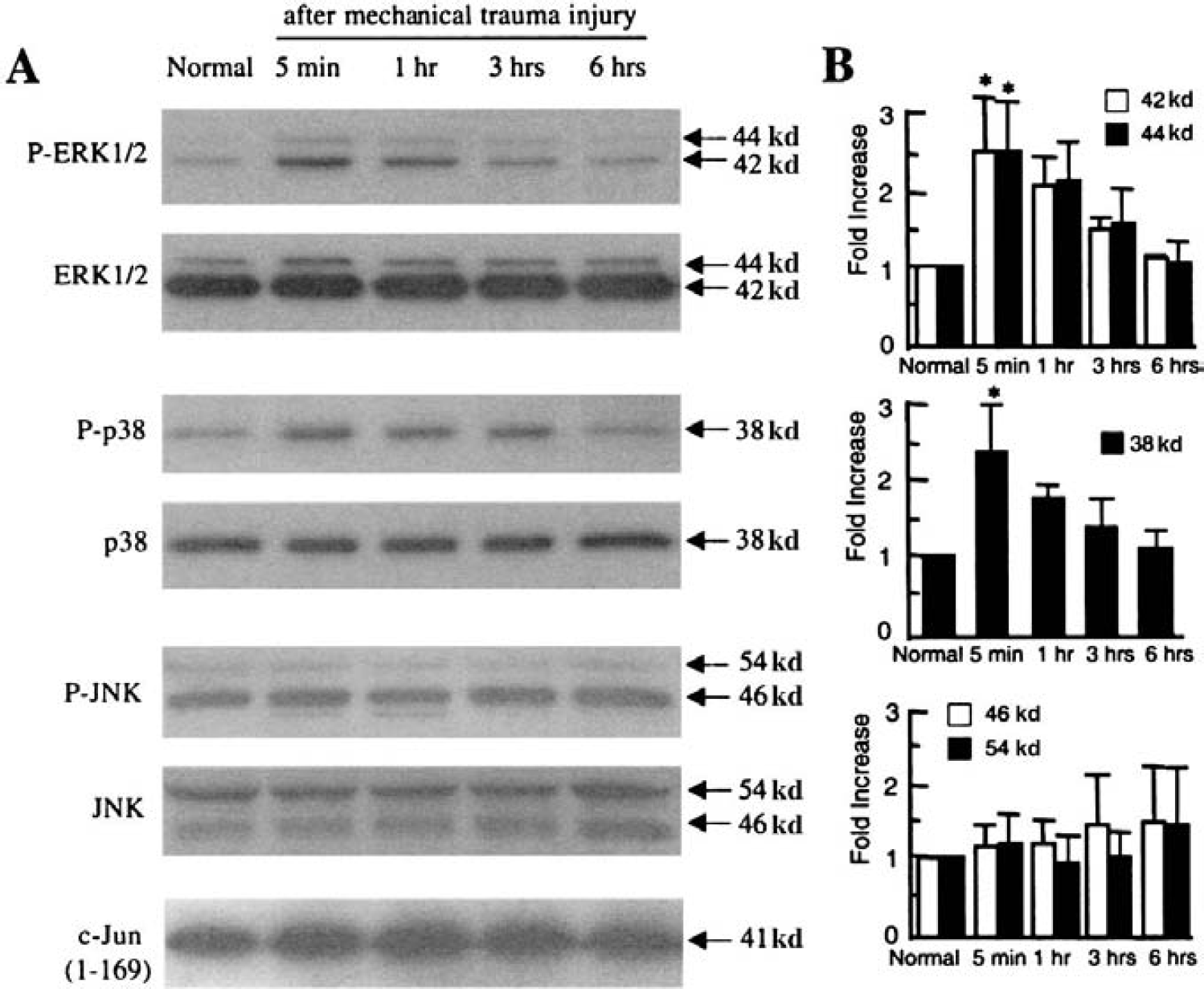
In vitro model of traumatic brain injury.
To assess how these MAPK pathways may mediate brain cell survival and death in our model of mechanical trauma, two pharmacologic inhibitors were used. PD98059 was used to inhibit MAPK/ERK kinase 1/2, the upstream activator of ERK (Cohen, 1997; Garrington and Johnson, 1999), whereas SB203580 was used to inhibit both p38 and JNK pathways (Cohen, 1997; Garrington and Johnson, 1999). Both drugs were added to the mixed cortical cultures 60 minutes before mechanical trauma. Cytotoxicity was assessed by measuring lactate dehydrogenase release at 24 hours. The efficacy of PD98059 was checked by assessing the level of ERK phosphorylation after trauma. Micromolar concentrations of PD98059 effectively inhibited ERK activation (Figs. 2A and 2B), and significantly protected against traumatic cytotoxicity (Fig. 3). The effectiveness of SB203580 was checked by assessing the phosphorylation state of ATF-2, a downstream substrate of both p38 and JNK pathways (Garrington and Johnson, 1999). After trauma, ATF-2 was rapidly phosphorylated according to a timeframe that closely followed that of p38 upregulation (data not shown). Micromolar concentrations of SB203580 inhibited the phosphorylation of ATF-2 (Figs. 2A and 2C), but had no detectable protective effects in terms of cell survival at 24 hours after mechanical trauma (Fig. 3).
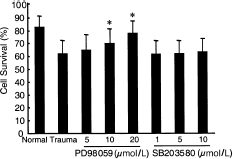
Efficacy of mitogen-activated protein kinase (MAPK) inhibitors in vitro. (A) Representative Western blots showing inhibitor efficacy. Twenty micromoles per liter PD98059, a MAP/ERK inhibitor of the extracellular signal-regulated kinase (ERK) pathway, reduces the phosphorylation of ERK after mechanical trauma. Ten micromoles per liter SB203580 reduces the phosphorylation of the p38/JNK (c-jun N-terminal kinase) substrate ATF-2. (B) Western blots were quantified from three independent culture experiments (with six culture wells per experiment) using standard densitometry techniques. Phospho-ERK was increased after mechanical trauma, and PD98059 significantly reduced trauma-induced phospho-ERK 1/2 levels. *P < 0.05 versus untreated mechanically damaged cultures. (C) Western blots were quantified from three independent culture experiments (with six culture wells per experiment) using standard densitometry techniques. Phospho-ATF-2 was increased after mechanical trauma, and SB203580 significantly reduced trauma-induced phospho-ATF-2 levels. *P < 0.05 versus untreated mechanically damaged cultures.
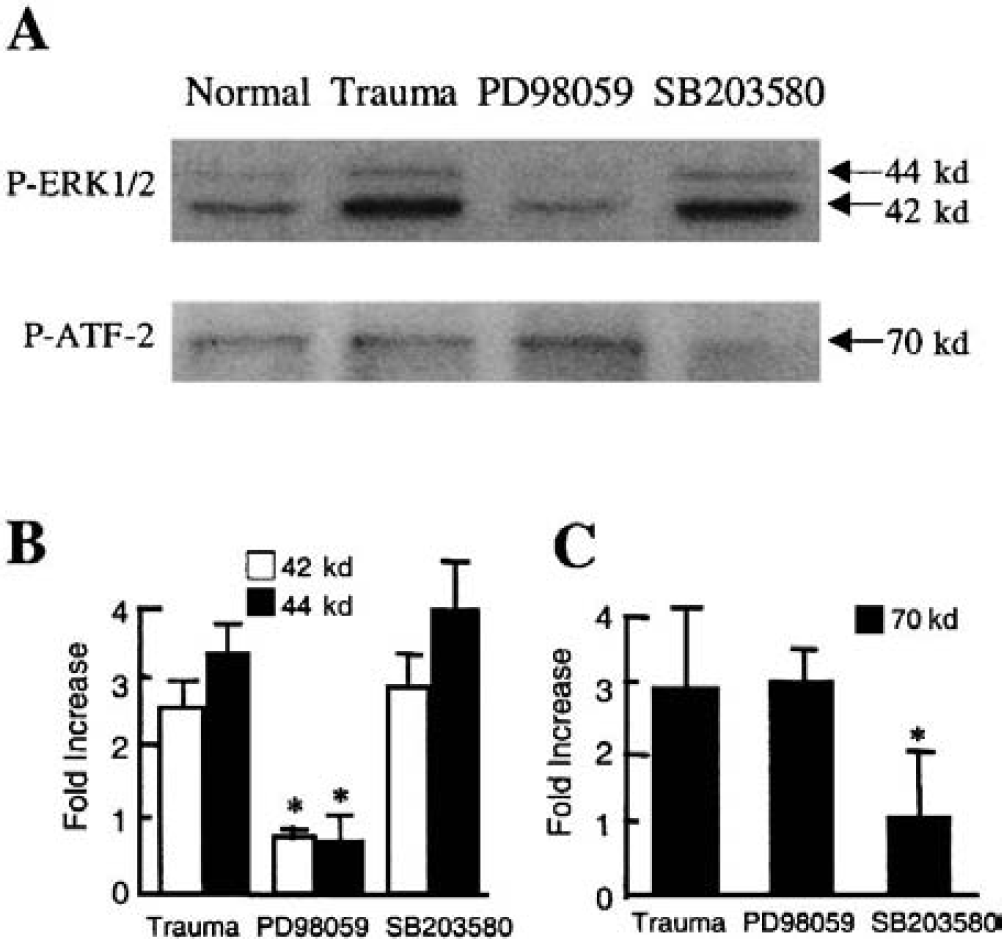
PD98059 significantly improves cell survival after mechanical trauma compared with untreated cultures (*P < 0.05). However, the p38/JNK (c-jun N-terminal kinase) inhibitor SB203580 has no detectable protective effects at the dose ranges studied. The “normal” group represents cultures subjected to medium change without trauma. Cultures maintained in high-glucose Dulbecco modified Eagle medium containing 10% horse serum was used as the baseline (100%) survival control. N = 4 independent culture experiments were performed (with six culture wells per experiment).
To further extend these in vitro findings, a standard mouse model of controlled cortical impact injury was used. Protein samples for Western blots were obtained from damaged cortical regions to assess the involvement of the various MAPK pathways. The ERK and p38 isoforms were rapidly phosphorylated after cortical impact trauma, indicating upregulation of these pathways in vivo (Figs. 4A and 4B). These rapid changes appeared to be sustained only transiently. By 1 to 3 hours after TBI, phospho-ERK and phospho-p38 had returned to approximately baseline levels. As in the previous in vitro system, no change was observed in the JNK pathway within the limits of our detection sensitivity (Figs. 4A and 4B). However, the antiphospho-JNK antibody also showed a cross-reactivity against p42 phospho-ERK; this was detected as the emergence of a new band close to the level of phospho-JNK on the Western blot (Fig. 4A). Nevertheless, an in-gel kinase activity assay showed no changes in the phosphorylation of c-jun, further confirming that the JNK pathway was not activated in this in vivo model of TBI (Fig. 4A).
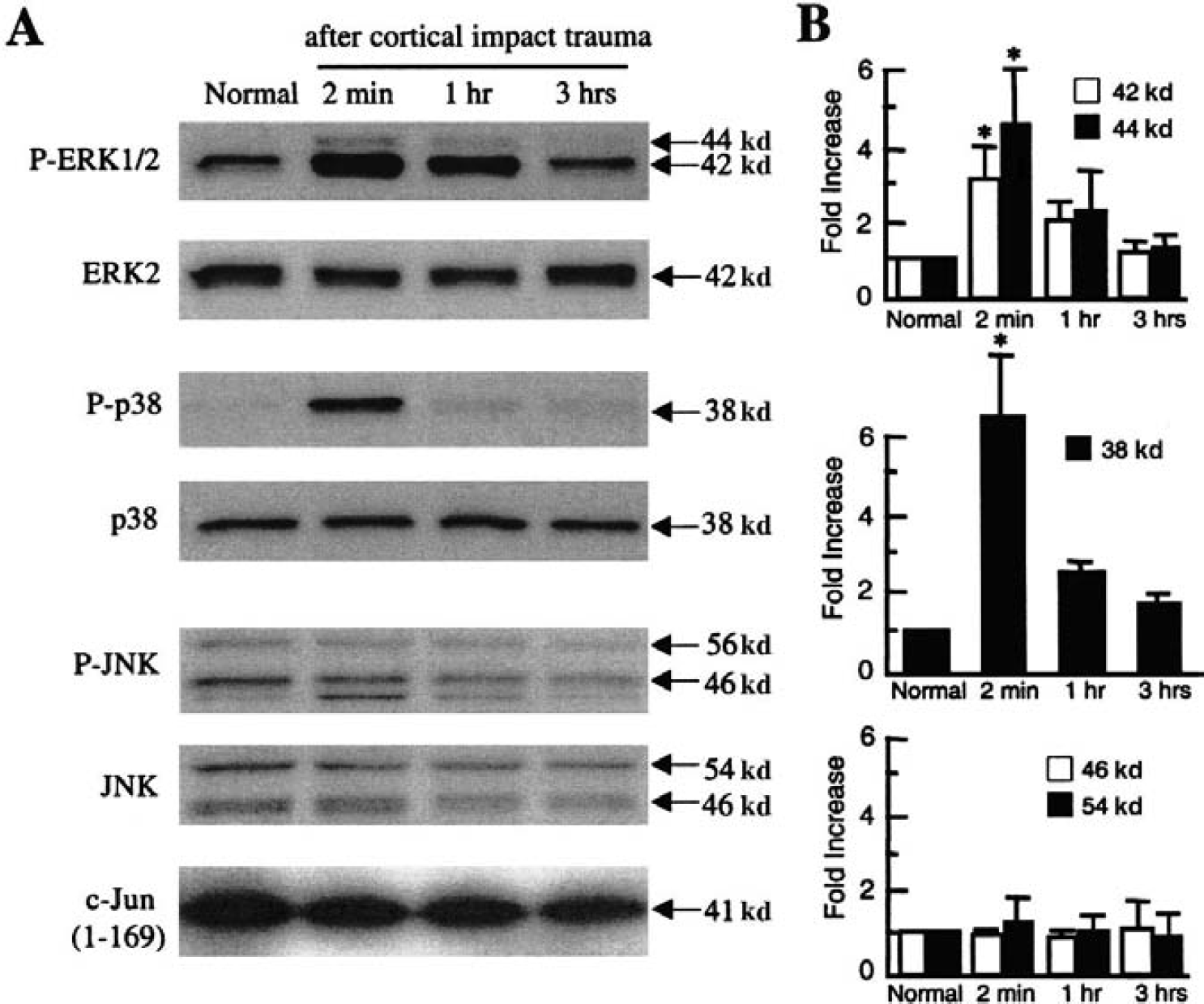
In vivo model of traumatic brain injury. (A) Representative Western blots show that extracellular signal-regulated kinase (ERK) 1/2 and p38 are rapidly phosphorylated after controlled cortical impact in mouse brain. Total (phosphorylated and nonphosphorylated) levels of ERK 2 and p38 remain constant. No changes in phosphorylation of c-jun N-terminal kinase (JNK) are detected. However, the antiphospho-JNK antibody cross-reacts with phospho-ERK so that a faint band at 42 kd is visible corresponding with the activation of the ERK pathway after trauma. An in-gel kinase assay using c-jun as the substrate further confirms that there was no detectable upregulation in the JNK pathway. (B) Western blots of phosphorylated forms of ERK, p38, and JNK were quantified using standard densitometry techniques. *P < 0.05 versus normal uninjured brain, N = 4 per group.
To confirm the cellular specificity of the phospho-ERK upregulation induced by trauma, confocal microscopy was used to assess immunohistochemical staining. These data showed that phospho-ERK in traumatized cortex approximately 2 minutes after injury colocalized with the neuronal marker NeuN (Figs. 5A, 5B, and 5C) but not the astrocytic marker glial fibrillary acidic protein (Figs. 5D, 5E, and 5F).
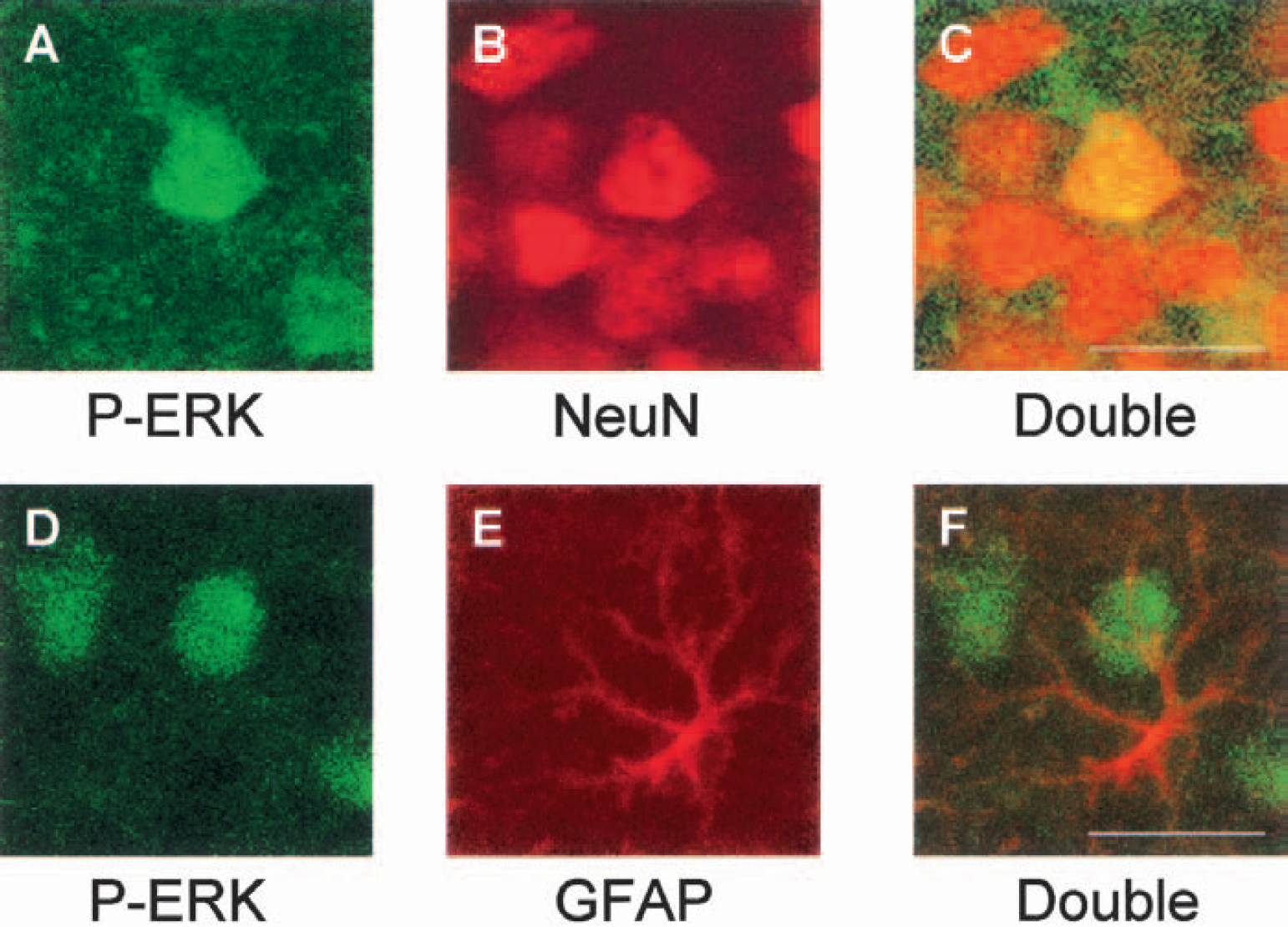
Fluorescent-stained cells in damaged cortex approximately 2 minutes after traumatic brain injury. Brain sections (40 μm) were stained for phospho-ERK (conjugated fluorescein isothiocyanate, green) (
Once again, PD98059 and SB203580 were used as pharmacologic inhibitors of the ERK and p38/JNK pathways, respectively. These drugs were injected into the lateral ventricles of the mouse brain 30 minutes before cortical impact injury (200-μmol/L PD98059 or 100-μmol/L SB203580 in 2-μL volumes). Control mice were treated with equal volumes of vehicle (0.4% dimethyl sulfoxide). PD98059 decreased the phosphorylation of ERK, and SB203580 decreased the phosphorylation of ATF-2, showing that pharmacologic inhibition was effective in vivo (Figs. 6A, 6B, and 6C).
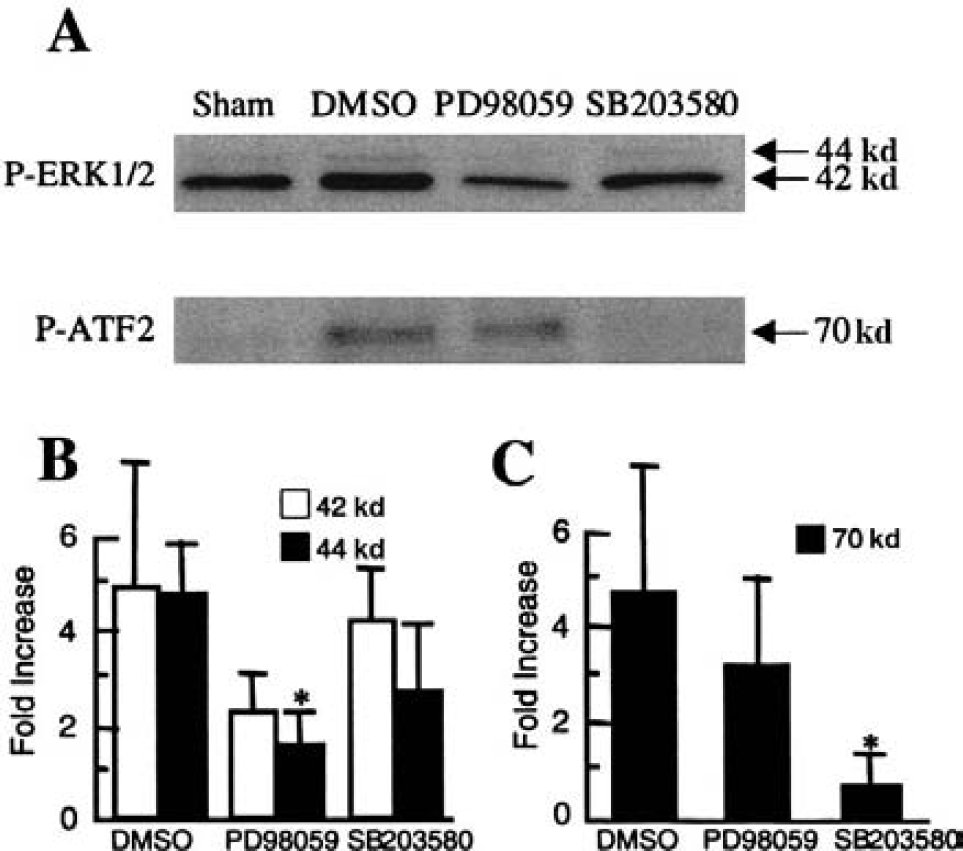
Efficacy of mitogen-activated protein kinase (MAPK) inhibitors in vivo.
To assess the contribution of these MAPK pathways to the pathophysiology of TBI, we examined motor and morphologic outcomes over 7 days after injury, and compared vehicle-treated mice with those treated with PD98059 or SB203580. Motor outcomes were assessed using the standard rotarod device, whereby the time (latency) that mice are able to run on a rotating rod was measured before and after TBI. Before cortical impact trauma, all mice had latencies on the order of 120 seconds, and there were no differences between the three treatment groups. At 1 day after TBI, latencies were significantly reduced to approximately 30% of preinjury baseline (Table 1). There were no statistically detectable differences between the drug-treated mice and vehicle-treated controls on days 1, 2, and 7 after injury (Table 1). By 7 days after injury, mice in all three groups had recovered motor function so that rotarod latencies approximated preinjury baselines (Table 1).
Rotarod latency after injury (seconds)
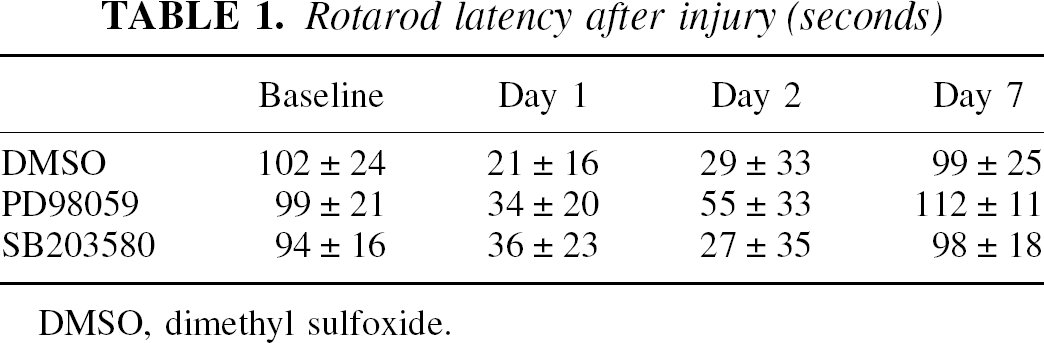
DMSO, dimethyl sulfoxide.
At 7 days after trauma, mice were killed and their brains removed for sectioning and cresyl-violet staining. In this controlled cortical impact model of brain injury, well-demarcated lesions occurred with cavitations that involved cortex and extended into hippocampal structures (Fig. 7A). Mice treated with PD98059 showed significantly decreased traumatic lesion volumes compared with vehicle-treated controls (Figs. 7A and 7B). However, there were no significant effects in the SB203580-treated mice (Figs. 7A and 7B).
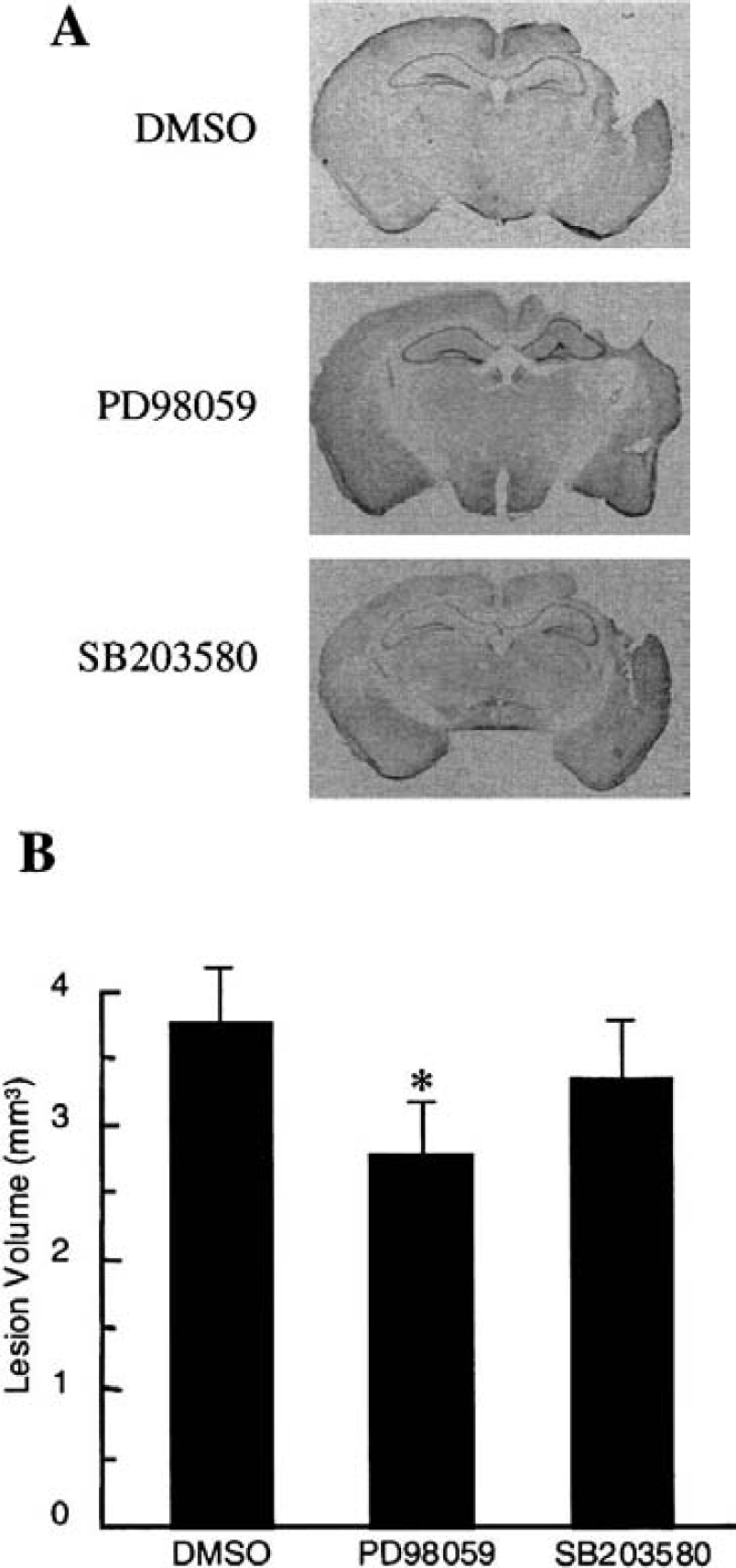
Representative cresyl-violet stained sections of mouse brains 7 days after controlled cortical impact trauma.
DISCUSSION
This study shows that ERK and p38 MAPK pathways are rapidly upregulated after TBI. The pathophysiology of TBI involves multiple downstream processes of excitotoxicity (Faden et al., 1989; Hayes et al., 1988; Palmer et al., 1993), oxidative stress (Arvin et al., 1996; Kontos and Povlishock, 1986; Xu et al., 1998), and abnormal apoptosis (Clark et al., 1997; Colicos and Dash, 1996; Conti et al., 1998; Crowe et al., 1997; Fox et al., 1998; Kaya et al., 1999; Liu et al., 1997; Pravdenkova et al., 1996; Rink et al., 1995; Springer et al., 1999; Yakovlev et al., 1997). Hence, the involvement of MAPKs is consistent with the idea that these pathways represent highly conserved upstream mechanisms for transducing stress signals in eukaryotic cells (Garrington and Johnson, 1999; Guyton et al., 1996; Mielke and Herdegen, 2000; Stanciu et al., 2000; Xia et al., 1995).
Extracellular perturbations and stress are transduced by MAPK cascades, resulting in alterations in transcriptional factor profiles and gene expression (Chang and Karin, 2001; Garrington and Johnson, 1999). However, the precise roles for the various MAPKs in determining the ultimate balance between cell survival and cell death remain to be fully defined. At present, beneficial versus detrimental effects appear to be dependent on model systems and the type of injury conditions used. For example, after serum withdrawal and oxidative stress in cerebellar neuronal cultures, the ERK pathway mediates protective effects and the p38/JNK pathways mediate deleterious effects (Xia et al., 1995). Extracellular signal-regulated kinase has been shown to be involved in preconditioning responses that protect against secondary insults (Gonzalez-Zulueta et al., 2000), and has been associated with neuroprotection in global cerebral ischemia (Hu et al., 2000; Sugino et al., 2000). For the most part, these protective effects of ERK appear to be related to the transcriptional upregulation of neuroprotective antioxidant systems (Gonzalez-Zulueta et al., 2000). Alternatively, sustained activation of ERK can be deleterious after hyperexcitation and focal ischemic injury (Alessandrini et al., 1999; Murray et al., 1998; Stanciu et al., 2000). The mechanisms that mediate these deleterious effects of ERK activation remain to be fully defined, but may include the fact that ERK can phosphorylate synapsin and amplify excitotoxic glutamate release after brain injury (Alessandrini et al., 1999; Murray et al., 1998). Another possible deleterious mechanism may involve the effects of extracellular proteolysis, because ERK is known to upregulate the extracellular matrix degrading enzyme matrix metalloproteinase 9 after brain injury (Wang et al., 2001). Extracellular signal-regulated kinase-mediated upregulation of matrix metalloproteinase 9 activity can degrade matrix components and interrupt critical cell-cell and cell-matrix interactions necessary for neuronal survival (Cuzner and Lo, 2001). We have previously shown that matrix metalloproteinase 9-deficient knockout mice are protected against TBI (Wang et al., 2000) and focal stroke (Asahi et al., 2000, 2001). Similarly, variable roles have also been described for p38 and JNK, depending on the models and injury paradigms used. For example, JNK appears to mediate Fas-induced apoptosis (Le-Niculescu et al., 1999) but prevents tumor necrosis factor–induced p38 activation and cell death (Minamino et al., 1999). Overall, questions of whether ERK or p38 is beneficial or detrimental may significantly depend on the model system and injury paradigm used. In the context of acute TBI, our present data suggest that ERK plays a deleterious role in vitro and in vivo.
In this study, we used PD98059 and SB203580 as pharmacologic probes to investigate the involvement of MAPK pathways. Inhibition of the ERK pathway with the MAPK/ERK kinase inhibitor PD98059 was protective against trauma in vitro and in vivo, whereas the p38/JNK inhibitor SB203580 had no detectable effect. At the micromolar doses used in this study, PD98059 is specific for the ERK pathway (Cohen, 1997), though crossinhibition against cyclooxygenase 1 and 2 in some model systems remains a possible caveat (Cohen, 1997; Davies et al., 2000). For the doses used here, SB203580 may inhibit both p38 and JNK (Cohen, 1997; Davies et al., 2000). However, because we did not observe changes in JNK after trauma, the effects of SB203580 may have been primarily mediated by p38 inhibition. The JNK– MAPK pathway is considered to be a general stress-response pathway (Davis, 2000; Mielke and Herdegen, 2000; Xia et al., 1995), so it was somewhat surprising that we did not observe changes in JNK in our in vitro and in vivo models of brain trauma. Upregulation in the JNK pathway has been described in some models of cerebral ischemia (Sugino et al., 2000), and tissue ischemia may accompany TBI under some conditions (Dietrich et al., 1998). The present data showing lack of JNK activation suggest that there may be critical differences between the pathophysiology of TBI and ischemia (McIntosh et al., 1998). Nevertheless, it is conceivable that our Western blot and in-gel kinase assays were not sensitive enough to low-level JNK phosphorylation.
There are a few caveats associated with the present study. First, although PD98059 significantly reduced morphologic lesion volumes, there were no detectable improvements in the motor outcomes. In part, this may be due to a lack of sensitivity in the rotarod test, because inherent plasticity mediating functional recovery in the untreated controls may mask subtle beneficial effects of the drug. More refined cognitive endpoints (e.g., water maze) using longer-term outcomes are warranted, and are being pursued in our laboratory. Second, we do not map the precise cerebral distributions of our intraventricular drug-delivery protocols. The Western blots show effective inhibition of the targeted MAPKs, but more data regarding brain drug distributions would be useful for future dose-optimization studies. Third, all treatments here were administered before traumatic injury, and our purpose was to establish proof of principle. In the clinical scenario, it will be important to determine the time windows available for therapy. Here, ERK and p38 activation was transient and returned to baseline by approximately 1 to 3 hours. Nevertheless, we cannot exclude the possibility that long-term changes may also occur as the brain recovers from trauma. Fourth, the focus in this study was limited to the cortex, where phospho-ERK was upregulated in neurons and inhibition of this pathway yielded protection against cortical damage. However, others have described complex patterns of differential upregulation of phospho-ERK and phospho-p38 in neurons and glia, depending on the type of insult and brain regions involved (Hu et al., 2000; Irving et al., 2000; Mandell et al., 2001; Namura et al., 2001; Sugino et al., 2000). It will be important to carefully assess the precise cellular patterns of MAPKs in multiple brain regions for this model of cortical impact trauma. Finally, we do not address other kinase pathways after brain injury. An important pathway that warrants investigation may be the serine threonine kinase pathway, which has been shown to mediate protective responses against a wide range of cell-death triggers (Datta et al., 1999). There may be significant cross talk between the various kinase pathways, so it will be important for future studies to integrate the responses of the wide spectrum of signal-transduction cascades after brain injury.
In summary, this report provides the first evidence that MAPK pathways contribute to the pathophysiology of TBI. Importantly, inhibition of the ERK pathway with PD98059 decreased cytotoxicity in vitro and reduced traumatic lesion volumes in vivo. Manipulation of the ERK pathways may represent a novel therapeutic target for ameliorating the devastating progression of tissue damage after TBI.
Footnotes
Acknowledgments:
The authors thank Drs. Tetsuyuki Teramoto, Minoru Asahi, and Michael Whalen for critical discussions, and Drs. Michael Moskowitz and Bruce Rosen for their unstinting intellectual support and advice.