
Abstract
TLR4 polymorphisms such as Asp299Gly and Thr399Ile related to Gram-negative sepsis have been reported to result in significantly blunted responsiveness to LPS. Our study group previously screened other TLR4 polymorphic variants by checking the NF-κB activation in comparison to wild type (WT) TLR4 in human embryonic kidney 293T cells. In this study, we found that the Lys694Arg (K694R) polymorphism reduced the activation of NF-κB, and the production of downstream inflammatory factors IL-1, TNF-α and IL-6, representing the K694R polymorphism, led to blunted responsiveness to LPS. Then, we examined the influence of the K694R polymorphism on total and cell-surface TLR4 expression by Western blotting and flow cytometry, respectively, but observed no differences between the K694R polymorphism and WT TLR4. We also used co-immunoprecipitation to determine the interaction of the K694R polymorphism and WT TLR4 with their co-receptor myeloid differentiation factor 2 (MD2) and their downstream signal adaptor MyD88. We found that K694R reduced the recruitment of MyD88 in TLR4 signalling but had no impact on the interaction with MD2.
Introduction
TLRs are important PRRs that play an essential role in the innate immune response and which recognise PAMPs.1,2 TLR4, the most researched receptor among the TLR family, is a transmembrane receptor which is composed of an extracellular domain, an intracellular domain and a leucine-rich repeat motif.3,4 The TLR4 agonist, toxic LPS, often consisting of O-antigen, core oligosaccharide and lipid A, constitutes the key component anchored in the outer membrane of various Gram-negative bacteria and is an important innate immune stimulator.5,6
The MyD88-dependent pathway is one of the most important TLR4 signalling pathways that leads to the activation of NF-κB and the production of inflammatory cytokines. 7 After stimulation with LPS, LPS-binding protein recognises the lipid A moiety of LPS, and the LPS complex is chaperoned to the extracellular myeloid differentiation factor 2 (MD2)-TLR4 heterodimer by another protein known as cluster of differentiation 14 (CD14).8–10 Subsequently, this MD2–TLR4 heterodimer dimerises with another MD2–TLR4 compound, and recruits the specific intracellular adaptor molecule MyD88 to induce the activation of NF-κB and the expression of inflammatory cytokine genes, such as TNF-α, IL-1, IL-6 and IL-18 (Figure 1).11–14
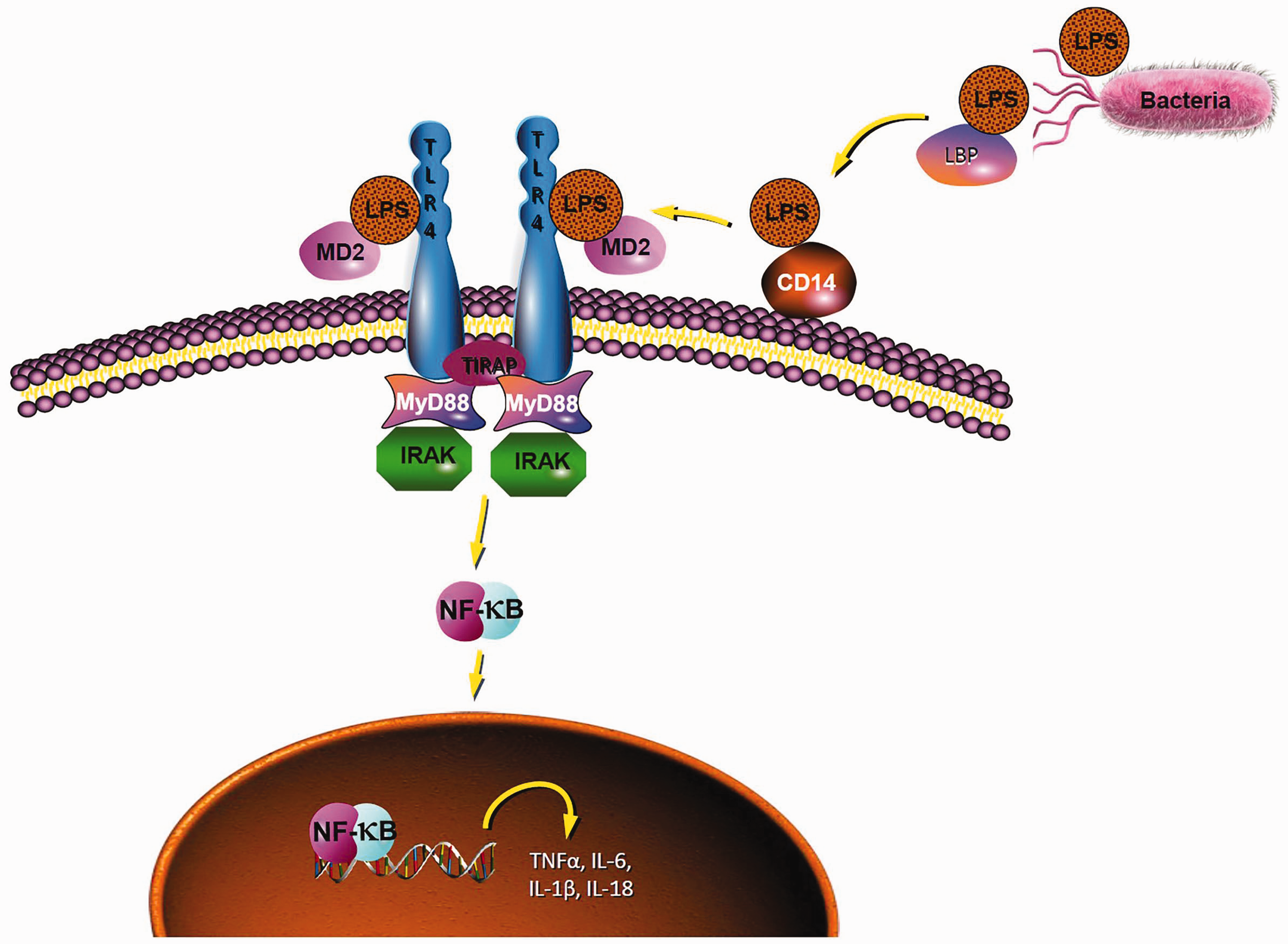
Diagram of the TLR4 signalling pathway.
It has been reported that TLR4 non-synonymous coding single nucleotide polymorphisms (cSNPs) such as Asp299Gly (D299G) and Thr399Ile (T399I) which encode single amino acid substitutions in the ectodomain of TLR4 could result in significantly blunted responsiveness to LPS.15–17 People with D299G/T399I polymorphisms are more vulnerable to bacterial infections, 15 periodontitis 18 and acute dental abscesses. 19 The current impact of non-synonymous cSNPs on TLR4 is mainly concentrated on two sites: D299G and T399I. However, there are 18 TLR4 polymorphisms in the UniProtKB database. In theory, non-synonymous cSNPs have an impact on the structure and function of the TLR4 protein, but there are few reports on the functional changes of other sites at present, and research on the mechanism is even scarcer. Our research group previously screened out the K694R polymorphism that also leads to blunted responsiveness to LPS, in addition to D299G and T399I. 20 In this study, we verify the impact of K694R that leads to the blunted response to LPS by checking the NF-κB activation and inflammatory factor level in comparison to wild type (WT) TLR4, and examine the influence of K694R on TLR4 expression and TLR4 interaction with MD2 and MyD88.
Methods
Plasmids, reagents, cell and culture
PHY-028-GFP-TLR4 (WT), PHY-028-GFP-TLR4 (K694R), pcDNA3.0-Flag-MD2, pcDNA3.0-CD14, pNF-κB-luc, pRL-SV40-N and pcDNA3.0-Flag-MyD88 were bought from Hanyin Biotechnology Limited Company (Shanghai, PR China). K694R TLR4 primers were: upper, 5′-
Cell transfection
For quantitative RT-PCR, Western blotting (WB) and flow cytometry, HEK293T cells were seeded onto six-well plates at a density of 4 × 105 cells/well and incubated in a 5%CO2 incubator until cell density reached 70–90%. Cells were co-transfected with plasmids PHY-028-GFP-TLR4 WT or K694R (1.5 µg), pcDNA3.0-Flag-MD2 (0.5 µg) and pcDNA3.0-CD14 (0.5 µg), as well as pNF-κB-luc (1 µg) and pRL-SV40-N (0.1 µg) for dual luciferase reporter assay. The transfection was conducted with 500 µl/well serum-free DMEM and 6 µl/well Lipofectamine 2000 transfection reagent (11668019; Invitrogen, Carlsbad, CA) for 6 h. The transfection medium was removed, and the complete DMEM containing 10% FBS was added to recover for 24 h. Between 36 and 48 h after transfection with plasmids conjugated with GFP, a fluorescence microscope cell image was taken to verify the efficiency of the transfection (Supplemental Figure S2). Cells were treated with LPS at 100 ng/ml for 6 h. For co-immunoprecipitation, HEK293T cells were seeded into 10 cm tissue culture dishes and co-transfected with pcDNA3.0-Flag-MD2 (2 µg), pcDNA3-CD14 (2 µg), PHY-028-GFP-TLR4 WT or K694R (6 µg) with 3 ml/dish serum-free DMEM and 20 µl Lipo2000 transfection reagent.
RNA isolation, reverse transcription and RT-PCR
Total RNA was extracted from HEK293T cells using 1 ml/well TRIzol (Takara, Shiga, Japan). Purified RNA was reverse transcribed using a PrimeScript RT Reagent Kit with gDNA Eraser (Takara) through the ProFlex PCR System (ABI, Singapore) to obtain complementary DNA (cDNA). cDNA (1 µl) was analysed by quantitative RT-PCR using 5 µl SYBR Green Supermix (Bio-Rad, Hercules, CA), 0.2 µl ROX enzyme, 0.8 µl gene-specific primers and 3 µl DEPC H2O (Sangon, Shanghai, PR China) on a Bio-Rad IQ5 real-time PCR instrument.
The following primers were synthesised by Sangon (Shanghai, China): human GAPDH forward, 5′-
The melting curve was analysed to ensure specific amplification, and data were processed using the 2−ΔΔCT method.
Western blotting
Whole cell lysates from HEK293T cells were prepared using a cold lysis buffer (100 µl/well) containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, sodium pyrophosphate, β-glycerophosphate, EDTA, Na3VO4 leupeptin and PMSF on ice for 15 min. The cell lysate concentrations were measured using an enhanced bicinchoninic acid (BCA) protein assay kit (P0010S; Beyotime, Beijing, PR China). Proteins (20 µg) were fractionated in 10% SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Merck Millipore, Darmstadt, Germany). Membranes were blocked with 5% BSA (B2064-100G; Sigma–Aldrich) in TBST for 1 h at room temperature (about 25°C). Then, they were probed with the primary Abs against GAPDH (1:10,000) and TLR4 (1:1000) at 4°C overnight (about 12 h). The membranes were washed five times with TBST (5 min/time) and incubated with secondary Ab (1:5000, 7076P2; Cell Signaling Technology) for 1 h at room temperature. Proteins were then visualised using the chemiluminescence method (SQ201; EpiZyme, Shanghai, PR China), analysed with a ChemiScope 60 image analyser (Clinx Science Instruments, Shanghai, PR China) and quantified using Image J software (National Institutes of Health, Bethesda, MD).
Dual luciferase reporter assay
Cells were lysed in reporter assay lysis buffer (Promega, Madison, WI), and firefly and Renilla luciferase activities were measured with the Dual Luciferase Reporter Assay system (Promega) on a FlexStation 3 (Molecular Devices, San Jose, CA). Firefly luciferase activity was normalised to Renilla luciferase activity, and values in cells transfected with K694R TLR4 polymorphism plasmids were normalised to those detected in cells transfected with WT TLR4 plasmids.
FACS
Cell-surface and total TLR4 expression levels of HEK293T cells were detected by flow cytometry using primary anti-TLR4 Ab (ab22048; Abcam) and secondary anti-mouse IgG (H+L), F(ab′)2 Fragment (Alexa Fluor® 647 Conjugate, 4410S; Cell Signaling Technology). Cells from each well (3 × 106) were extracted using 0.25% trypsin (15090-046; Gibco, Grand Island, NY) in PBS, and centrifuged at 300 g at room temperature for 5 min, and the supernatant was removed. Following washing, 200 µl PBS was added to each tube, and the cells were flicked gently. Subsequently, 3 µl primary anti-TLR4 Ab was added to each tube, which were flicked gently again and allowed to stand for 30 min. Then, the secondary anti-mouse IgG (H+L), F(ab′)2 Fragment (Alexa Fluor® 647 Conjugate) was added and allowed to stand for 30 min in the dark. Flow cytometry (FlowJo V Software, Ashland, OR) was used to detect and measure the TLR4 levels on the cell surface. For total TLR4 staining, before the incubation with anti-TLR4 Ab, cells were first permeabilised and fixed using a BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (556547; BD, Franklin Lakes, NJ). Subsequently, these permeabilised cells were incubated with 3 µl primary anti-TLR4 Ab for 30 min and secondary anti-mouse IgG (H+L), F(ab′)2 Fragment (Alexa Fluor® 647 Conjugate) for 30 min in the dark.
Co-immunoprecipitation
Cells were fully lysed with 1 ml/10 cm dish of lysis buffer, which was the same as in WB, with protease inhibitors, phosphatase inhibitors and PMSF on ice added for 15 min. After 90 min of rotation for full lysis, the samples were centrifuged at 13,800 g for 15 min at 4°C to remove the precipitate, and 5% of protein solution (50 µl) was saved as input. The rest of the cell extract was divided into two parts, both of which were rotated with 20 µl Protein A/G Agarose beads (sc-2003; Santa Cruz Biotechnology, Texas, USA) for 3 h at 4°C. Pre-cleared extracts were then rotated with 6 μg of the appropriate anti-TLR4 Ab (14-9917-82; Thermo Fisher Scientific) and mouse IgG (B900620; Proteintech) for 3 h at 4°C, after which extracts were rotated with 30 μl Protein A/G agarose beads overnight at 4°C. After overnight rotation, the Protein A/G agarose beads were washed five times with 1 ml/tube ice pre-cooled lysis buffer, following which 20 µl of 2× loading buffer was used to elute the proteins which were mixed gently. Following centrifugation at 13,800 g for 1 min at 4°C, the IP product was assessed using the aforementioned WB protocol. The primary Ab of Flag was used to detect Flag-labelled content in the IP products.
Statistical analysis
Graphs are presented as the mean ± SD. Data were analysed using GraphPad Prism v7 (GraphPad Software, La Jolla, CA). Statistical differences between two groups were evaluated using Student’s t-test. A P value of < 0.05 was considered to indicate a statistically significant difference.
Results
K694R polymorphism does not affect total TLR4 levels and TLR4 cell-surface expression
The TLR4 expression level determines LPS susceptibility. 21 To explore whether the K694R mutant affects the expression of TLR4, we examined total and cell-surface TLR4 expression in mRNA and protein levels. TLR4 mRNA levels of cells transfected with K694R or WT TLR4 were significantly increased compared to those transfected with empty vector (Figure 2b). We found no difference in TLR4 mRNA expression between WT TLR4 and the K694R variant (Figure 2b). In protein levels examined by WB, TLR4 of cells transfected with K694R or WT TLR4 showed a prominent increase compared to those transfected with empty vector. However, the difference in TLR4 expression of cells transfected with K694R or WT TLR4 was not significant (Figure 2c and d), indicating that TLR4 plasmid overexpression was successfully constructed at protein level. During the research, we found that LPS could induce a slight increase in TLR4 protein levels in HEK293T cells without transfection of TLR4 plasmids (Figure 2c and d). However, after transfection of WT or K694R plasmids, TLR4 expression did not alter significantly when stimulated with LPS (Figure 2c and d). In flow cytometry analysis, we observed that the K694R polymorphism did not influence the percentage cell-surface (Figure 3a and b) and total (Figure 4a and b) TLR4-positive HEK293T cell population in the GFP-positive HEK293T cell population. Our results indicated that the K694R polymorphism did not influence TLR4 expression, and the low reactivity of LPS caused by the K694R polymorphism was not affected by TLR4 expression levels.
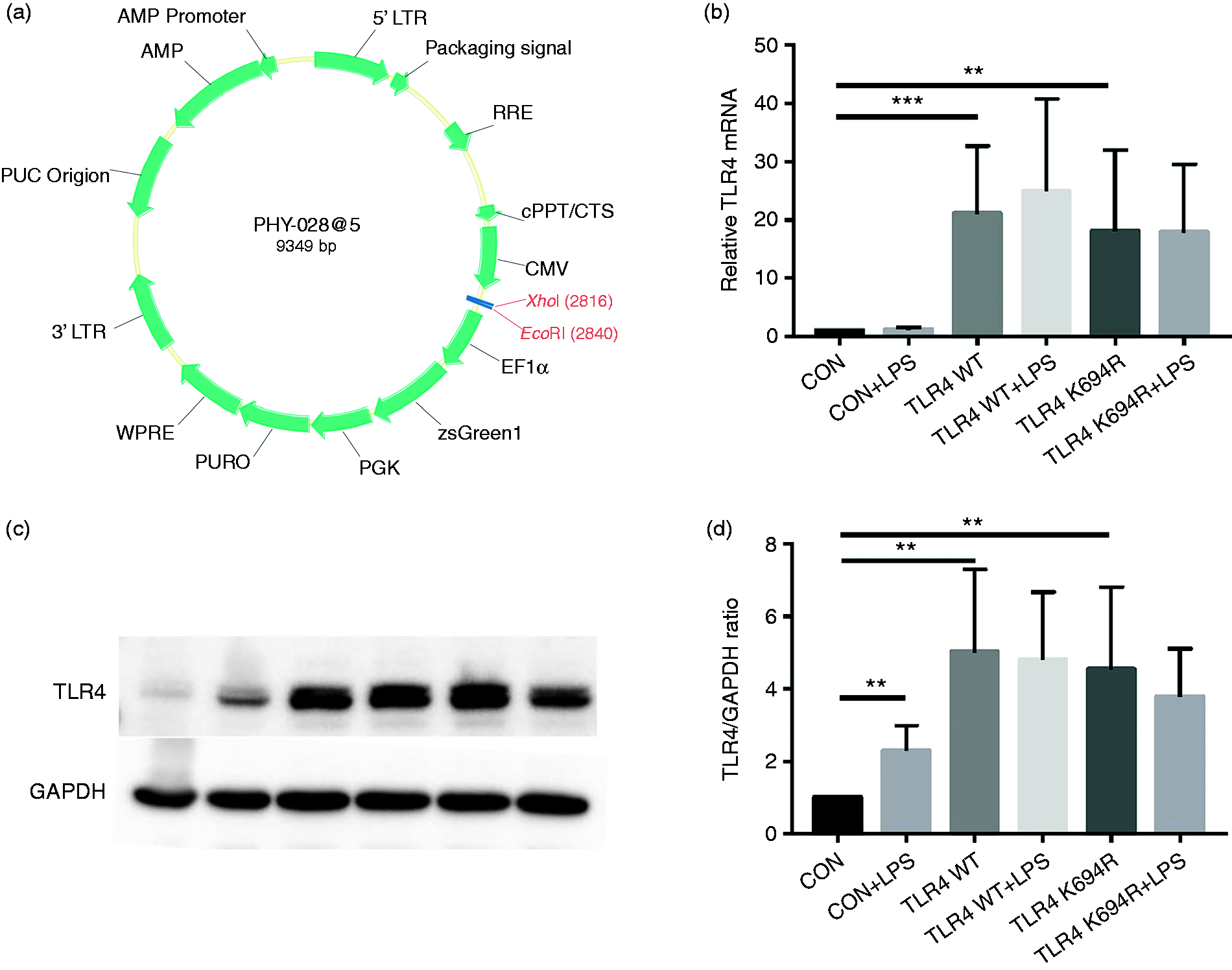
The K694R polymorphism does not affect TLR4 expression levels. (a) Diagram of wild type (WT) and K694R (K694G) TLR4 plasmid vector. (b) mRNA expression levels for the genes TLR4 in human embryonic kidney 293T (HEK293T) cells transiently transfected with WT or K694R TLR4 plasmid or empty vector as control, treated with or without LPS for 6 h. (c) Western blot (WB) analysis of TLR4 expression levels in HEK293T cells transiently transfected with WT, K694R TLR4 plasmid or empty vector treated with or without LPS for 6h. (d) Quantification of TLR4 expression levels in (c). Graphs are the mean ± SD, n = 6, *P < 0.05, Student’s t-test.
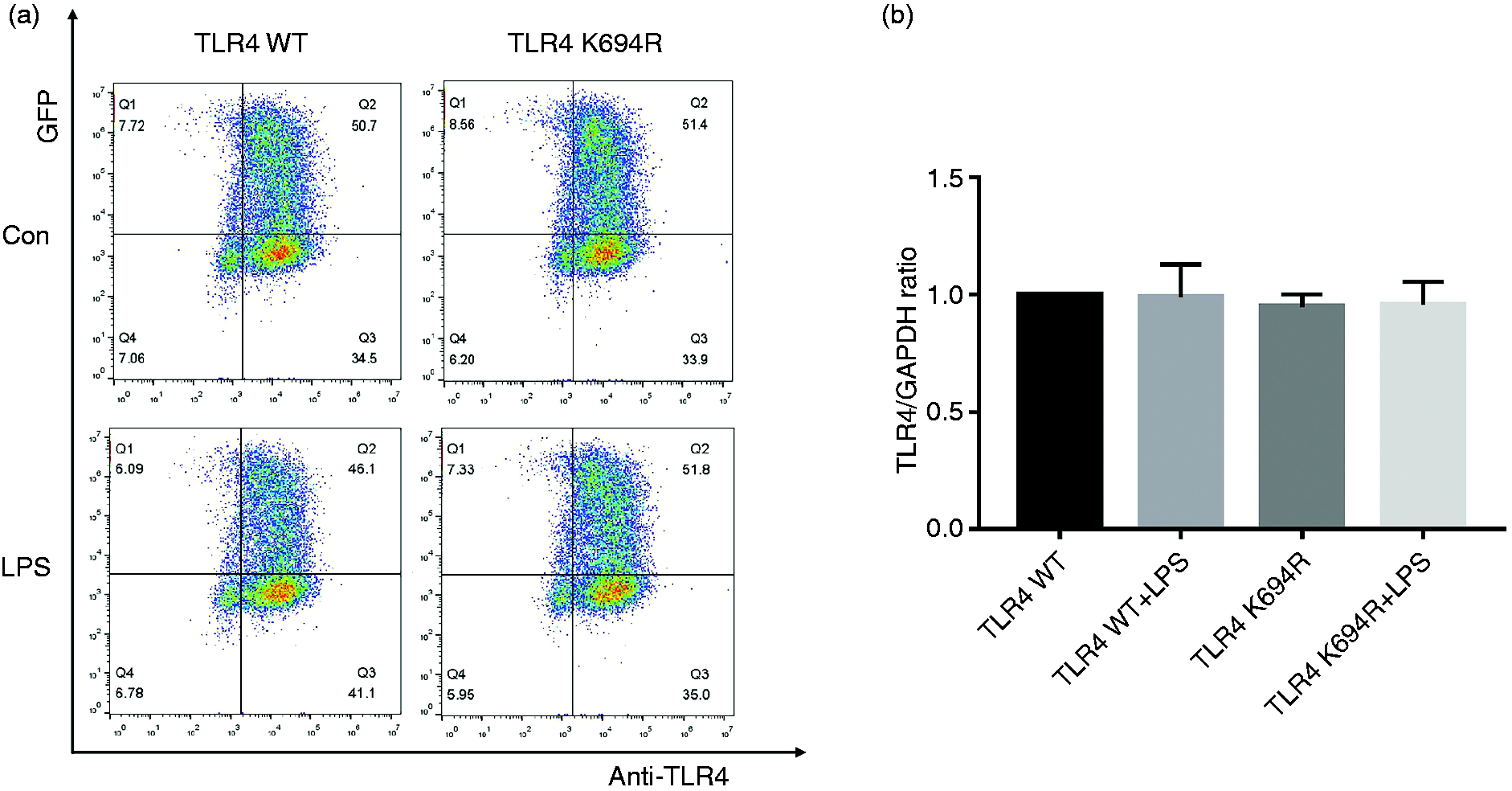
The K694R polymorphism does not affect cell-surface TLR4 expression. (a) Representative flow cytometric identification of HEK293T cells transiently transfected with K694R or WT TLR4 plasmid, treated with or without LPS for 6 h, respectively, gated on GFP+ cells. The cell-surface TLR4 was stained with the anti-TLR4 Ab. Anti-mouse IgG (H+L), F(ab′)2 Fragment (Alexa Fluor® 647 Conjugate) was used as the second Ab. (b) Quantification of cell-surface TLR4 stained with anti-TLR4 Ab in GFP+ cells in the Q2 quadrant. Graphs are the mean ± SD, n = 6, *P < 0.05, Student’s t-test.
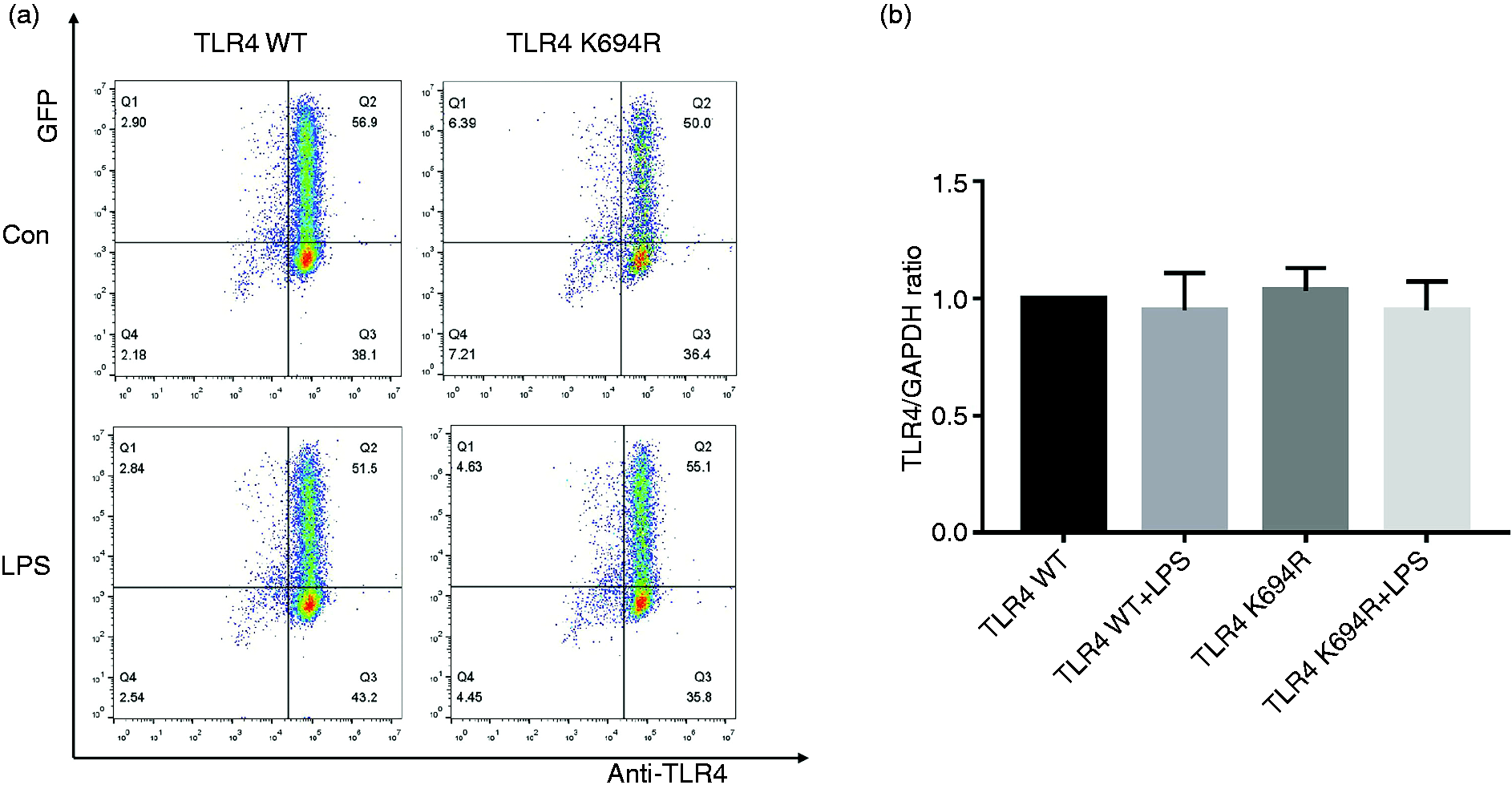
The K694R polymorphism does not affect total TLR4 levels. (a) Representative flow cytometric identification of HEK293T cells transiently transfected with K694R or WT TLR4 plasmid, treated with or without LPS for 6 h, respectively, gated on GFP+ cells. Total TLR4 was stained with anti-TLR4 Ab. Anti-mouse IgG (H+L), F(ab′)2 Fragment (Alexa Fluor® 647 Conjugate) was used as the second Ab. (b) Quantification of total TLR4 stained with anti-TLR4 Ab in GFP+ cells in the Q2 quadrant. Graphs are the mean ± SD, n = 6, *P < 0.05, Student’s t-test.
K694R TLR4 polymorphism does not affect TLR4 interaction with MD2
MD2 is an essential co-receptor and LPS-binding component for TLR4 signalling after LPS stimulation.22,23 Here, we investigated the influence of the K694R polymorphism on TLR4–MD2 interactions. Our results showed that the amounts of Flag-MD2 immunoprecipitated by anti-TLR4 Ab were not significantly different between WT and K694R groups (Figure 5a and b). Whole-cell lysates obtained from cells complemented with WT or K694R TLR4 showed comparable total levels of Flag-MD2 proteins (Figure 5a). Thus, under conditions of similar total expression of the interacting proteins, similar amounts of Flag-MD2 interacted with WT or K694R GFP-TLR4 variants, indicating that the K694R polymorphism did not affect TLR4–MD2 interactions.
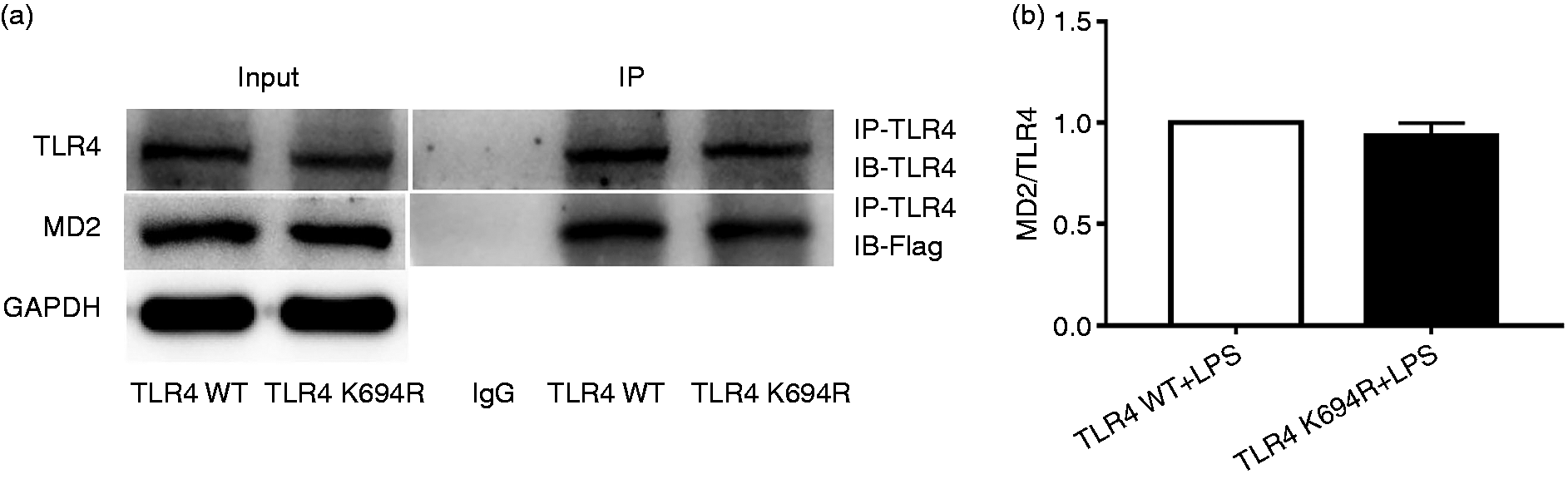
The K694R polymorphism does not affect TLR4 interaction with myeloid differentiation factor 2 (MD2). (a) HEK293T cells transiently transfected with WT or K694R TLR4, CD14 and Flag-MD2 plasmids, treated with LPS, were subjected to co-immunoprecipitation with anti-TLR4 Ab, and analysed by WB with anti-Flag Ab. (b) Densitometric quantification of the data shown in (a). The results are presented as ratios of MD2 and TLR4. Graphs are the mean ± SD, n = 3, *P < 0.05, Student’s t-test.
K694R polymorphism impairs the ability of TLR4 to recruit MyD88
MyD88 is an important downstream signal adaptor of TLR4 after LPS stimulation. 7 TLR4 downstream signal transduction through the MyD88-dependent pathway initiates transcription of cytokines such as TNF-α, IL-1β and IL-6 to amplify inflammatory reactions in a cascade manner. 7 To determine the impact of the K694R TLR4 polymorphism on the recruitment of MyD88, we used co-immunoprecipitation to study the LPS-induced association of transfected Flag-MyD88 with WT or K694R TLR4. Our results proved that K694R polymorphism reduced TLR4 to recruit Flag-MyD88 in comparison with WT TLR4 after induction by LPS (Figure 6a and b). Similar total levels of TLR4 and Flag-MyD88 (Figure 6a) suggested differences in MyD88 recruitment to K694R versus WT TLR4 which were not due to the diversity in total expression of TLR4 or MyD88. Our results demonstrated that the K694R polymorphism impaired the ability of TLR4 to recruit MyD88.
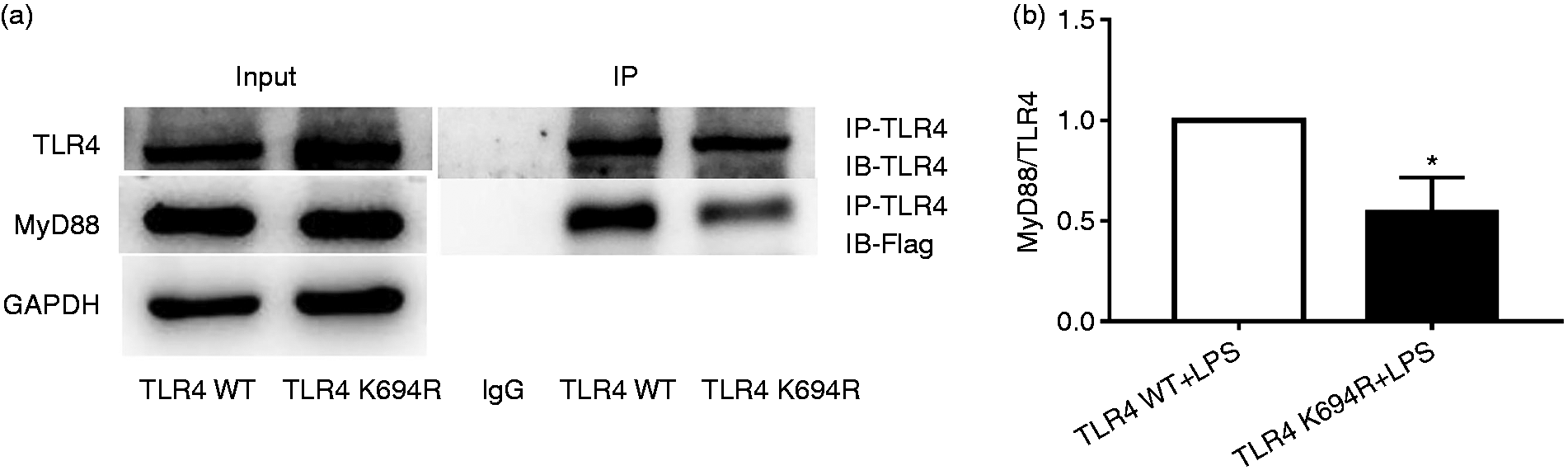
The K694R polymorphism impairs the ability of TLR4 to recruit MyD88. (a) HEK293T cells transiently transfected with WT or K694R TLR4, CD14, MD2 and Flag-MyD88 plasmids, treated with LPS, were subjected to co-immunoprecipitation with anti-TLR4 Ab, and analysed by WB with anti-Flag Ab. (b) Densitometric quantification of the data shown in (a) The results are presented as ratios of MyD88 and TLR4. Graphs are the mean ± SD, n = 3, *P < 0.05, Student’s t-test.
K694R polymorphism TLR4 impairs NF-κB activation and production of TNF-α, IL-1β, IL-6 and IL-18
Our study group previously investigated 18 naturally mutated TLR4 SNPs utilising the UniProtKB database (Supplemental Table S3), and screened out the K694R polymorphism, which also leads to blunted responsiveness to LPS in addition to D299G and T399I, by examining the activity of NF-κB. 20 In this study, we conducted the dual luciferase reporter assay and quantitative RT-PCR to verify the blunted response of the K694R polymorphism TLR4. Our results showed that the activity of NF-κB of K694R TLR4 detected by dual luciferase reporter assay was reduced (Figure 7a) in comparison to WT TLR4. Downstream pro-inflammatory TNF-α, IL-1β, IL-6 and IL-18 mRNA of K694R TLR4 regulated by NF-κB was decreased significantly (Figure 7b) compared to WT TLR4. Our results revealed that K694R TLR4 polymorphism could lead to blunted responses to LPS.
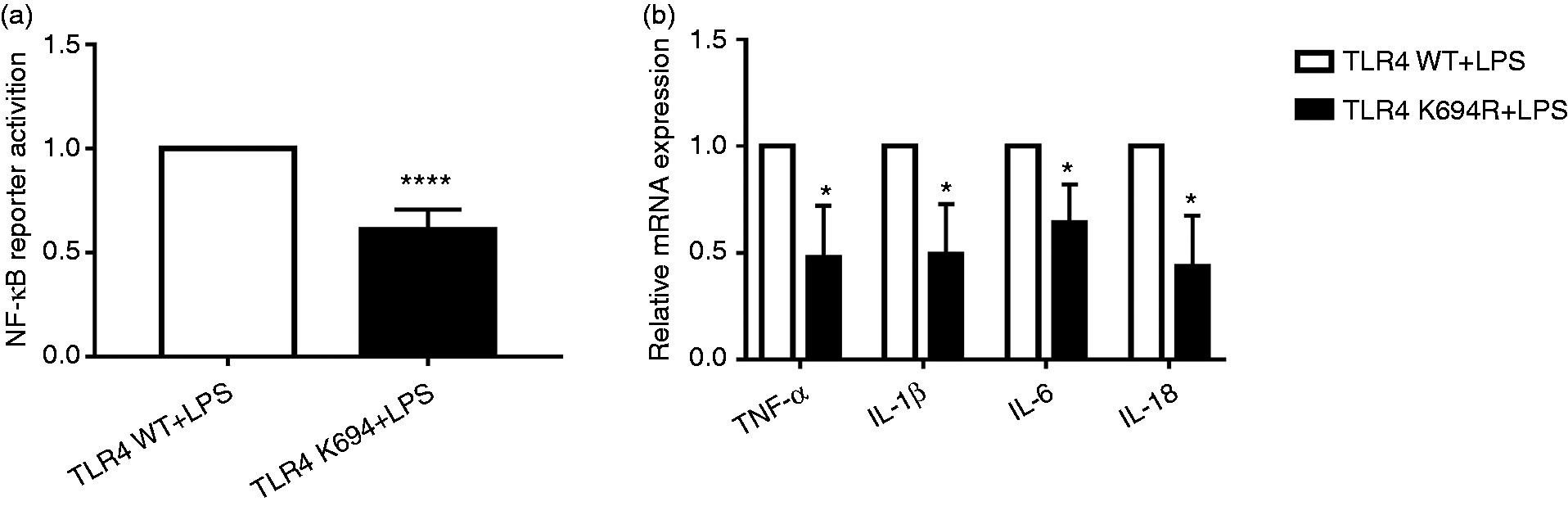
The K694R polymorphism impairs NF-κB activation and production of TNF-α, IL-1β, IL-6 and IL-18. (a) HEK293T cells transiently transfected with WT or K694R TLR4 plasmids were co-transfected with Flag-MD2, CD14, pNFκB-luc and pRL-SV40-N. Cells were treated with LPS for 6 h, and cell lysates were analysed for firefly and Renilla luciferase activities. Graphs are the mean ± SD, n = 6, ****P < 0.0001, Student’s t-test. (b) mRNA expression levels for the genes TNF-α, IL-1β, IL-6 and IL-18 in HEK293T cells transiently transfected with WT or K694R TLR4 plasmids and treated with LPS for 6 h were measured by quantitative RT-PCR. Graphs are the mean ± SD, n = 6, *P < 0.0001, Student’s t-test.
Discussion
Lys694Arg is another new TLR4 polymorphism that exhibits blunted responses to LPS, leading to the reduced activation of NF-κB and the decreased production of pro-inflammatory factors. The base A at this position is mutated to G, resulting in a change in the aa at position 694 from lysine to arginine. This site is located in the Toll/IL-1 receptor (TIR) conserved domain of TLR4, as identified by Blast software, and is very similar in different mammals. We predicted the effect of this site mutation using SIFT and Polyphen software,24–26 both of which identified significant changes in the structure and function of the protein. Therefore, we speculated that K694R might result in changes in the structure and function of the TLR4 protein. However, our results demonstrated that the K694R polymorphism did not influence total TLR4 protein levels and cell-surface expression of HEK293T cells in comparison to WT TLR4. Other previously investigated TLR4 polymorphisms (e.g. D299G and T399I) also showed no significant impact on the expression of TLR4 protein. 27
During this research, we found that LPS could induce the expression of TLR4 on the HEK293T cell population. It has been reported that LPS stimulation could enhance TLR4 expression of enterocytes, rat pulmonary artery smooth-muscle cells, human aortic smooth-muscle cells and renal inner medullary collecting duct cells significantly in a concentration- and time-dependent manner.28–32 Up-regulation of TLR4 expression accompanied by LPS activation is the beginning of the TLR4 signalling pathway.
MD2 is an extracellular principal co-receptor of TLR4 carrying a hydrophobic pocket for LPS recognition and conferring molecular specificity for LPS interaction and TLR4 signalling.8,10,33 LPS binding induces the formation of an m-shaped receptor multimer composed of two copies of the TLR4–MD2–LPS complex arranged symmetrically. 10 LPS interacts with the large hydrophobic pocket in MD2 and directly bridges the two components of the multimer. 34 Five of the six acyl chains of LPS are buried deep inside the pocket, and the remaining chain is exposed to the surface of MD2, forming a hydrophobic interaction with the conserved phenylalanines of TLR4. 34 MD2–/– mice do not respond to LPS, and in MD2–/– embryonic fibroblasts, TLR4 is not able to reach the plasma membrane and predominantly resides in the Golgi apparatus, whereas TLR4 is distributed at the leading edge surface of cells in WT embryonic fibroblasts. 35 It is hypothesised that D299G and T399I polymorphisms distantly impose conformational changes, affecting TLR4–MD2 interactions. 36 We speculate that the K694R polymorphism may also affect the interplay between TLR4 and MD2. However, our results identified no significant differences in TLR4–MD2 interactions between WT and K694R TLR4s, suggesting that the blunted response of LPS induced by K694R TLR4 cannot be explained by its insufficient binding with MD2.
Subsequent to recognition of LPS, TLR4 recruits MyD88 which possesses a C-terminal TIR domain that interacts with TLR4.37,38 The N-terminal death domain of MyD88 that binds other death domains such as the IL-1 receptor–associated kinase is responsible for the activation of NF-κB.39,40 MyD88-deficient (MyD88−/−) mice insufficiently produce pro-inflammatory factors such as TNF-α and IL-6. 41 The silence of MyD88 prevents a LPS-induced increase in intestinal tight junction permeability, which induces intestinal inflammation. 42 It has been reported that the D299G TLR4 polymorphism, which reduces the LPS response, alters the TLR4 signalling pathway because of its impaired recruitment of MyD88. 27 For the first time, we found another TLR4 polymorphism, namely K694R, that altered TLR4 signalling due to impaired recruitment of MyD88 to TLR4. Deficient recruitment of MyD88 to K694R TLR4 is linked with impaired activation of NF-κB and decreased LPS-induced production of TNF-α, IL-1, IL-6 and IL-18.
The innovation of this research is that we have verified a new TLR4 polymorphism, Lys694Arg, exhibiting a blunted response to LPS. We have also studied the mechanism of the blunted response to LPS, which offers complementary research on TLR4 non-synonymous cSNP-related mechanisms and provides potential clinical research value for the discovery and treatment of the genetic factors of sepsis.
Conclusion
Our results show that the Lys694Arg variant could reduce the LPS response, represented by reduced NF-κB activation and decreased production of pro-inflammatory factors, by the impaired recruitment of MyD88 to TLR4.
Supplemental Material
sj-pdf-1-ini-10.1177_1753425920927479 - Supplemental material for Lys694Arg polymorphism leads to blunted responses to LPS by interfering TLR4 with recruitment of MyD88
Supplemental material, sj-pdf-1-ini-10.1177_1753425920927479 for Lys694Arg polymorphism leads to blunted responses to LPS by interfering TLR4 with recruitment of MyD88 by Yajie Yang, Yan Hu, Yile Zhou, Tao Liang, Haihong Tang, Huihui Ju, Qiqing Shi and Hao Fang in Innate Immunity
Footnotes
Acknowledgements
The authors are indebted to Professors Miao Changhong and Jiao Jing for their preliminary experimental basis and to Professor Shi Yi for her experimental guidance during this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was supported by Shanghai Minhang District Natural Science Research Project (no. 2019MHZ028).
Supplemental material
Supplemental material is available for this article online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.