
Abstract
An elevated pro-inflammatory cytokine response is the primary cause of death by toxic shock after exposure to staphylococcal enterotoxin B (SEB). Identifying an intracellular signal mediator that predominantly controls the pro-inflammatory response is important for developing a therapeutic strategy. We examined the role of the signaling adaptor MyD88 in cell culture and in a mouse model of toxic shock. Our results indicated that elevated tumor necrosis factor-α, interferon-γ, interleukin (IL)-1α/β and IL-6 production from mouse spleen cells treated with SEB alone or in combination with lipopolysaccharide (LPS) was regulated by MyD88. Elevated levels of MyD88 protein in spleen cells, as well as in CD11c+ or Mac3+ cells, and activation of nuclear factor-κB in spleen cells were observed in mice treated with SEB. An SEB-dose dependent lethality was observed in LPS-potentiated and in D-galactosamine-sensitized mice. D-Galactosamine treatment of spleen cells had no effect in cytokine induction but rather increased the sensitivity to toxic shock in mice. Our results demonstrated an impaired pro-inflammatory cytokine production by spleen cells of MyD88–/– mice in response to SEB or SEB plus LPS. Most importantly, MyD88–/– mice were resistant to SEB-induced death. These results demonstrate that MyD88-dependent pro-inflammatory signaling is responsible for SEB intoxication. In addition, our studies also demonstrated that LPS potentiation, in comparison to D-galactosamine sensitization, contributes to a stronger SEB–induced lethality. This is due to the pro-inflammatory cytokine response elicited by MyD88 after exposure to SEB and LPS. These findings offer an important insight upon SEB intoxication and subsequent therapy targeting MyD88.
Introduction
The profound clinical consequences of staphylococcal enterotoxin B (SEB)-induced toxic shock are known to stem from an excessive pro-inflammatory cytokine response. The SEB cross-links major histocompatibility complex (MHC) class II molecules present on antigen presenting cells (APCs) to T-cell receptor (TCR) β-chains, triggering a release of pro-inflammatory cytokines from both cell types. Secretion of tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), interleukins-1 (IL-1) and -6 (IL-6) subsequently leads to toxic shock. 1 – 5 In a mouse model, the biological effects of SEB are potentiated by lipopolysaccharide (LPS), a bacterial factor that binds to Toll-like receptor 4 (TLR4) on the surface of cells. Staphylococcal enterotoxin B and LPS synergistically amplify the pro-inflammatory cytokines that lead to severe toxicity.6,7 Although LPS enhances the lethality of SEB in vivo, 6 – 8 the mechanism underlying this powerful synergistic effect on pro-inflammatory cytokine responses still remains unclear. Earlier studies suggest that the signal transduction mechanism driving IL-1 and TNF-α expression after SEB binding to MHC class II in human monocytes involves protein kinase C (PKC) and protein tyrosine kinase (PTK). 9 – 11 Recent results indicate that IFN-γ as well as IL-1β use the myeloid differentiation factor 88 (MyD88)-dependent pathways for signaling. 12 The adaptor protein MyD88 integrates and transduces intracellular signals generated by the TLR and the IL-1 receptor (IL-1R) superfamily that are critical for innate immune regulation and host defense.13,14 However, the impact of MyD88-mediated inflammatory signaling with respect to toxic shock remains unknown.
In general, ligand binding to IL-1R/TLR family receptors triggers recruitment of MyD88 to the receptor complex through the Toll interleukin recognition (TIR) domain. MyD88 subsequently recruits downstream signaling molecules known as IL-1R-associated protein kinase (IRAK). Activation of IRAK leads to a series of ‘downstream’ signaling cascades that activate nuclear factor-κB (NF-κB), p38 mitogen-activated protein kinase (MAPK), and other regulators of gene expression, permitting the trans-activation of pro-inflammatory cytokine genes. 15 – 17 It was reported that ligation of MHC class II with SEB up-regulates monocyte membrane TLR4 expression, 18 a receptor for LPS. This scenario may, in part, explain the pathology of septic patients who are simultaneously exposed to multiple bacterial products from Gram-negative and Gram-positive pathogens. An understanding of how multiple host response pathways interact is important for finding potential treatments.
A MyD88 deficiency reportedly improves resistance against sepsis caused by polymicrobial infection. 19 Despite the distinct known functions of MyD88 as a regulator of pro-inflammatory responses, an important question remains: does SEB activate MyD88 and co-operate with the TLR signaling cascade to amplify inflammatory response? Earlier studies showed that mice lacking MyD88 have defective pro-inflammatory signaling and provide a useful model for evaluating innate immunity and overall host defense.20,21 In this study, we investigated the activation of MyD88-dependent pro-inflammatory cytokine responses to SEB in a LPS-dependent and LPS-independent D-galactosamine (d-GalN) sensitized model. We characterized MyD88 up-regulation, pro-inflammatory cytokine response, and lethality by investigating MyD88–/– mice after exposure to SEB in primary cells and in a mouse model of toxicity.
Materials and methods
Reagents
Staphylococcal enterotoxin B was purchased from Porton Down (Salisbury, UK), and was prepared under GMP condition and endotoxin free. Escherichia coli LPS (O55:B5) was purchased from Difco Laboratories (Detriot, MI, USA). Mouse CD90 (Thy 1.2), anti-Mac3 and anti-CD11c monoclonal antibodies (mAbs) conjugated with magnetic beads were purchased from Milteneyi Biotec Inc. (Auburn, CA, USA). D-Galactosamine and bovine serum albumin (BSA, fraction V) were purchased from Sigma Aldrich (St Louis, MO, USA). A cytometric bead array (CBA) kit for cytokine assay was purchased from BD Biosciences Pharmingen (San Diego, CA, USA). An RNA-extracting reagent (Tri-Reagent) was obtained from Molecular Research Center Inc. (Cincinnati, OH, USA) and Maloney murine leukemia virus reverse transcriptase from Perkin Elmer (Waltham, MA, USA). Primary anti-MyD88 antibody was obtained from (AnaSpec, Inc. (San Jose, CA, USA). β-Actin antibody was purchased from Cell Signalling (Danvers, MA, USA). The [methyl-3H]-thymidine was purchased from Amersham Life Sciences (Arlington Heights, IL, USA).
Mice
Pathogen-free BALB/c and C57BL/6 female mice (6–8 weeks old, 18–20 g) were obtained from Charles River (NCI-Frederick, Frederick, MD, USA). The MyD88 gene knockout (MyD88–/–) mice of C57BL/6 background were obtained from Oriental Bio-service, Inc. (OBS, Ukyo-ku, Kyoto, Japan) and bred at Charles River Laboratories, (Wilmington, MA, USA).
Inoculation of mice
To determine levels of cytokines in serum and in vivo up-regulation of MyD88 and NF-κB activation, mice were injected intraperitoneally (i.p.) with varying amounts of SEB and, 2 h later, with LPS. Blood samples were collected through tail vein at 30 min, 1 h, and 2 h and used for measuring serum cytokines, after which mice were euthanized and spleens were removed. Monocytes, macrophages, and dendritic cells were isolated as described below.
Spleen cell isolation and cultures
Spleens were removed aseptically from euthanized mice. Single-cell suspensions were prepared by lysing red blood cells using ammonium chloride-potassium lysing (ACK) buffer from Lonza-Cambrex (Walkersville, MD, USA), followed by three washes with media. CD11c+ cells were isolated as described elsewhere. 22 Briefly, spleen cells were resuspended (108 cells/400 µl) in PBS (4°C), supplemented with 2 mm EDTA and 0.5% BSA. Paramagnetic beads coated with anti-CD11c were incubated for 15 min (4°C) with spleen cells (108 cells/100 µl). The antibody-labeled cells were washed and passed through a type LS or MS iron-fiber column placed within a strong magnetic field (Miltenyi Biotec). The CD11c+ cells bound to the column were eluted with buffer after withdrawal from the magnetic field. All cells were cultured in RPMI 1640 medium containing L-glutamine and 5% fetal bovine serum. Spleen cells from BALB/c, C57BL/6, or MyD88–/– mice were cultured with SEB (200 ng/ml) for 2 h, before adding LPS (1 µg/ml). Cultures were incubated (37°C, 5% CO2) for 16 h, supernatant was collected, and cells were harvested for measuring transcriptional activation of the MyD88 gene and intracellular expression of MyD88 protein.
RT-PCR
For measuring transcriptional activation of MyD88, total RNA was extracted from spleen cells using Tri-Reagent and reverse transcribed into cDNA with Maloney murine leukemia virus reverse transcriptase according to the manufacturer’s instructions (Perkin Elmer). Amplification of cDNA was performed with gene-specific primers, MyD88 (forward 5′-CACTCGCAGTTTGTTGGATG-3′; reverse 5′-CGCAGGATACTGGGAAAGTC-3′), and β-actin (forward 5′-TCCTGTGGCATCCACGAAACT-3′; reverse 5′-GAAGCATTTGCGGTGGACGAT-3′) using platinum TAQ DNA polymerase (Invitrogen, Carlsbad, CA, USA) as described elsewhere. 23 The PCR products were run on a 1.5% agarose gel, stained with ethidium bromide, and photographed.
Cytokine analysis
Cytokines in culture supernatants were measured by a CBA kit using capture beads coated with antibodies specific for cytokines and flow cytometry analysis according to the manufacturer’s method, as described elsewhere. 24 Cytokine measurement was confirmed by dilution of culture supernatant using Inflammation and Th1/Th2 CBA kits by acquiring 1800 beads. Interleukin-1α was measured by an ELISA kit according to the manufacturer’s procedure (R&D Systems, Minneapolis, MN, USA).
Nuclear factor-κB assay
Nuclear factor-κB was examined in nuclear extract of spleen cells. To prepare nuclear extract, spleen cells (4 × 106) were washed and resuspended with 1 ml of cold PBS and placed on ice for 15 min to allow the cells to swell. Nonidet P-40 (10%, 50 µl) was added to the cells and briefly centrifuged. The nuclear pellet was resuspended in lysis buffer (Active Motif) containing dithiothreitol (DTT) and protease inhibitor for 30 min on ice, centrifuged for 10 min at 14,000 g at 4°C and extract collected for NF-κB assay. The TransAm Chemi kit (Active Motif, Carlsbad, CA, USA) had NF-κB oligonucleotides immobilized to a 96-well plate. To the 96-well plate, nuclear extract of spleen cells were placed in duplicates to the plate for an hour. The plates were then washed several times and diluted NF-κB antibody was added to the plate for 1 h at room temperature. The plate was washed and a diluted horseradish peroxidase (HRP)-conjugated secondary antibody was added to the plate for an hour at room temperature. After incubation, the plate was washed with wash buffer and a chemiluminescent working solution was added to detect NF-κB recognition of an epitope on p65 subunit.
Flow cytometry
To examine intracellular expression of MyD88 in Mac3+ cells, spleen cells were labeled with FITC conjugated anti-Mac3 antibody, washed briefly by centrifugation, permeabilized, and then incubated with primary MyD88 antibody followed by secondary Alexa Fluor 568-conjugated to goat anti-rabbit-IgG antibody. Unbound antibody was removed by washing the cells with HBSS (4°C) and centrifugation. After two additional washes, the labeled cells were fixed with 1% paraformaldehyde in PBS, and intracellular immunofluorescence was measured by a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Secondary anti-rabbit IgG was used as an isotype control and analyzed by flow cytometry.
Confocal microscopy
In vivo up-regulation of MyD88 in TLR4 CD11c+ dendritic cells was detected by intracellular staining. Spleen cells from BALB/c mice injected with different doses of SEB and LPS were isolated. The CD11c+ cells were positively selected from total cells by anti-CD11c mAbs conjugated with magnetic beads (Miltenyi Biotec). Cells were fixed by cytospin, permeabilized, and labeled with FITC-conjugated anti-TLR4 and anti-MyD88 antibodies (AnaSpec). Cells were next exposed to a secondary Alexa Fluor 568-conjugated to goat anti-rabbit-IgG antibody and finally examined by confocal microscopy.
Western blots
Mouse spleen cells (5 × 106 cells/ml) were stimulated with LPS (1 µg/ml) or SEB (200 ng/ml) or left untreated for 2 h. Cells were collected into fresh 1.5-ml centrifuge tubes and chilled on ice for 5 min before centrifuging. Membrane and cytoplasm separation was done by resuspending the pellets in 50 µl of lysis buffer (Active Motif) in the presence of DTT, protease inhibitors and phosphatase inhibitors and incubated on ice for 30–60 min. Membrane fraction was collected by centrifuging the lysates at 14,000 g for 20 min. The supernatant contained the cytoplasmic fraction and pellet contained membrane fraction. Samples containing 10 µg of total cytoplasmic proteins were separated by gel electrophoresis and transferred to nitrocellulose membranes. Membranes were blocked overnight in Tris-buffered saline containing 0.1% Tween-20 and 3% BSA at 4°C. Blots were extensively washed and probed with anti-MyD88 polyclonal antibody followed by HRP-conjugated secondary Ab. Blots were washed extensively and developed with chemiluminescent substrate in the presence of hydrogen peroxide using Immun-Star Western© Chemiluminescent Kit (BioRad, Hercules, CA, USA). An imaging system VersaDoc Model 4000 (BioRad) was used to capture the image.
Proliferation assay
Total spleen cells (1 × 106) were cultured in a 96-well plate with SEB or SEA (10, 100 and 200 ng/ml) in a total 200 µl volume for 3 d at 37°C. After 3 d, cultures were pulsed with 1 µCi [ 3 H]-thymidine/well for 10 h and proliferation was measured by harvesting the cells onto microplate unifilters and measuring radioactivity in a liquid scintillation counter (Packard, Meriden, CT, USA).
Statistical analysis
The SAS program v9.2 (Cary, NC, USA) was used for statistical analysis. The original cytokine data were plotted using t-tests with step-down Bonferroni adjustment performed on log values to determine statistical significance. Kaplan–Meier survival analysis (dependent variables: survival status, time to death) and log-rank test to compare survival curves (with step-down Bonferroni adjustment for pair-wise comparisons) among groups were performed.
Results
Staphylococcal enterotoxin B and LPS-induced cytokine production by spleen cells correlated with the up-regulation of MyD88
To examine SEB-induced MyD88-mediated signaling, we treated BALB/c mouse spleen cells with SEB or SEB with LPS for 4 h, 16 h, or 36 h and measured cytokine release into the culture supernatant. Our results revealed increased production of TNF-α, IFN-γ, and IL-6 in culture supernatants from cells treated with SEB versus untreated controls (Fig. 1A). While TNF-α appeared to be released earlier and dropped after 16 h, the release of IFN-γ and IL-6 appeared late and continued for up to 36 h after treatment (Fig.1A). Along with cytokine production, we also examined functional up-regulation of MyD88. MyD88 is generally recruited as a dimer to the receptor complex. Results shown in Fig 1B indicate up-regulation of MyD88 mRNA after exposure to SEB and/or LPS compared to untreated cells. Results shown in Figure 1C indicate an increase in MyD88 protein in spleen cells 2 h after stimulation with SEB or LPS and SEB plus LPS compared to unstimulated (Unt) spleen cells. These results suggest that, along with transcriptional up-regulation of MyD88 mRNA, de novo synthesis of MyD88 increased with SEB stimulation, both of which correlated with pro-inflammatory cytokine production.
Pro-inflammatory cytokine production by murine spleen cells in response to SEB and LPS correlates with up-regulation of MyD88. Spleen cells from BALB/c mice were cultured with SEB (200 ng/ml, optimum dose) or LPS (1 µg/ml) or SEB plus LPS. Culture supernatants were collected and analyzed at 4, 16 and 36 h. (A) Release of TNF-α, IL-6, and IFN-γ in culture supernatants. Cytokines in serum were measured by a CBA and flow cytometry as described elsewhere.
24
Results are shown from one representative experiment of two with similar results. The cytokine assay was repeated by dilution and two independent assay kits such as Th1/Th2 and inflammatory cytokines. (B) Transcriptional up-regulation of MyD88 in spleen cells cultured with SEB, LPS or SEB plus LPS at 2 h. The RNA was reverse transcribed and amplified with specific primers. The RT-PCR experiments were performed at least three times on independent samples with similar results. (C) Up-regulation of MyD88 protein in spleen cells after SEB or LPS stimulation. BALB/c spleen cells were cultured with SEB or LPS for 2 h or kept untreated. β-Actin (bottom panel) was used as loading control. The figure shows one representative experiment of three with similar results.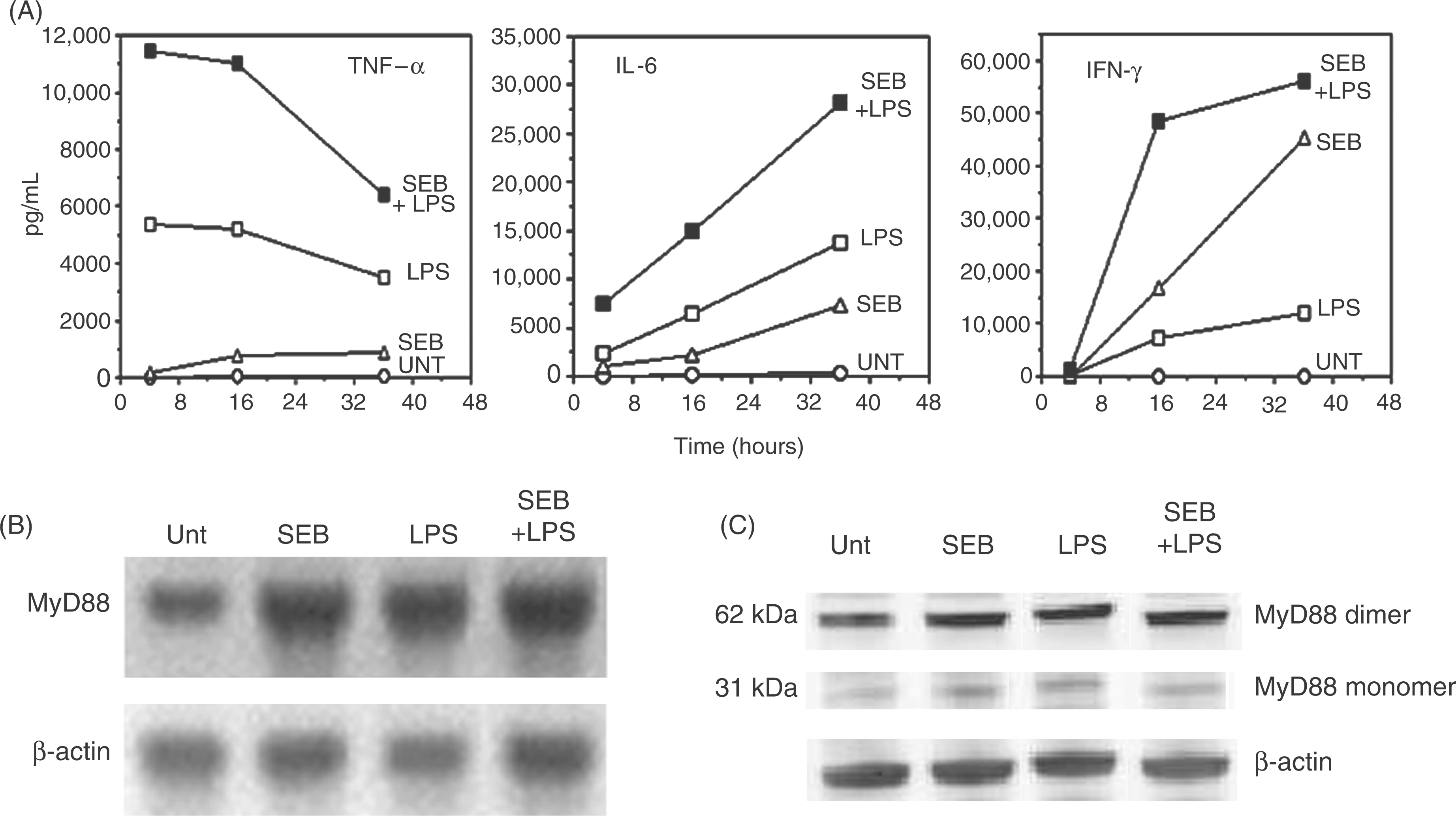
In vivo serum cytokine responses correlated with MyD88 up-regulation in response to SEB challenge dose
We next examined in vivo responses by co-administering SEB and LPS to BALB/c mice and measuring serum cytokines at 30 min, 1 h and 2 h as well as activation of MyD88. In previous experiments (data not shown), we established that inoculation of 1 µg of SEB (followed by 60 µg of LPS) is equivalent to LD90. Similarly, 0.25 µg of SEB was the equivalent to LD10, and 0.05 µg was equivalent to LD0. In this experimental set up (Fig. 2A), we show that, within 1 h, SEB plus LPS greatly increased TNF-α and IL-6 in serum relative to those mice injected with only SEB or LPS. The highest total levels of IFN-γ, IL-6, and IL-1α released in vivo corresponded to the highest dose, LD90 within 1 h. However, serum levels of TNF-α and IL-6 dropped by 2 h despite the higher doses of SEB plus LPS. This may be due to the exhaustion of target cells responding rapidly to SEB plus LPS (double hit) with a robust cellular activation and cytokine responses. Interferon-γ was induced by SEB alone and continued to increase past 1 h (Fig. 2A).
Serum cytokine response correlates with SEB challenge dose and up-regulation of MyD88. BALB/c mice were inoculated as follows: saline (control); SEB only (1 µg); LPS only (60 µg); SEB (0.05 µg) +LPS (60 µg), LD0; SEB (0.25 µg) +LPS (60 µg), LD10; SEB (1 µg) +LPS (60 µg), LD90. Data represent (A) serum TNF-α, IFN-γ, and IL-6 at 30 min, 1 h, and 2 h after LPS injection; serum IL-1α was measured only at 2 h. (B) In vivo activation of TLR4 and MyD88 in CD11c+ cells. The intracellular level of MyD88 was detected in positively selected CD11c+ cells using confocal microscopy. Bar 20 µm. (C) In vivo up-regulation of MyD88 in Mac3+ cells. Cells were labeled with FITC-conjugated anti-Mac3 antibody, and appropriate isotype control and analyzed by surface marker phenotype and flow cytometry. (D) Presence of NF-κB p65 subunit in nuclear extract of spleen cells. Data from one representative experiment of two are shown.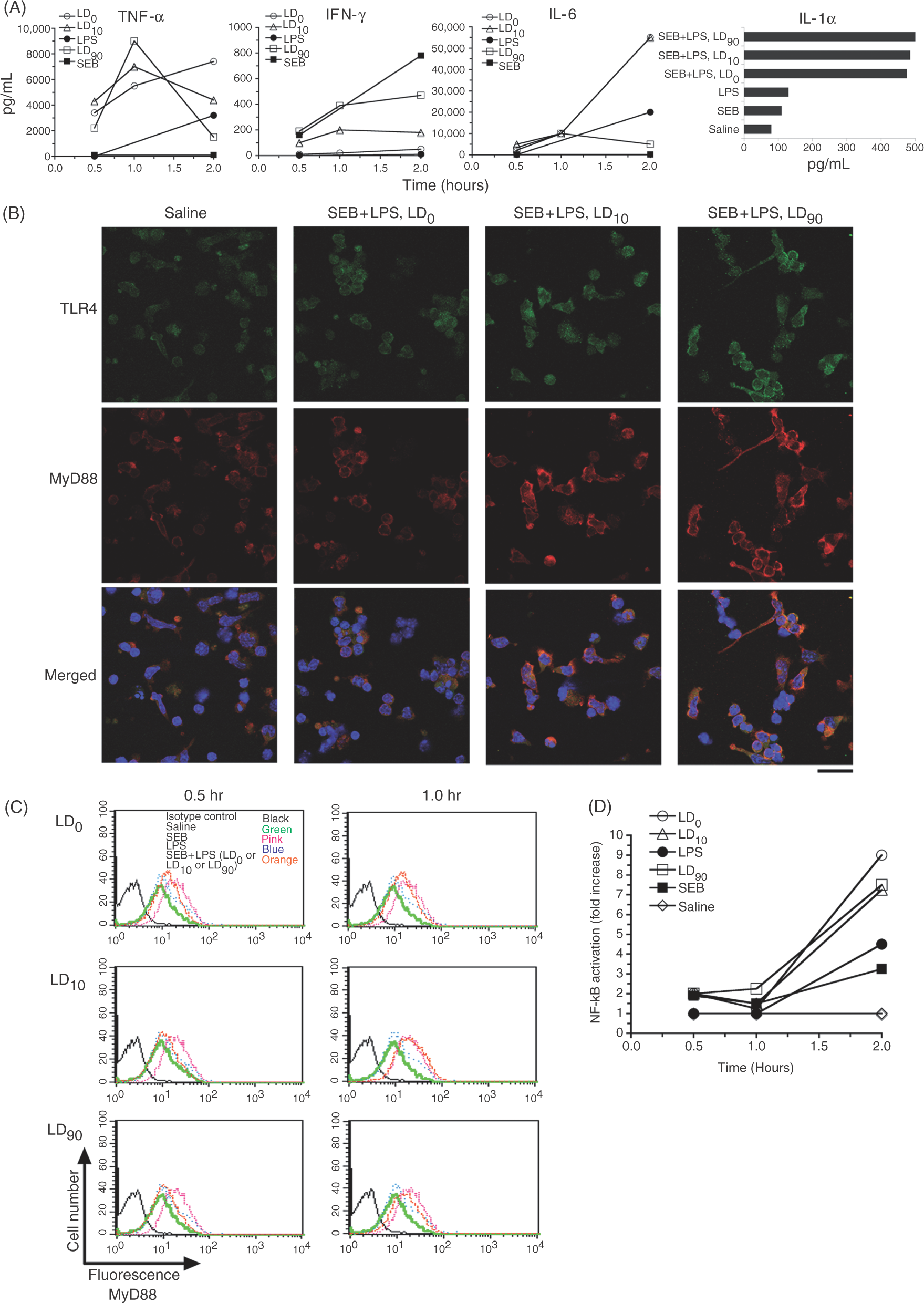
In addition to those cytokine responses, we examined up-regulation of TLR4 and MyD88 in MHC class II+ APCs. The CD11c+ cells were purified from the spleens of BALB/c mice that were previously inoculated with SEB (0.05 µg, 0.25 µg, and 1 µg/mouse corresponding to LD0, LD10 and LD90 and LPS (60 µg). Similar to transcriptional up-regulation and de novo synthesis of MyD88 in SEB-treated spleen cells (Fig. 1B, C), results shown in Fig 2B indicate that expression of MyD88 corresponded with increasing concentrations of SEB in the presence of LPS as compared to saline-treated control mice. It is likely that the high dose of SEB (1 µg/mouse) increased the up-regulation of MyD88, which may precede the cytokine responses. The activation of MyD88 was also examined in Mac3+ cells from mice injected with a high dose of SEB or SEB plus LPS after 30 min and 1 h. Results shown in Fig 2C indicate an increase in expression of MyD88 in cells compared to saline-treated mice. In addition, increased transcriptional activation of MyD88 in spleen cells of treated mice was also observed (data not shown). We also examined the transcription factor NF-κB since it is a key component of the signal transduction pathway downstream to MyD88. A strong increase in nuclear NF-κB p65 subunit was observed in spleen cells after an SEB challenge in mice (Fig. 2D). Taken together, these results suggest that up-regulation of MyD88 in vivo as well as NF-κB activation was induced as a consequence of SEB exposure in mice.
Staphylococcal enterotoxin B, but not D-GalN, induced pro-inflammatory cytokine production in mouse spleen cell culture
Lipopolysaccharide is known to activate pro-inflammatory signaling via TLR4–MyD88 and synergize SEB toxicity. We next examined SEB toxicity independent of LPS. D-Galactosamine has also been shown to sensitize mice to SEB–induced toxic shock.
25
Prior to using d-GalN in vivo, we wanted to test the effects of d-GalN on pro-inflammatory cytokine production in cultured spleen cells. Results shown in Figure 3 indicate that up-regulation of cytokine production was not affected by d-GalN treatment. These results suggest that, unlike LPS, d-GalN is a viable potentiator of SEB-induced toxicity since it would not contribute to cytokine production.
Staphylococcal enterotoxin B but not D-GalN induced cytokine production by spleen cells. Spleen cells (5 × 106 cells/well) from BALB/c mice were cultured with SEB (200 ng/ml) alone or with D-GalN (0.1 or 1 mg/ml) for 16 h. Culture supernatants were tested for the presence of pro-inflammatory cytokines. Results shown are one representative experiment of two with similar results.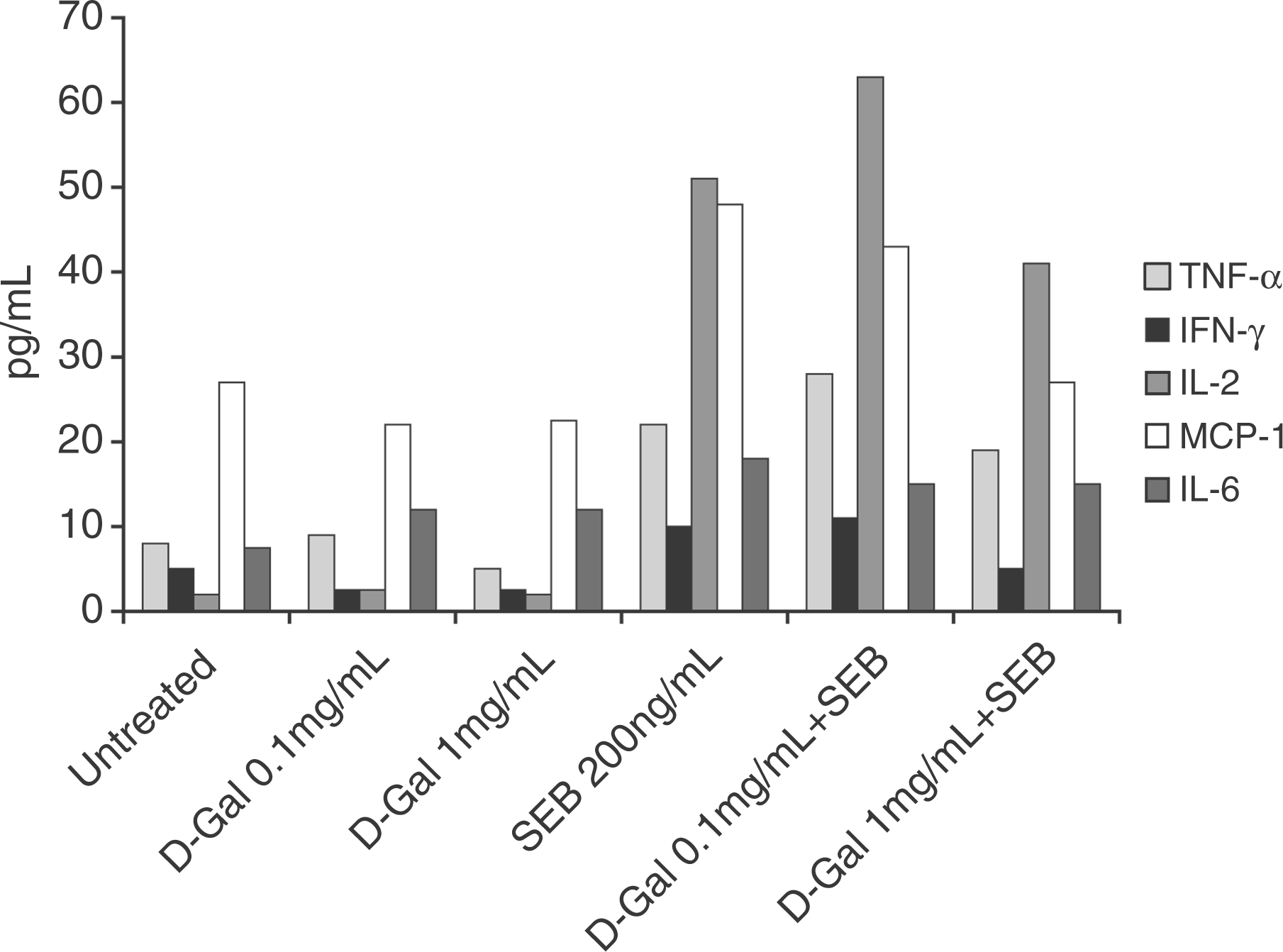
Staphylococcal enterotoxin B dose-dependent toxicity in LPS and d-GalN sensitized mice
Mice are more resistant than humans to the pathogenic effects of enterotoxins. D-Galactosamine is a hepatotoxic agent that has been used to demonstrate the applicability of d-GalN-sensitized mice in studying the pathogenesis of bacterial exotoxin mediated lethal shock.
25
To evaluate further the role of MyD88 in mediating SEB toxicity, we examined d-GalN sensitized mice that were challenged with varying doses of SEB. For this, we compared survival to SEB intoxication in LPS and d-GalN sensitized-BALB/c mice (Fig. 4A, B). For LPS-potentiated SEB toxicity, BALB/c mice were inoculated (i.p.) with increasing doses of SEB (0.5, 5, 25 and 50 µg/mouse which are equivalent to 1 × LD50, 10 × LD50, 50 × LD50 and 100 × LD50, respectively) followed by LPS (60 µg) 2 h later. For d-GalN-sensitized experiment, BALB/c mice were inoculated (i.p.) with d-GalN (20 mg/mouse) 30 min prior to SEB (2, 20 and 100 µg/mouse). Mice were observed for 4 d. Control mice inoculated with d-GalN or LPS survived. In both mouse models of SEB toxicity, SEB dose dependent lethality was noted (Fig. 4A,B). In both cases, LPS-potentiated or d-GalN sensitized mice, there was a significant SEB-dose dependent lethality (P ≤ 0.05). These results suggest that, in our model, SEB toxicity is driven by SEB-induced production of pro-inflammatory cytokines.
Staphylococcal enterotoxin B dose-dependent toxicity in LPS-potentiated and LPS-independent d-GalN sensitized BALB/c mice. (A) LPS-potentiated lethal toxicity to SEB challenge. Mice (n = 6/group) were inoculated (i.p.) with SEB (0.5, 5, 25 or 50 µg/mouse which is equivalent to 1 × LD50, 10 × LD50, 50 × LD50 and 100 × LD50, respectively) followed by LPS (60 µg/mouse), or LPS alone. Mice were observed for survival. Data presented as a percentage of surviving toxic shock-induced death. Control mice inoculated with 60 µg of LPS survived. (B) D-Galactosamine-sensitized mice toxicity to SEB challenge. BALB/c mice (n = 6/group) were inoculated (i.p.) with d-GalN (20 mg/mouse) 30 min prior to SEB (2, 20 and 100 µg/mouse) inoculation or with SEB or d-GalN alone and observed for 4 d. Data represent two separate experiments.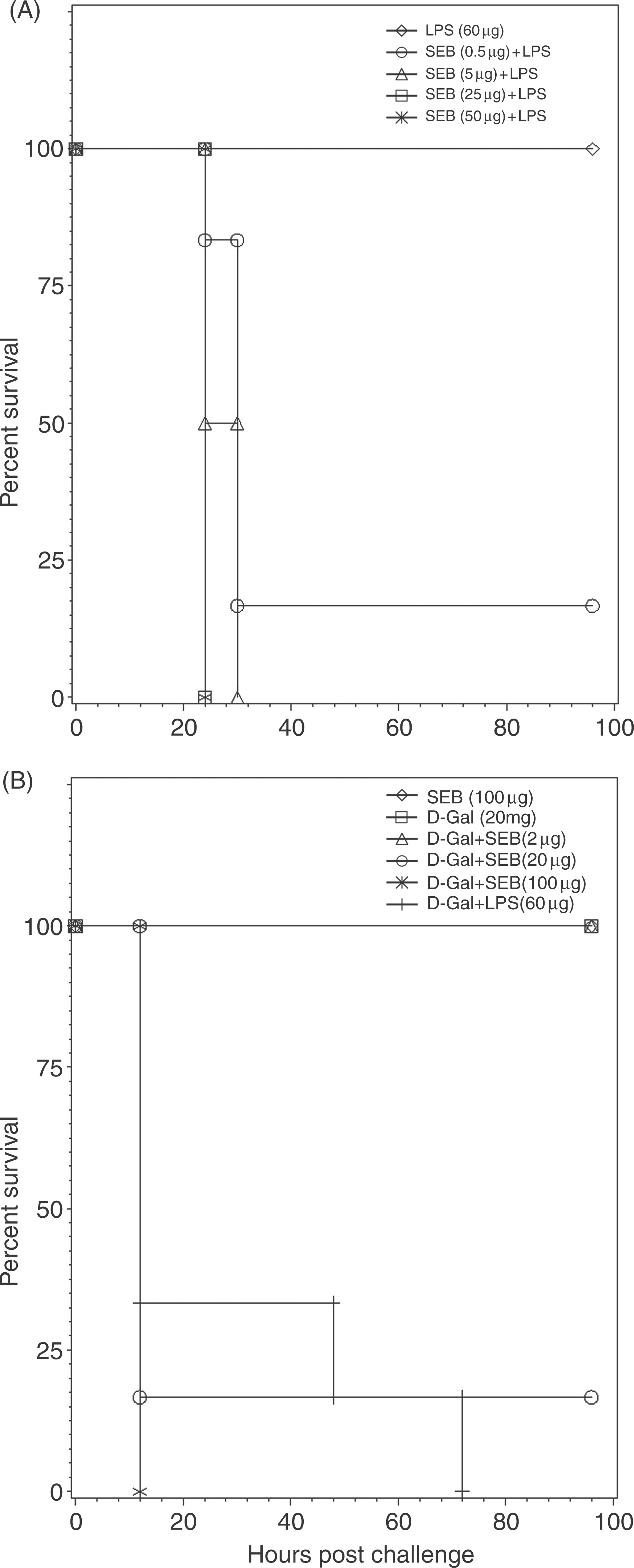
The MyD88 deficiency impaired pro-inflammatory cytokine production by spleen cells in response to SEB and LPS
It was previously reported that pro-inflammatory cytokines and chemokine responses to LPS are impaired in MyD88-deficient macrophages.26,27 Staphylococcal enterotoxin B activation of human monocytes was also reported to cause increased TLR4 expression through ligation of MHC class II molecules. 18 The results presented in Figure 2 showed a marked increase in in vivo up-regulation of MyD88 in MHC class II-positive CD11c+ and Mac3+ cells after SEB and LPS stimulation in BALB/c mice which correlate MyD88 up-regulation with cytokine production in BALB/c mice. To demonstrate further that SEB-induced cytokine production by spleen cells is MyD88 dependent irrespective of MHC haplotype, we used BALB/c, C57BL/6 and MyD88 gene knockout (MyD88–/–, C57BL/6 background) in ex vivo culture.
Earlier results from our laboratory indicated C57BL/6 mice (H-2b) and BALB/c mice (H-2d) background are particularly sensitive to SEA and SEB, respectively.
7
To examine whether SEB-induced toxic shock is only relevant to BALB/c, we measured SEB-induced proliferation of spleen cells from BALB/c and C57BL/6 mice. These experiments were necessary in order to use properly the MyD88 gene knockout mouse (MyD88–/–) model of C57BL/6 background. Spleen cells from both BALB/c and C57BL/6 mice underwent significantly increased proliferation in response to SEB in a dose-dependent manner (Fig. 5A). We also examined in vitro cytokine production of spleen cells in response to SEB by comparing MyD88-deficient mice (C57BL/6 background) to wild-type mice C57BL/6. Figure 5B shows significant reduction in TNF-α, IFN-γ, and IL-2 production in MyD88-deficient total spleen cells in response to SEB. These results suggest that cytokine production was MyD88 dependent in C57BL/6 mice. To extend this observation further, we also examined cytokine production by adherent and non-adherent cells of MyD88-deficient mice and wild-type mice after SEB stimulation. Isolated adherent MHC class II-positive macrophages (adherent cells) from MyD88-deficient and T-cells (non-adherent) from C57BL/6 (wild-type) or MyD88–/– mice were cultured with SEB. Adherent or non-adherent cell types alone were not capable of releasing cytokine in response to SEB in culture suggesting that both cell types are required for SEB response. Interestingly, it was noted that adherent monocyte/macrophages from wild-type mice responded to SEB by increasing TNF-α production, which was significantly reduced in MyD88-deficient monocyte/macrophages. This was not observed with T-cell derived cytokines such as IFN-γ or IL-2. We also investigated in vitro cytokine response after SEB plus LPS treatment. While SEB plus LPS had a strong TNF-α, IFN-γ or IL-6 in wild-type cells, the response was significantly reduced in MyD88-deficient spleen cells (Fig. 5C). In addition, macrophage chemotactic protein (MCP-1) and IL-12 were also significantly reduced in MyD88–deficient mice. These results suggest that, in both cell types, MyD88-mediated pro-inflammatory signaling is involved.
Impairment of pro-inflammatory cytokine responses after SEB and LPS stimulation of spleen cells from MyD88 gene knockout mice (MyD88–/–). (A) Proliferative response to SEB by spleen cells from BALB/c and C57BL/6 mice. Spleen cells from BALB/c and C57BL/6 mice were cultured with SEB at 10, 100 and 200 ng/ml and proliferative response was measured as described in Materials and Methods. Results are expressed as mean ± SD and significance assigned as *P = 0.0215 and xP = 0.0205. (B). Impairment of pro-inflammatory cytokine response after SEB stimulation of spleen cells from MyD88–/– mice or in reconstituted cultures. Spleen cells from MyD88–/– or C57BL/6 (wild-type) mice were in vitro cultured with SEB (200 ng/ml) for 16 h. Results are expressed as mean ± SD and significance was assigned as P = 0.0002. (C) TNF-α, IFN-γ, IL-6, MCP-1 and IL-12 production by spleen cells from C57BL/6 and MyD88–/– mice in response to SEB (200 ng/ml) + LPS (1 µg/ml). Spleen cells were cultured for 16 h. Graphs show mean values ± SD, significance was assigned as *P = 0.0002 and xP = 0.037.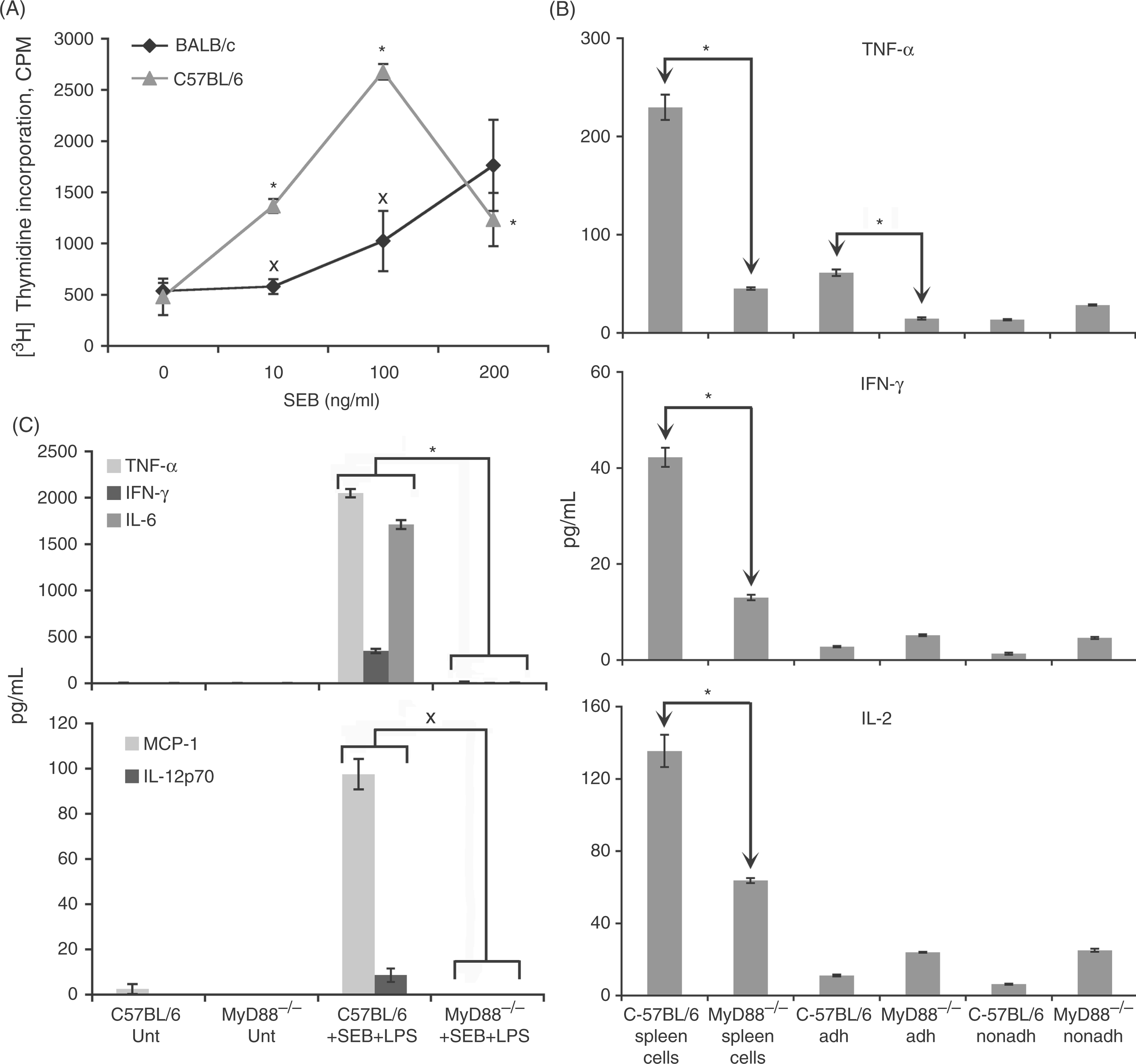
MyD88 is required for SEB toxicity with LPS potentiation in C57BL/6 mice
The in vivo results demonstrated SEB-induced toxicity and death in LPS and d-GalN sensitized BALB/c mice. To validate further the requirement of MyD88 in SEB intoxication, we compared the survival of LPS sensitized C57BL/6 and MyD88–/– mice using different doses of SEB challenge. Our results (Fig. 6A,B) showed the dose-dependent toxicity of SEB in C57BL/6 mice (P = 0.0001), while none of the MyD88–/– mice showed mortality. These results suggest that, irrespective of the genetic background (different MHC-haplotype), MyD88-mediated pro-inflammatory signaling contributed to SEB induced lethality in mice whereas MyD88–/– mice were resistant to SEB induced toxic shock.
MyD88–/– mice resistant to SEB intoxication. Lipopolysaccharide-potentiated lethal SEB challenge of C57BL/6 and MyD88–/– mice was used to evaluate MyD88-mediated toxicity to SEB in vivo. (A) C57BL/6 mice (n = 6/group) were inoculated (i.p.) with SEB (5, 25 or 50 µg/mouse) followed by LPS (110 µg/mouse), or SEB or LPS alone. (B) MyD88–/– mice (n = 6/group) were inoculated (i.p.) with SEB (5, 25 or 50 µg/mouse) followed by LPS (110 µg/mouse), or SEB or LPS alone. Mice were observed for survival. Data presented as the percentage survival. Control mice injected with 110 µg of LPS or 50 µg of SEB survived. MyD88 is required for SEB toxicity in d-GalN sensitized mice. MyD88–/– mice were protected from SEB toxic shock induced death after high doses of SEB challenge. C57BL/6 (n = 6) and MyD88–/– mice (n = 6) were injected with d-GalN (20 mg/mouse) 30 min prior to SEB (100 µg/mouse). Mice were examined for survival. All control C57BL/6 and MyD88–/– mice survived d-GalN treatment.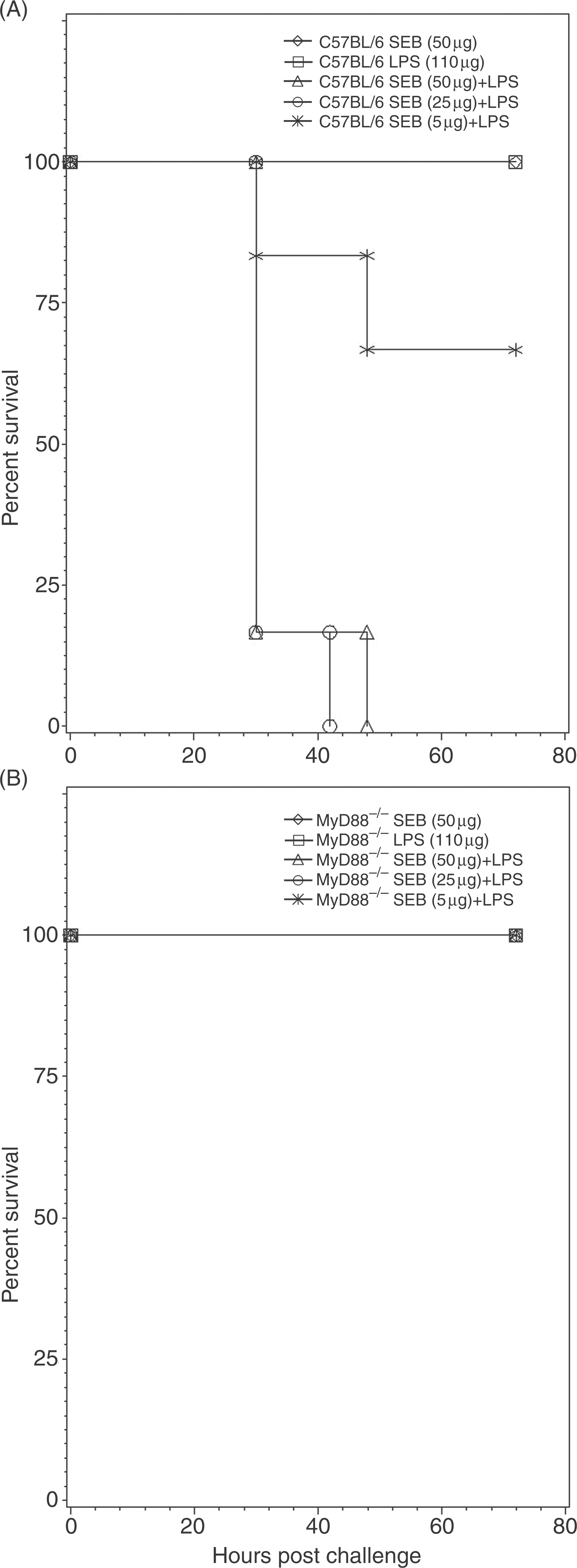
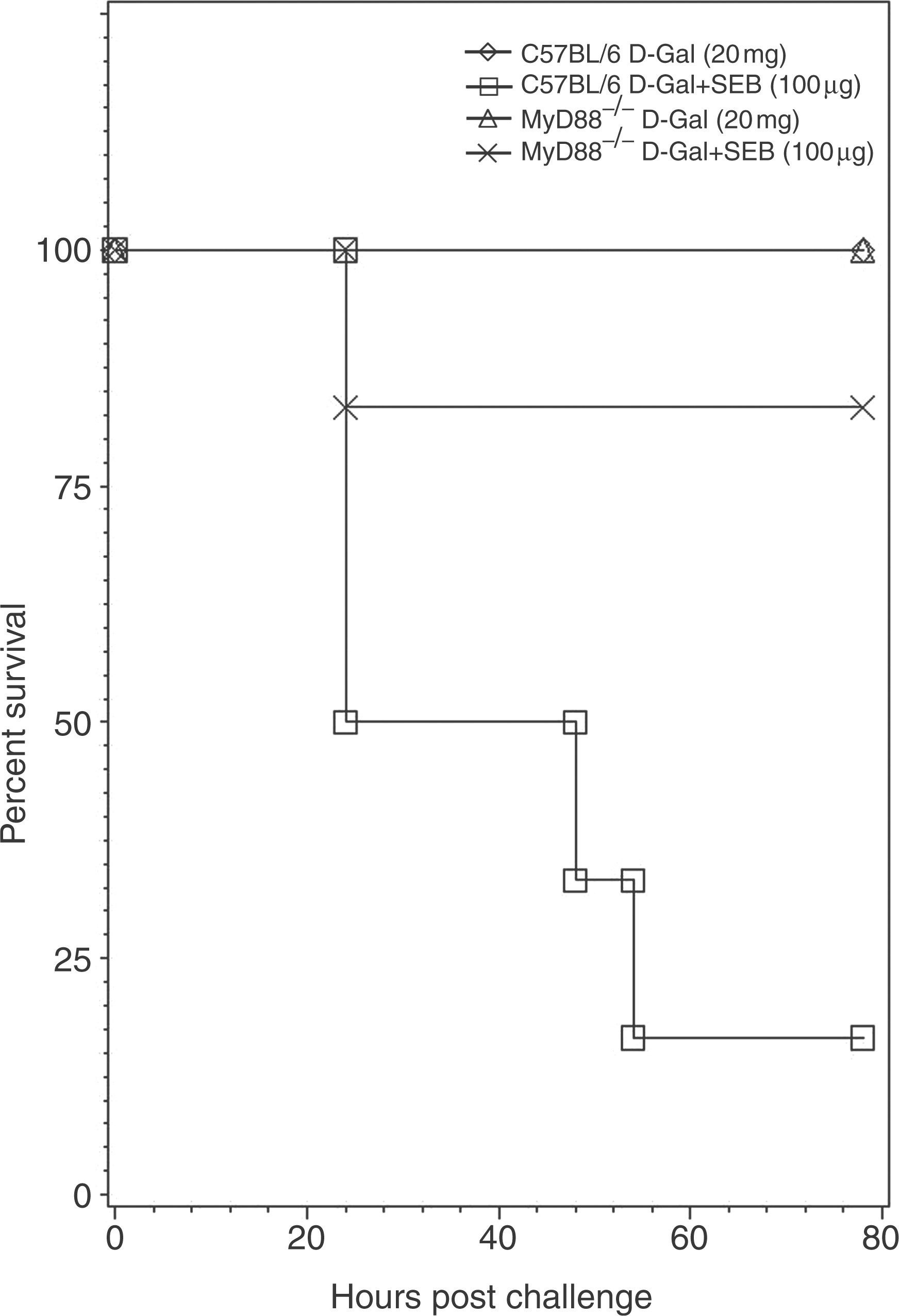
MyD88 is required for SEB toxicity independent of LPS in C57BL/6 mice
To demonstrate further that MyD88 is required for SEB toxicity in the absence of LPS, we compared SEB toxicity in wild-type and MyD88–/– mice using a high dose of SEB (100 µg/mouse) in a more appropriate d-GalN sensitization model. At 24 h, we observed only 50% survival in wild-type mice while 83% of MyD88–/– mice survived. By 54 h, only 17% of wild-type mice survived whereas 83% of MyD88–/– mice were alive (P = 0.0303). These results point to the significant contribution of MyD88 to SEB-induced toxicity.
Discussion
We have shown here that SEB-induced lethality in a mouse challenge model depends on MyD88–mediated pro-inflammatory signaling mechanism. Our data demonstrate that spleen cells from MyD88–/– mice had impaired inflammatory cytokine production in response to SEB. MyD88–/– mice were protected from a lethal SEB challenge. These data are compatible with previous reports that peritoneal macrophages from MyD88-deficient mice are unable to produce any detectable levels of TNF-α and IL-6 in response to Staphylococcus aureus infection. 28 In addition, our data also confirmed LPS potentiation of SEB toxicity that synergized with MyD88-mediated pro-inflammatory signaling and caused strong lethality compared to LPS-independent system. d-GalN-sensitized mice succumbed to death when exposed to SEB while d-GalN did not affect cytokine production. D-Galactosamine likely aided SEB toxicity primarily through hepatocytes which have been characterized by a marked decrease in the UTP levels after d-GalN treatment. 25 The signal transduction mechanism for IL-1 and TNF-α expression after SEB binding to MHC class II molecules on human monocytes has been partially characterized and involves protein kinase C (PKC) and protein tyrosine kinase (PTK).9–11 However, it was not known whether the elevated pro-inflammatory response associated with SEB toxic shock is solely controlled by these pathways. Our data suggest that MyD88–mediated signaling dominantly influences cytokine production involved in SEB toxic shock.
In general, MyD88 integrates and transduces intracellular signals generated by the TLR and IL-1R superfamily. This is done through TIR domain interactions that are critical for several aspects of innate immune regulation.29,30 However, recent reports suggest that not only IL-1- but also IFN-γ-induced signaling regulates MyD88-dependent pathways. 12 It is also known that MyD88 can associate with signaling proteins by means other than homophilic TIR or death-domain interactions. Interactions have been detected between MyD88 and other proteins lacking TIR and death domains such as Bruton’s tyrosine kinase, 31 phosphatidyl-inositol-3-OH kinase, 32 and IFN regulatory factor 7. 33 Our results indicate that binding of SEB to MHC class II molecules, which were expressed at high levels on the surface of APCs, activated MyD88-mediated signaling. The MyD88-mediated pro-inflammatory cytokine expression was also observed in MHC class II positive primary monocytes in isolation, (i.e. in the absence of T-cells) or selected after stimulation of total peripheral blood mononuclear cells with SEB exposure (manuscript submitted). Previous studies indicated that SEB binding to MHC class II molecules on monocytes elicits rapid transcription of IL-1β and TNF-α gene(s)4,11 and increased membrane expression of TLR4. 18 Furthermore, attenuation of several cytokines and chemokines released in response to LPS by MyD88-defective cells was also reported.25,26 In line with these observations, our results provide a possible link between MyD88–mediated signaling and toxic shock. 7 Thus, our results demonstrated that MyD88 was a required component of intracellular signaling due to exposure of SEB as well as SEB plus LPS. This signaling intersects at a number of points that include activation of both APCs and T-cells, leading to the release of pro-inflammatory cytokines and toxic shock. In this study, our results demonstrated that, upon d-GalN application, mice developed sensitivity to lethal shock induced not only by SEB but also LPS. However, the basic difference between both toxins is that macrophages are mediating the endotoxin reactions while the lethal shock triggered by the exotoxin SEB is mediated by SEB-reactive T-cells, as well as activated APCs, such as CD11c+ monocytes and macrophages. Our study uncovers a key process in the severe immunological phenomenon associated with exposure to SEB. In line with this observation, there was no obvious difference in cytokine production in both d-GalN-treated and untreated spleen cell cultures. D-Galactosamine-sensitized mice, however, succumbed to SEB-induced lethal shock. Staphylococcal enterotoxin B-induced death is associated with MyD88-mediated induction of pro-inflammatory cytokines. Similar to TLR4–MyD88 signaling with LPS binding to primary monocytes/macrophages, our data demonstrated that SEB plus LPS stimulation (dual effects) induced robust inflammatory responses. Reduced expression of TNF-α, IFN-γ, IL-6 by spleen cells from MyD88–/– mice treated with SEB, as well as SEB plus LPS stimulation was observed. Most importantly, our data demonstrates that MyD88–/– mice survived SEB induced lethal shock. Similar results were also observed with SEA (i.e. MyD88–/– mice were resistant to SEA toxic shock induced death). 34 This validates the significance of MyD88-mediated signaling mechanism that plays a critical role in Gram-positive toxic shock. Thus, this study highlights the dominant role MyD88 plays in imparting SEB intoxication.
Conclusions
These results suggest that MyD88 may represent an important target for therapeutic intervention for toxic shock syndrome following exposure to SEB. Targeting MyD88 to disrupt pro-inflammatory responses with an appropriate inhibitor would likely provide critical leads towards the development of efficacious therapeutics against toxic shock.
Footnotes
Acknowledgements
The authors thank Amy Egnew and Thomas Plummer for technical assistance, Lorraine Farinick for figure preparation, Diana Fisher for statistical analysis and Dr Bradley Stiles for critical review of the manuscript.
Research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996. The facility where this research was conducted is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Views expressed in this paper are those of the authors and do not purport to reflect official policy of the US Government USAMRIID administrators.
Funding
This work was funded by the Defense Threat Reduction Agency (KUS).