
Abstract
Sepsis is a life-threatening event predominantly caused by Gram-negative bacteria. Bacterial infection causes a pronounced macrophage (MΦ) and dendritic cell activation that leads to excessive pro-inflammatory cytokine IL-1β, IL-6 and TNF-α production (cytokine storm), resulting in endotoxic shock. Previous experimental studies have revealed that inhibiting NF-κB signaling ameliorates disease symptoms; however, the contribution of myeloid p65 in endotoxic shock remains elusive. In this study, we demonstrate increased mortality in mice lacking p65 in the myeloid lineage (p65Δmye) compared with wild type mice upon ultra-pure LPS challenge. We show that increased susceptibility to LPS-induced shock was associated with elevated serum level of IL-1β and IL-6. Mechanistic analyses revealed that LPS-induced pro-inflammatory cytokine production was ameliorated in p65-deficient bone marrow-derived MΦs; however, p65-deficient ‘activated’ peritoneal MΦs exhibited elevated IL-1β and IL-6. We show that the elevated pro-inflammatory cytokine secretion was due, in part, to increased accumulation of IL-1β mRNA and protein in activated inflammatory MΦs. The increased IL-1β was linked with heightened binding of PU.1 and CCAAT/enhancer binding protein-β to Il1b and Il6 promoters in activated inflammatory MΦs. Our data provide insight into a role for NF-κB in the negative regulation of pro-inflammatory cytokines in myeloid cells.
Introduction
Sepsis is a life-threatening event that can be complicated by shock, coagulopathy and multiorgan dysfunction, causing millions of deaths globally each year. 1 Sepsis is predominantly caused by Gram-negative bacterial infection, 2 and the host–microbial interaction promotes an acute immune/inflammatory response involving both humoral and cellular components that can cause endotoxic shock and death within a few days. 3
The major outer surface membrane component of the Gram-negative bacteria cell envelope is LPS. LPS interacts with the LPS-binding protein and CD14 and binds TLR4/MD2 on mononuclear cells, including dendritic cells (DCs), monocytes and macrophages (MΦs). 4 The LPS–TLR4/MD2 interaction leads to the recruitment of the adaptor proteins myeloid differentiation primary response protein 88 (MyD88) and Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF), stimulating the subsequent activation of the IκBα kinase (IKK) complex (IKKβ, IKKα and NEMO).5,6 The activated IKK complex phosphorylates IκB, leading to IκB ubiquitination and proteasomal degradation, freeing NF-κB/Rel complexes, and enabling NF-κB translocation to the nucleus and transcription of NF-κB-responsive pro-inflammatory cytokines (e.g. IL-1, IL-6 and TNF-α).5–8 The mammalian NF-κB family comprises five members: RelA (p65), RelB, c-Rel, p105/p50 (NF-κB1) and p100/p52 (NF-κB2). The NF-κB pathway plays a critical functional role in LPS-induced inflammatory gene expression and LPS-induced lethality.5,6,8–10 The importance of p50 and p65 of the canonical pathway in the pathophysiology of sepsis has been demonstrated by increased nuclear binding activity of each subunit in both animal models and PBMCs of patients with sepsis. 11 Increased NF-κB activity is associated with increased mortality and worse clinical outcomes, 12 and inhibiting NF-κB activity protects mice from endotoxin-induced shock.13,14
The cytokines IL-1β, IL-6 and TNF-α are thought to promote many of the immunopathologic features of LPS-induced shock.4,15 Elevated levels of plasma IL-1β, IL-6, and TNF-α are associated with the onset of septic shock.15–18 Administering LPS into the bloodstream results in a rapid increase in serum IL-1β and TNF-α, the latter of which correlated with the pathophysiologic changes (fever, tachycardia and slight hypertension) in humans. 19 Furthermore, intravenous administration of TNF-α and/or IL-1β in mice and humans results in a septic shock-like state.15,20–26 Though IL-6 has been shown to not have a causal role in septic shock, plasma IL-6 levels correlate with mortality from septic shock. 18 Clinical and preclinical murine studies suggest that IL-1β, IL-6 and TNF-α have differential roles in immunopathogenesis of the septic shock reaction, including modulating thermoregulation, food intake and lethality of sepsis.27,28
There are several lines of clinical and experimental evidence suggesting that pro-inflammatory cytokines derived from MΦs are important in the LPS-induced shock. 4 LPS stimulation of human myeloid cell lines (THP-1 and U937) and PBMC-derived MΦs results in the induction of pro-inflammatory cytokine genes. 29 Furthermore, transfer of MΦs from C3H/HeN mice to LPS-resistant C3H/HeJ mice reconstituted high sensitivity to the lethal effects of LPS. 30 Though there are substantial data independently linking NF-κB signaling and MΦ-derived pro-inflammatory cytokine response to LPS-induced shock, the requirement of p65 NF-κB signaling in myeloid cells in LPS-induced shock has not been directly demonstrated. As studying the individual contributions of the NF-κB subunits in LPS-induced shock in vivo has been limited by the embryonic lethality of mice lacking p65, 31 we have recently generated LysMCrep65fl/fl mice. 32 In mice lacking p65 in the myeloid lineage (p65Δmye), we demonstrate increased mortality, upon ultra-pure LPS (U-LPS) challenge, that was associated with elevated serum levels of IL-1β and IL-6. We show that loss of p65 leads to increased IL-1β and IL-6 levels in peritoneal ‘activated’ MΦs in response to U-LPS, which was linked to increased binding of the transcription factors C/EBP-β and PU.1 to the associated genes' regulatory elements. This study reveals an important role for NF-κB in restraining PU.1 and CCAAT/enhancer binding protein β (c/EBP-β)-driven pro-inflammatory cytokine production in myeloid cells, which confers protection from LPS-induced pro-inflammatory cytokine production and shock.
Materials and methods
Mice
Male and female lysozyme M (LysM)cre/cre RelA/p65fl/fl (RelA/p65Δmye, C57BL/6/129/SvEv) and LysMcre/cre RelA/p65+/+ [wild type (WT) control line for RelA/p65Δmye mice]. 32 All mice were housed under specific pathogen-free conditions, animal care was performed by experienced veterinary technicians and all experimental techniques were approved by the Institutional Animal Care and Use Committee (IACUC) of Cincinnati Children's Hospital Medical Center (CCHMC).
LPS-induced shock
WT and RelA/p65Δmye mice received one i.p. injection of U-LPS from Escherichia coli K-12 (LPS-EK Ultrapure; InvivoGen, San Diego, CA, USA) (40 mg per kg body mass) diluted in pyrogen-free saline, and evidence of LPS-induced shock was examined every 12 h. In initial dose–response experiments 40 mg/kg U-LPS was shown to generate a robust inflammatory response (cytokine production IL-1β, TNF-α and IL-6) in the absence of a rapid shock response in the WT C57BL6 mice. In accordance with CCHMC IACUC-approved protocols, we designated mice as experiencing irreversible LPS-induced shock via the following criteria: hunched posture, lethargy, labored breathing, no righting reflex response and hypothermia (>5℃ temperature loss). When designated as experiencing LPS-induced shock, mice were immediately euthanized by CO2 inhalation.
Bone Marrow Derived-Macrophages (BMDM)
Bone marrow was obtained from mice femurs using 5 ml harvesting buffer (25 mM HEPES/50 µg/ml gentamycin in 1 × HBSS). Cells were passed through a 70-μM filter and centrifuged at 300 g before being re-suspended in 20 ml BMDM media (20% M-CSF in 10% FBS containing complete DMEM) and cultured for 5 d (37 ℃, 5% CO2).
Peritoneal MΦ
Mice were injected i.p. with 1 ml 3.85% thioglycollate as previously described. 33 Three d after the i.p. injection, a peritoneal lavage was performed, and cells were centrifuged and re-suspended in 25 ml 10% FBS complete DMEM and plated for 4 h for adherence. Cells were then washed with PBS and detached with trypsin/EDTA (Life Technologies, Carlsbad, CA, USA) at 37 ℃ for 5 min and re-suspended in 10% FBS complete DMEM. Cells were then gently scraped off and re-suspended in 10% FBS complete DMEM for analyses.
MΦ stimulation
BMDM (1 × 105 cells/250 μl) were plated in a 96-well plate and incubated for 18 h at 37 ℃, 5% CO2. BMDM or peritoneal MΦs (2 × 106 or 2.5 × 105) were stimulated with 0, 10 or 100 ng/ml U-LPS for the indicated hours according to experiments and then stimulated for 1 h with 2 mM ATP (InvivoGen, San Diego, CA, USA) for inflammasome activation. Supernatant and mRNA were collected and analyzed for cytokine induction.
ELISA
IL-1β, IL-6 and TNF-α levels were measured in the supernatant after cell culture using the ELISA Duo-Set kit according to the manufacturer's instructions (R&D System, Minneapolis, MN, USA). Serum type I IFN levels were determined with the IFN-β bioassay as previously described. 34
FACS analysis
Single-cell suspensions were washed with FACS buffer (PBS/1% BSA) and incubated for 30 min at 25 ℃ (room temperature) with combinations of the following Abs: PE anti-mouse F4/80 (clone CI:A3-1; AbD Serotec, Raleigh, NC, USA), PE-Cy7 anti-mouse CD11b (clone M1/70; BD Pharmingen, San Jose, CA, USA), Alexafluor-647 anti-mouse Ly6C (clone ER-MP20; AbD Serotec) and FITC anti-mouse Ly6G (clone 1A8; BD Pharmingen, San Jose, CA, USAs). Cells were washed once with FACS buffer and analyzed on a FACSCanto II (BD Immunocytometry System, San Jose, CA, USA), and analysis was performed using FlowJo software (Tree Star, Ashland, OR, USA). For experiments conducted to determine immune cell populations in the peritoneal cavity, the following Abs were used: APC anti-mouse CD19 (clone 1D3), PE anti-mouse CD23 (clone B3B4), PE-Cy7 anti-mouse IgM (clone R6-60.2) or CD3 (clone 145-2C1) or FcɛRIα (clone MAR-1), FITC anti-mouse B220 (clone RA3-6B2), PerCp-Cy5.5 anti-mouse CD11b (clone M1/70) or CD11c (clone HL3), AlexaFluor750 anti-mouse c-KIT (clone 2B8) or CD4 (clone GK1.5; BD Pharmingen), and PE anti-mouse MHC class II (clone NIMR-4; eBioscience; San Diego, CA, USA). All Abs were used in a 1:200 dilution in FACS buffer.
Western blot
BMDM or peritoneal MΦs after stimulation were lysed using mammalian protein extraction reagent (M-PER; Life Technologies, Carlsbad, CA, USA). Approximately 40 µg protein extract was separated on a 4–12% Bis-Tris gel and transferred to a nitrocellulose membrane (Life Technologies). The following Abs were used: rabbit anti-mouse caspase-1 p10 (M20; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and goat anti-mouse IL-1β/IL-1F2 (R&D Systems) followed by goat anti-rabbit (Calbiochem, Darmstadt, Germany) or sheep anti-goat (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) peroxidase-conjugated Abs. Rabbit anti-mouse β-actin (Sigma, St. Louis, MO, USA) was used as loading control, and ECL-plus detection reagent (Life Technologies) was used for detection.
Chromatin immunoprecipitation (ChIP) assay
Peritoneal MΦs (2 × 107 cells) were cross-linked with 0.8% formaldehyde for 10 min at room temperature. Cells were collected, lysed and then sonicated for 9 min (peak power 175 W, duty factor 5%, cycles/burst 200 count) at 4 ℃ using a Covaris S series Sonicator (Covaris, Woburn, MA, USA), yielding chromatin fragments with an average size of around 400 bp. To reduce unspecific binding, each chromatin sample was pre-cleared with 10 μl Dynabeads® Protein G magnetic beads (Life Technologies). One µg primary Ab for C/EBP-β (sc-150X; Santa Cruz Biotechnology) or PU.1 (Spi-1) (sc-352X; Santa Cruz Biotechnology) was bound to 5 μl Dynabeads® Protein G magnetic beads (Life Technologies) for 1 h at room temperature on a rotating wheel. Five µg chromatin complexes was then added to the Ab/beads mix and immunoprecipitated for 14–16 h at 4 ℃ on a rotating wheel. The precipitates were washed with 1 ml twice for each of the following buffers in succession: low-salt wash buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 150 mM NaCl), high-salt wash buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, 400 mM NaCl), LiCl wash buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 250 mM LiCl, 0.5% sodium deoxycholate, 0.5% Nonidet P-40) and Triton X-100 buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 0.2% Triton X-100). Chromatin complexes were eluted in 100 μl elution buffer (10 mM Tris-HCl pH 7.6, 1 mM EDTA, 250 mM NaCl, 0.3% SDS) and incubated at 37 ℃ for 1 h with shaking. To degrade proteins and reverse cross-link, 5 μl of Proteinase K solution (Life Technologies) was added to the eluates and incubated at 37 ℃ for 1 h followed by incubation at 65 ℃ for 6 h. DNA was purified with a QIAquick® PCR purification kit (Qiagen, Valencia, CA, USA), eluted in 30 μl molecular biology-grade water (Sigma) and analyzed with quantitative real-time PCR.
Real-time PCR analysis
The RNA samples (1 µg) were subjected to reverse transcription analysis using SuperScript II reverse transcriptase (Life Technologies) according to the manufacturer's instructions. Mouse Il1b, caspase-1, Ifnb, Ccl12 and Cxcl10 were quantified by real-time PCR using the iQ5 multicolor real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA, USA) with iQ5 software V2.0 and LightCycler FastStart DNA Master SYBR Green I. The sequences of primers and probes used for quantitative RT-PCR are described in Supplemental Table S1. Gene expression was determined as relative expression on a linear curve based on a gel-extracted standard and was normalized to Hprt amplified from the same cDNA mix. Results were expressed as the gene of interest/Hprt ratio that had been normalized for each group to the unstimulated p65Δmye group. For analyzing the ChIP samples, a region of the Il1b and Il6 promoters containing putative binding sites for C/EBP-β and PU.1 and an intronic region of Gapdh were quantified with real-time quantitative PCR using the Taqman® Universal PCR Mastermix chemistry (Applied Biosystems by Life Technologies) with Prime Time 5' 6-FAM/ZEN/3' IBFQ taqman probes (IDT, Coralville, IA). Primer sets used for either expression or ChIP analysis are reported in Supplemental Table S1.
MTT assay
Cells (1 × 106 or 5 × 104) were plated in a 96-well plate and stimulated with U-LPS at the indicated concentrations and lengths of time. Supernatant was poured off after stimulation, and 100 μl 4 mg/ml MTT (Sigma-Aldrich) was added and followed by 1 h of incubation at 37 ℃. The formazan crystals were dissolved using 100 μl MTT lysis buffer (10% SDS in isopropanol), and absorbance was measured at 570 nm and the background (OD 630 nm) subtracted. Results are presented as percentage of survival with the control (untreated cells) representing 100% survival.
Statistical analysis
Data are presented as mean ± SEM. Statistical significance was determined by means of ANOVA, followed by the Tukey's post-hoc test or a two-tailed unpaired t-test with GraphPad Prism 5 (GraphPad Inc., San Diego, CA, USA). P < 0.05 was considered significant.
Results
U-LPS challenge decreases survival rate in p65Δmye mice
To determine whether myeloid-specific p65 regulates LPS-induced shock, p65Δmye (LysMCre p65fl/fl) mice and littermate WT (LysMCre p65WT/WT) control mice were administered U-LPS (40 mg/kg) by i.p. injection. In accordance with CCHMC IACUC-approved protocols, we designated mice as experiencing irreversible LPS-induced shock via the following criteria: hunched posture, lethargy, labored breathing, no righting reflex response and hypothermia (>5℃ temperature loss). When designated as experiencing LPS-induced shock, mice were immediately euthanized. WT mice did not demonstrate evidence of LPS-induced shock 18 h after U-LPS challenge (Figure 1). In contrast, >30% (n = 7/24) of the littermate age-, mass- and strain-matched p65Δmye mice died within 12 h, and 90% (n = 21/24) died within 18 h of U-LPS injection (Figure 1; P < 0.0005). These data indicate that myeloid p65 deletion renders mice more susceptible to U-LPS challenge.
In vivo U-LPS challenge results in increased pro-inflammatory cytokine production and mortality in p65Δmye mice. (a). U-LPS-challenged survival of p65Δmye and littermate WT mice. Serum cytokine levels of (b) IL-1β, (c) IL-6 and (d) TNF-α at indicated time points (h) in WT and p65Δmye mice following one i.p. injection with U-LPS (40 mg/kg). Data represent the mean ± SEM of two independent experiments with (a) n = 7–11 mice per group and (b–d) n = 3–11 mice per group. *P < 0.05 between groups.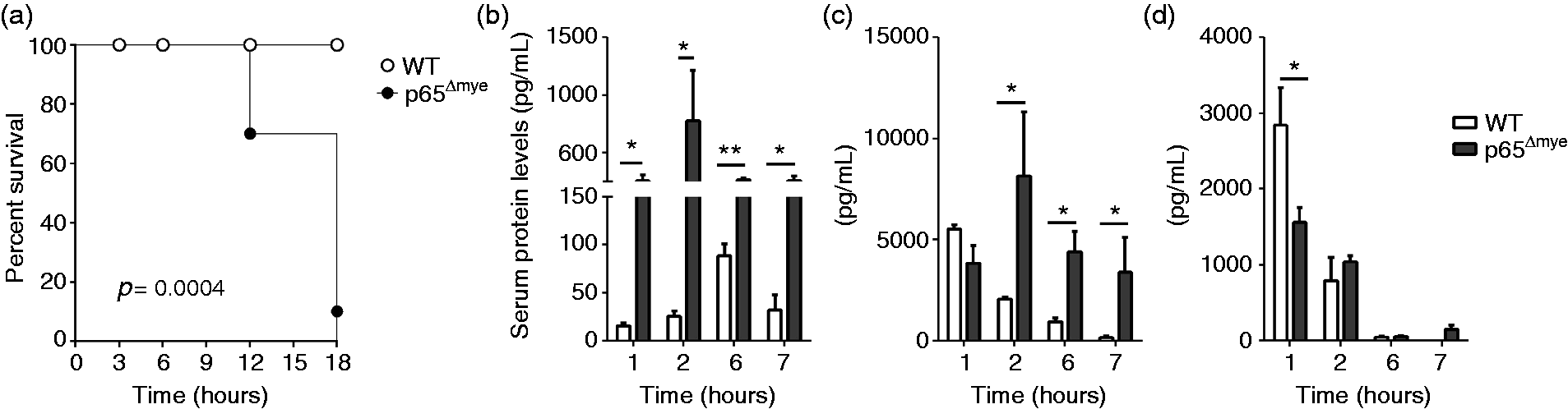
U-LPS challenge increases pro-inflammatory cytokines and alters cell recruitment in p65Δmye mice
As LPS-induced shock is associated with innate immune activation, we examined pro-inflammatory cytokine levels in the sera and inflammatory cell populations in the peritoneal cavity of U-LPS-challenged WT and p65Δmye mice. U-LPS challenge of WT mice induced a potent pro-inflammatory cytokine response characterized by increased serum IL-1β, IL-6 and TNF-α (Figure 1b–d). Consistent with previous observations, we observed a rapid rise in serum IL-6 and TNF-α within 1–2 h and an IL-1β response by 6 h after U-LPS challenge in WT mice (Figure 1b–d). Notably, serum cytokine levels tapered by 6 h after U-LPS challenge in WT mice (Figure 1b–d). In p65Δmye mice, we observed a significantly more potent initial IL-1β- and IL-6-specific response (Figure 1b, c). Moreover, U-LPS challenge induced a significantly more rapid, stronger and sustained IL-1β and IL-6 response, with serum IL-1β and IL-6 levels 30-fold and 4-fold higher, respectively, in the p65Δmye than WT mice at 2 h (Figure 1b, c). Though serum TNF-α levels 1 h after U-LPS challenge were reduced in the p65Δmye mice compared with WT mice, levels were comparable between groups throughout the rest of the time course (Figure 1d).
Cell populations in peritoneal cavity of WT and p65Δmye mice at steady state and following UP-LPS exposure.
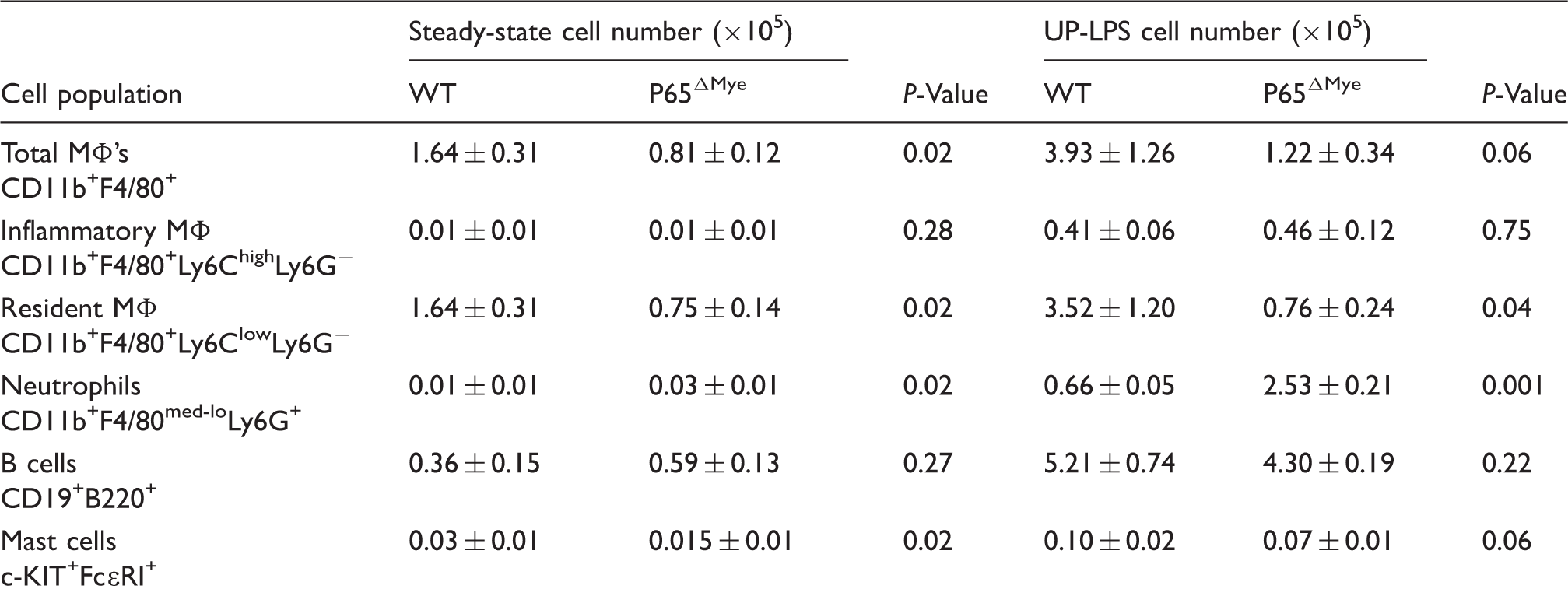
Data represent the total cell numbers (×105) in the peritoneal cavity at steady state (n = 7–8 mice per group) and 60 min following U-LPS challenge (n = 3–4 mice per group). Data represented as mean ± SD are representative of three separate experiments.
Decreased inflammation and viability in p65Δmye BMDM upon U-LPS stimulation
As MΦs have long been associated with mortality in sepsis,
35
we examined U-LPS responses in BMDM from WT and p65Δmye mice to determine whether the exaggerated phenotype in p65Δmye mice can be attributed to an inherent defect in myeloid cells after p65 deletion (Figure 2). We initially examined BMDM that were generated from bone marrow cells after in vitro stimulation with M-CSF for 5 d,
32
as these cells represent a homogenous myeloid cell population that possess most characteristics of macrophages.
36
We have previously demonstrated efficient and specific LysM-Cre-mediated deletion of RelA/p65 in the myeloid compartment by (1) loss of total RelA/p65 protein in RelA/p65Δmye BMDM in the presence of normal levels of total RelA/p65 in the spleen and colonic epithelium of RelA/p65 Δmye mice; (2) loss of RelA/p65 activation in RelA/p65Δmye BMDM following LPS stimulation; and (3) normal levels of IKK-α, c-Rel and p105 in RelA/p65Δmye BMDM; these data support the selective RelA/p65 deletion in myeloid cells and that this is independent of effects of other components of the NF-κB signaling pathway.
32
Flow cytometry analyses of WT and p65Δmye BMDM revealed that the majority of the cultured cells were large in size with granular morphology (SSChiFSChi) (Figure 2a, b, gate a). This population was identified as F4/80+CD11b+ cells that consisted of both Ly6Clo and Ly6Chi cell subsets, representing both mature and immature MΦs, respectively. The comparable level of Ly6Clo and Ly6Chi cell subsets and level of expression of the BMDM maturation marker F4/80
37
on WT and p65Δmye BMDM populations (Figure 2a, b) indicate that p65 is dispensable for the development and maturation of BMDM in vitro. U-LPS stimulation of WT BMDM induced a pronounced pro-inflammatory cytokine response (IL-1β, IL-6 and TNF-α) that was evident at 3 h and 6 h post-stimulation (Figure 2c). In contrast, U-LPS stimulation of p65Δmye BMDM did not induce a pronounced IL-1β, IL-6 and TNF-α response. Examining the BMDM viability after U-LPS stimulation did reveal a significant reduction in viability in p65Δmye BMDM compared with WT BMDM after high U-LPS (10 ng/ml or higher) stimulation (Figure 2d); however, we only observed ∼10% reduction in viability in p65Δmye BMDM compared with WT BMDM, suggesting that the absence of a pro-inflammatory cytokine response to U-LPS in p65Δmye BMDM is not likely attributed to decreased myeloid cell viability. Collectively, these data suggest that myeloid p65 deletion decreases a BMDM's capability to produce pro-inflammatory cytokines after U-LPS stimulation.
In vitro U-LPS stimulation decreases pro-inflammatory cytokine expression and viability in p65Δmye BMDM. (a, b) Flow cytometry analyses of F4/80, CD11b and Ly6C expression in WT and p65Δmye BMDM, respectively. (a–d) Indicated gatings. (c) Pro-inflammatory cytokine IL-1β, IL-6 and TNF-α secretion from BMDM pretreated for 0, 3 or 6 h with 100 ng/ml U-LPS followed by 1 h of 2 mM ATP stimulation. (d) Viability of BMDM determined by MTT assay. Cells were treated with 0, 1, 10 100 or 1000 ng/ml U-LPS for 24 h prior to MTT assay. Data represent the mean ± SEM from n = 2–3 separate experiments. Significant differences indicated (*P < 0.05; **P < 0.01; ***P < 0.005) between groups. N.D.: not detectable.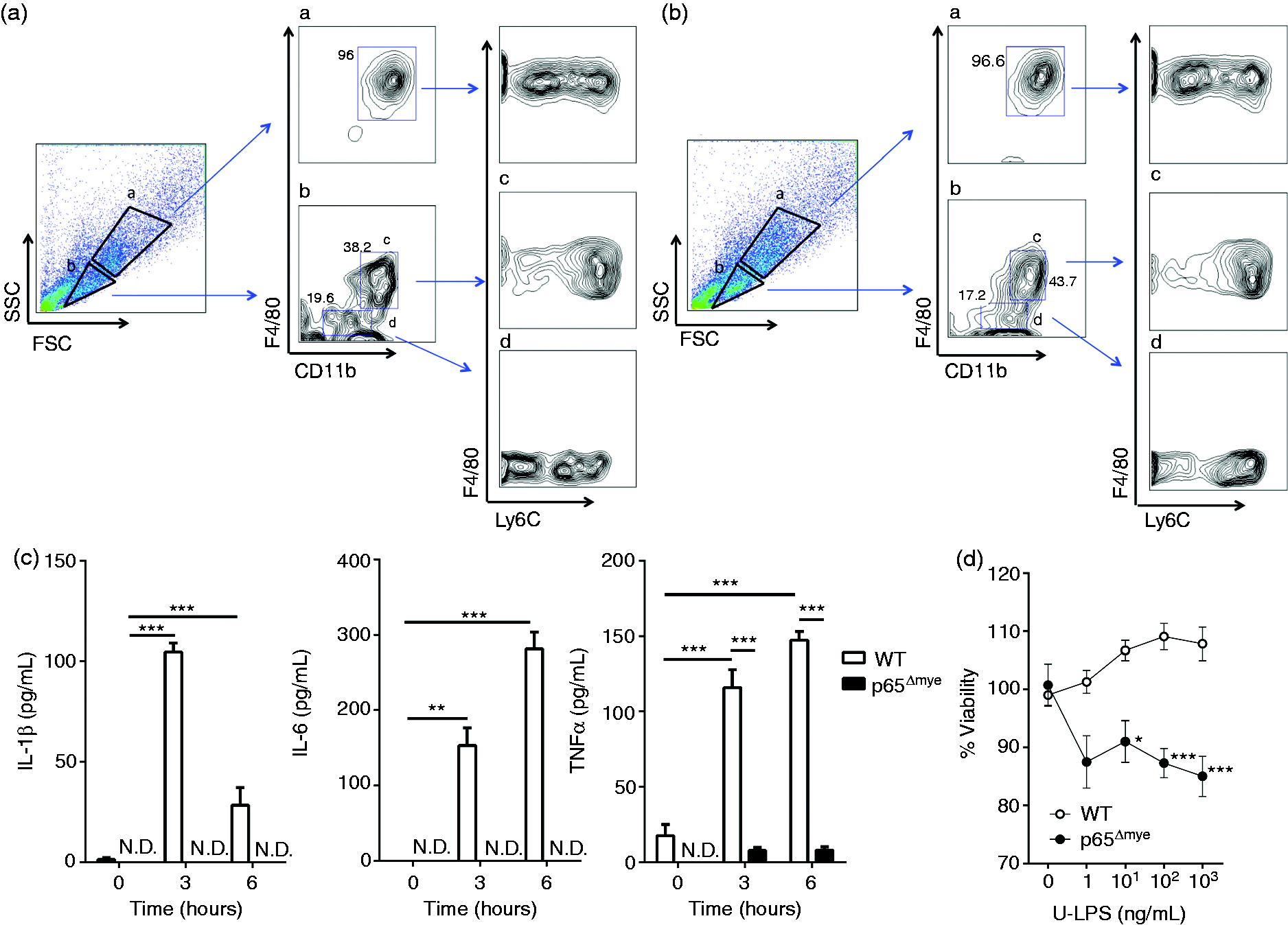
U-LPS stimulation results in increased pro-inflammatory cytokine production in p65Δmye peritoneal MΦs
As our BMDM studies did not reconcile our in vivo observations, we next examined the effect of p65 deletion on inflammatory MΦs. We assessed the responsiveness of thioglycollate-elicited MΦs to U-LPS. Peritoneal cells were acquired from WT and p65Δmye mice 3 d after i.p. thioglycollate injection and purity assessed after 4 h adherence (Supplemental Figure S1). Flow cytometry analyses of peritoneal cells revealed that myeloid deletion of p65 did not alter the inflammatory cell recruitment into the peritoneal cavity of mice after thioglycollate exposure (Supplemental Figure S1). Moreover, flow cytometry profiles demonstrated the presence of similar proportions of SSChiFSClo F4/80+CD11b+ Ly6Clow and Ly6Chigh cells (Supplemental Figure S1). U-LPS stimulation of thioglycollate-elicited WT and p65Δmye MΦs resulted in a significant increase in IL-1β, IL-6 and TNF-α secretion (Figure 3a). Notably, the IL-1β and IL-6 levels derived from p65Δmye thioglycollate-elicited MΦs were significantly higher than those of WT cells (Figure 3a; 2-fold IL-1β, P < 0.05; and 4-fold IL-6, P < 0.05), whereas the levels of TNF-α were comparable between groups (Figure 3a). This was strikingly similar to the pro-inflammatory cytokine response observed in vivo after U-LPS administration to WT and p65Δmye mice (Figure 1).
In vitro U-LPS challenge increases pro-inflammatory cytokine expression in p65Δmye peritoneal MΦs. (a) Pro-inflammatory cytokine IL-1β, IL-6 and TNF-α secretion. (b) Il1b and caspase-1 gene expression analyzed by quantitative real-time PCR. (c) Western blot of pro-IL-1β expression in unstimulated peritoneal inflammatory MΦs. (d) Western blot of pro-IL-1β, IL-1β, pro-caspase-1 and caspase-1 expression of peritoneal MΦs pretreated for 6 h with 0, 10 or 100 ng/ml U-LPS followed by a 1-h stimulation with 2 mM ATP. Data represent the mean ± SEM of two independent experiments with n = 2–3 mice per group. Cytokine expression is normalized to the expression of the reference gene (hprt). Significant differences (*P < 0.05; **P < 0.01; ***P < 0.005) between groups. N.D.: not detectable.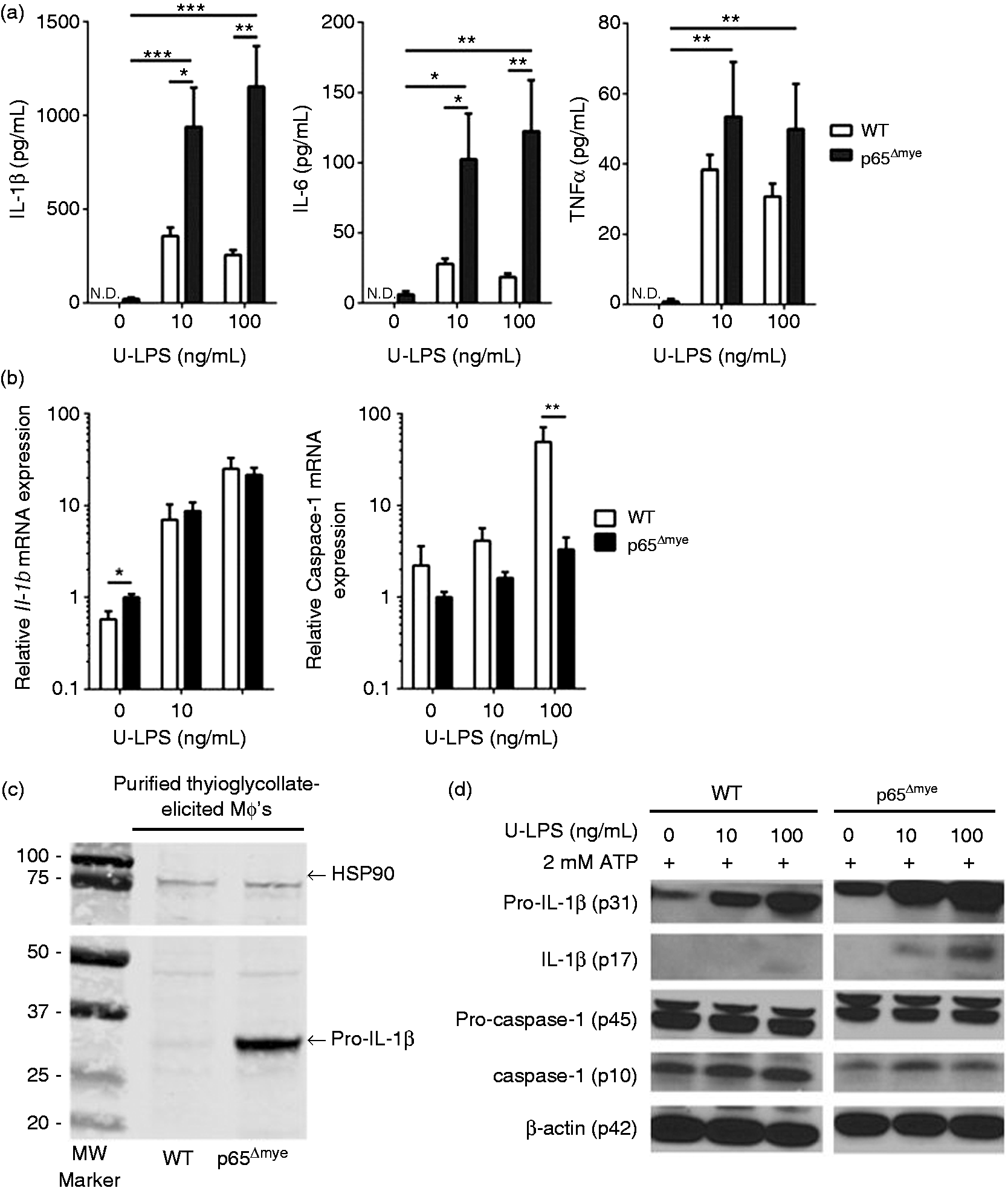
To begin to elucidate the molecular basis of the increased IL-1β and IL-6 secretion in p65Δmye MΦs, we examined mRNA and protein levels of IL-1β and inflammasome activation (caspase1) in WT and p65Δmye MΦs after U-LPS stimulation. We show that U-LPS and ATP stimulation of WT MΦs stimulated an increase in Il1b and caspase-1 mRNA (Figure 3b) and pro-IL-1β and cleaved IL-1β protein (Figure 3a, D). U-LPS and inflammasome activation (2 mM ATP) did not have an observable effect on pro-caspase-1 levels but appeared to increase activated caspase-1 (p10) (Figure 3d). U-LPS and ATP stimulation of p65Δmye MΦs led to a comparable increase in IL-1β mRNA; however, the level of induction of caspase-1 mRNA was reduced in p65Δmye MΦs compared with WT MΦs (Figure 3b). Consistent with this observation, the protein levels of pro-caspase-1 and active caspase-1 in p65Δmye were reduced in comparison with WT MΦs stimulated with U-LPS and ATP (Figure 3d). Surprisingly, the level of pro-IL-1β mRNA and protein in unstimulated p65Δmye MΦs was greater than that observed in unstimulated WT MΦs (Figure 3b, c). Furthermore, we observed increased pro-IL-1β and cleaved IL-1β after U-LPS and ATP stimulation (Figure 3a, d). These data suggest that p65Δmye inflammatory MΦs possess a reduced level of inflammasome machinery compared to that of WT; however, steady-state levels of pro-IL-1β mRNA and protein appear to be significantly elevated.
p65 deletion does not alter surface TLR4 expression, MyD88-independent TLR4 signaling or anti-inflammatory responses in thioglycollate-elicited MΦs
Following our observation of heightened IL-1β and IL-6 cytokine production in p65Δmye peritoneal MΦs after U-LPS stimulation, we were interested in delineating the underlying molecular basis for the exaggerated p65Δmye peritoneal MΦ response. We speculated that heightened U-LPS response by thioglycollate-elicited p65Δmye peritoneal MΦs could be attributed to: (1) differential TLR expression conferring differential signaling magnitude and cytokine levels; (2) heightened activation of the TLR4-MyD88-independent pathway through IRF3; or (3) counter-inflammatory signaling that may dominate the pro-inflammatory cytokine response such that an immunosuppressive state exists in which inflammatory effects may be inadequate.
To test these possibilities, we looked for evidence of altered TLR4 expression, TLR4–MyD88–IRF3 signaling, and IL-10 responses between WT and p65Δmye peritoneal MΦs. We demonstrate that mean fluorescence intensity (MFI) of TLR4 protein and Tlr2 and Tlr4 mRNA expression by peritoneal MΦs post-thioglycollate activation between WT and p65Δmye mice were comparable (Supplementary Figure S2, and results not shown). These data suggest that the increased pro-inflammatory cytokine response in p65Δmye mice cannot be explained by altered TLR4 expression by MΦs. To assess differences in the MyD88-independent signaling pathway, we examined Ccl12 and Cxcl10 mRNA fold induction in peritoneal MΦs following U-LPS stimulation (Figure 4). We show a ∼10-fold increase in Cxcl10 and ∼10–25-fold increase in Ccl12 mRNA following U-LPS stimulation of p65Δmye peritoneal MΦs, suggesting that MyD88-independent signaling pathway in p65Δmye MΦs remains intact (Figure 4). U-LPS-induced MΦ chemokine expression is predominantly driven by type I IFN, IFN-β, in an autocrine fashion. We show that steady-state level of IFN-β mRNA and protein was not different between WT and p65Δmye peritoneal MΦs (Figure 5c, and results not shown). Consistent with our observed increase in chemokine production, LPS-stimulation induced an increase in IFN-β production in both WT and p65Δmye peritoneal MΦs; however, the level of induction in p65Δmye peritoneal MΦs was greater than that observed in WT peritoneal MΦs (Figure 4c). Finally, we show increased IL-10 secretion in both WT and p65Δmye peritoneal MΦs after U-LPS stimulation and that the level of induction was equivalent between WT and p65Δmye MΦs (Figure 4). These data indicate that the elevated inflammatory state in p65Δmye mice is not due to decreased anti-inflammatory responses.
MyD88-independent signaling pathway is intact in p65Δmye peritoneal MΦs following in vitro U-LPS challenge. Fold change in (a) Cxcl10 and (b) Ccl12 gene expression and (c) IFN-β and (d) IL-10 secretion in peritoneal MΦs pretreated for 6 h with 0, 10 or 100 ng/ml U-LPS followed by 1 h 2mM ATP stimulation. Data represent the mean ± SEM of two independent experiments with n = 2–3 mice per group. Significant differences (*P < 0.05; **P < 0.01; ***P < 0.005) between groups. Western blot of C/EBP-β and PU.1 expression in thioglycollate-activated WT and p65Δmye purified MΦs. Representative Western blot of C/EBP-β and PU.1 expression in thioglycollate-activated, purified WT and p65Δmye MΦs.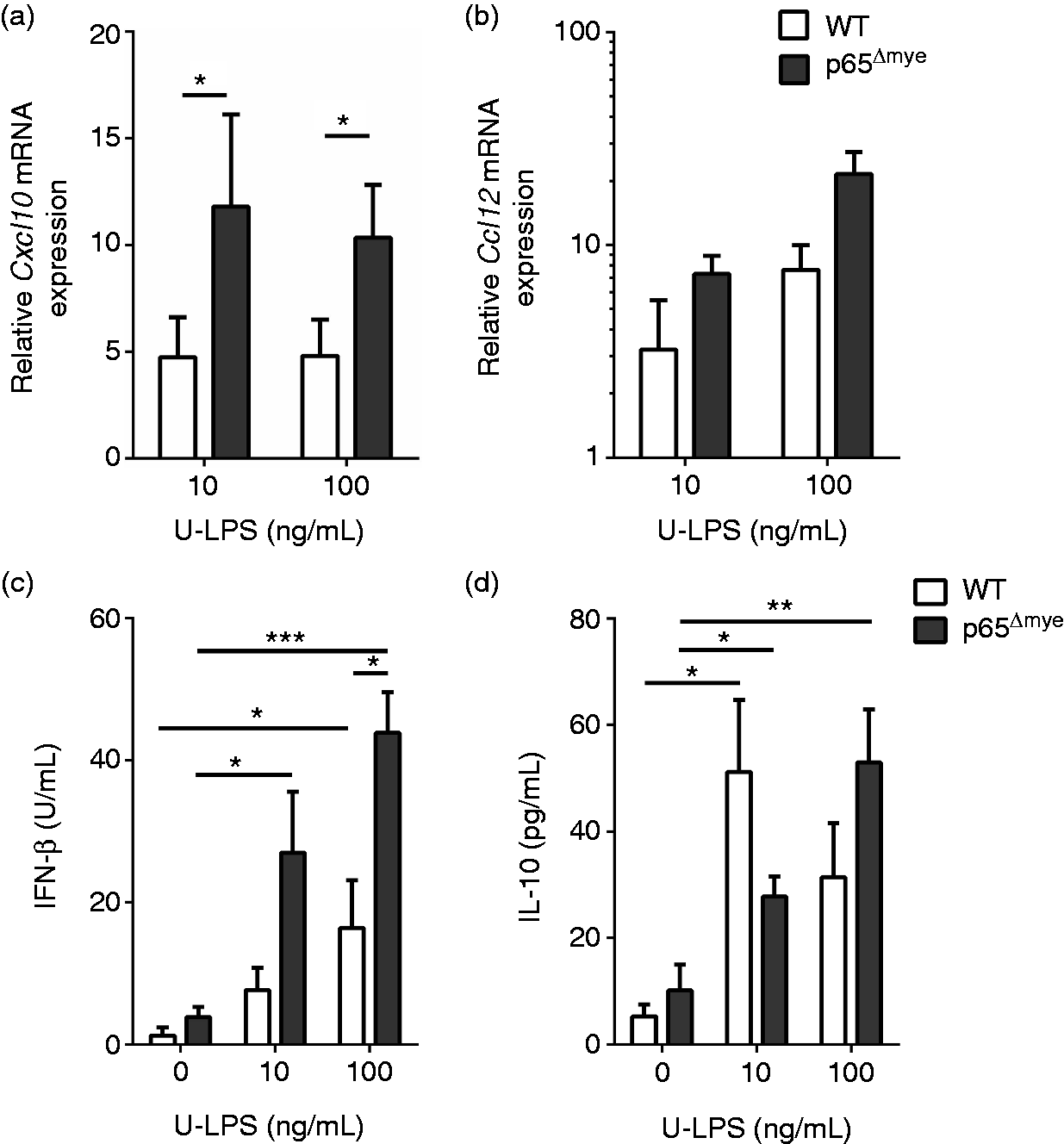
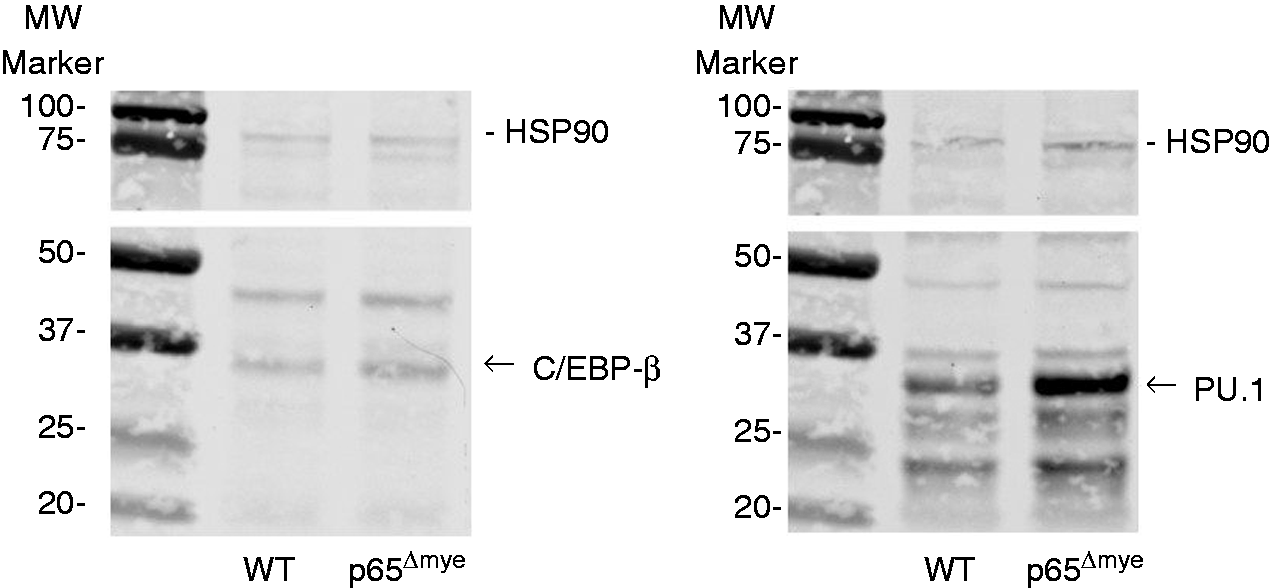
Binding of C/EBP-β and PU.1 to the 5' promoter region of Il1b and Il6 genes is increased in p65Δmye peritoneal MΦs
It was particularly striking to us that in the absence of stimulation, the levels of Il1b mRNA and pro-IL-1β were elevated in p65Δmye MΦs compared with WT MΦs (Figure 3). To confirm this observation, we purified thioglycollate-elicited WT and p65Δmye MΦs and examined pro-IL-1β levels by Western blot. Indeed, pro-IL-1β was significantly increased in unstimulated, purified, thioglycollate-elicited p65Δmye MΦs compared with WT cells (Figure 3c). Given the observed elevated levels of IL-1β mRNA and protein in the unstimulated p65Δmye peritoneal MΦs in the absence of altered steady-state cytokines such as IFN-β and inflammasome levels, we speculated that loss of p65 in the myeloid compartment led to altered transcriptional regulation of these cytokines. Inflammatory gene expression (Il1b and Il6) in activated myeloid cells is predominantly regulated by three transcription factors (NF-κB, C/EBP-β and PU.1). C/EBP-β directly binds to a motif in the Il6 promoter, whereas PU.1 is known to bind to the Il6 and Il1b promoters, with the latter promoter binding being via a protein–protein tether with C/EBP-β. 38
Binding of C/EBP-β and PU.1 on the 5' promoter region of Il1β and Il6 in WT and p65Δmye peritoneal MΦs.
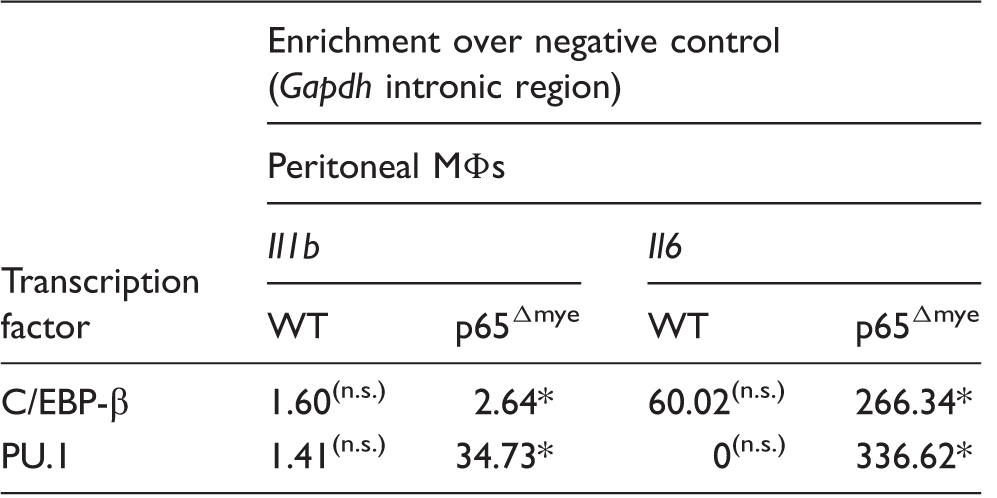
ChIP data reveal that there is a significantly increased binding of C/EBP-β and PU.1 on Il1b and Il6 promoters in p65Δmye peritoneal MΦs. The amount of immunoprecipitated DNA was analyzed by real-time PCR and then normalized over the respective input control. Data represent the fold increase of immunoprecipitated DNA for Il1b or Il6 over a negative control DNA fragment (Gapdh intronic region). *P < 0.05 vs. negative control; n = 2 for each group. n.s.: not significant.
Discussion
In the present study, we investigated whether the NF-κB RelA/p65 subunit in myeloid cells regulates LPS-induced shock responses. We show (1) that p65Δmye mice had increased susceptibility to LPS-induced cell death and that this susceptibility was associated with an enhanced inflammatory response, as evidenced by increased pro-inflammatory cytokines (IL-1β and IL-6); (2) that LPS stimulation of ‘activated’ (thioglycollate-elicited), but not bone marrow-derived, MΦs deficient in p65 led to increased secretion of IL-1β and IL-6; and (3) that p65-deficient MΦs possess increased steady-state C/EBP-β and PU.1 and Il1b mRNA and pro-IL-1β protein levels and that this was associated with heightened C/EBP-β and PU.1 binding to Il1b and Il6 promoters. These studies identify a previously undescribed transcription-based, anti-inflammatory mechanism whereby p65 regulates the transcription factors C/EBP-β and PU.1 binding to Il1b and Il6 promoters and myeloid pro-inflammatory cytokine production.
Though it may seem unexpected and counterintuitive, there is significant evidence supporting an anti-inflammatory role for NF-κB signaling in the myeloid cell compartment and promoting a protective response against LPS-induced shock. For example, the p50 homodimer, which lacks a transactivation domain, has been shown to repress expression of NF-κB target genes and inhibit inflammation in MΦs. 39 Previous studies have reported increased sensitivity of p50-deficient mice that are heterozygous for p65 (p50−/− p65+/−) to LPS-induced shock, suggesting anti-inflammatory roles of the p50 homodimer and p65:p50 heterodimers in septic shock. 40 Similarly, mice deficient in IKKβ in the myeloid cell compartment were more susceptible to LPS-induced shock. 41
Endotoxin is thought to activate the LPS receptor TLR4/MD2, which is largely expressed on myeloid cells, 4 leading to production of the pro-inflammatory cytokines (IL-1β, IL-6, TNF-α and type I IFN-β).5,6,8 These cytokines promote many of the immunopathologic features of LPS-induced shock.4,15 IL-1β and IL-6 have both been linked to cell death and septic shock. 42 IL-6 induces phosphorylation and redistribution of VE-cadherin, which leads to vascular leakage, 43 and is associated with increased mortality in patients with endotoxic shock. 44 IL-1β is thought to promote the shock-like state, including hypotension, neutropenia and thrombocytopenia, which, via indirect generation of cyclooxygenase products, stimulation of nitric oxide and PAF production and through TNF-α synergism.22,45 The importance of IL-1β in the LPS-induced shock is highlighted by the demonstration that neutralizing IL-1β activity with Anakinra [IL-1 receptor antagonist (IL-1Rα)] completely protected against LPS-induced mortality. 41 Furthermore, a randomized placebo-controlled open-label phase II study revealed a dose-dependent improvement in 28-d mortality in individuals who received the IL-1 inhibitor IL-1Rα infusion. 46 LPS-induced IFN-β is thought to be the primary driver of the LPS-induced shock. 47 IFN-β, through paracrine and autocrine loops, stimulates both MyD88-dependent and MyD88-independent pathways that augment production of critical chemokines and cytokines, such as IL-6, TNF-α, IL-12, IL-1β and NO. 48 IFN-β−/− and IFN-αR1−/− mice are resistant to lethal endotoxemia.49,50 We show that loss of p65 in the myeloid cell compartment does not affect ‘steady-state’ IFN-β production but increases U-LPS-induced IFN-β production. These studies suggest that p65 in myeloid cells is not involved in the regulation of basal IFN-β production but may affect type I IFN autocrine effects. Consistent with this argument, we observed increased chemokine production in p65Δmye peritoneal MΦs 24 h following U-LPS stimulation. Given the kinetics of the observed rapid rise in IL-1β and IL-6 in the p65Δmye myeloid cell compartment, the altered type I IFN autocrine loop is not likely to explain the observed elevation in these cytokines.
In the present study, we show that enhanced levels of pro-inflammatory cytokines appeared to be related to heightened C/EBP-β and PU.1 interactions with the Il1b and Il6 promoters in activated myeloid cells. Moreover, we observed increased binding of C/EBP-β and PU.1 with the Il1b and Il6 promoters and increased Il1b and Il6 mRNA in thioglycollate-elicited MΦs but not BMDM following p65 deletion. PU.1 is a winged helix-turn-helix (wHTH) factor of the ETS family of proteins and is essential for the myeloid cell development.51,52 PU.1 binds to purine-rich sequences (the PU box) on the promoter of the target genes and, in collaboration with other transcription factors (C/EBP family C/EBP-α and C/EBP-β), induces gene expression. 51 In particular, the Il1b promoter is transactivated by C/EBP-β through the transcription factor PU.1, which acts as a tether in order to allow C/EBP-β to bind the DNA. 38 Interestingly, a naturally occurring polymorphism in the human IL1B promoter has been identified, and this SNP alters PU.1 and C/EBP-β interactions and controls IL-1β production. 53
Recent studies defining PU.1 and C/EBP family member transcriptional regulation of cell fate decisions have identified an important contribution from antagonistic and cooperative relationships between transcription factors. 54 The underlying molecular processes involved in the antagonism–cooperative switch are not yet clear; however, the concentration and ratio of various transcription factors seems to be important. Studies in U937 and PUER cells have revealed that increased C/EBP-α levels are sufficient to modulate PU.1-induced myeloid cell differentiation.55,56 Similarly, G-CSF modulation of C/EBP-α and PU.1 ratio can modulate granulocyte differentiation. 55 Furthermore, PU.1 has been shown to modulate T-cell and natural killer cell gene expression in a dose-dependent manner. 57 Interestingly, we show that increased PU.1 interaction with the IL-1β and IL-6 promoters was associated with heightened total PU.1 levels in the p65Δmye MΦs, whereas the increased binding of C/EBP-β was not associated with increased levels of total C/EBP. Thus, we speculate that the dose-dependent increase in PU.1 drives Il1b expression in p65Δmye cells. One possible explanation for the increased PU.1 levels is altered expression of the microRNA miR-155. Moreover, miR-155 levels were significantly decreased in thioglycollate-elicited p65Δmye peritoneal MΦs compared with WT MΦs (miR-155/U6 ratio; 1.4 ± 0.9 vs. 0.5 ± 0.4, WT vs. p65Δmye peritoneal MΦs; mean ± SD; n = 3 per group; *P < 0.05). MiR-155 is a negative regulator of proteins involved in LPS signaling, including Fas-associated death domain protein, IκB kinase epsilon (IKKɛ) and the receptor (TNF-R superfamily)-interacting serine-threonine kinase 1 (Ripk1), and of PU.1 (Spi1) expression.58,59 Previous studies using an activated B-cell tumor cell line have revealed that NF-κB induces the up-regulation of miR-155, which leads to down-regulation of PU.1 protein levels and subsequently to reduced expression of PU.1-response genes. 59 Indeed, an NF-κB p50/p65-responsive site has been identified in the human MIR155HG promoter. 60
Previously, Greten et al. demonstrated enhanced IL-1β secretion and increased susceptibility to endotoxin-induced shock in the IkkbΔmye mice; however, the enhanced IL-1β production was attributed to altered NF-κB-mediated signaling pathways. 41 Moreover, the authors observed that loss of IKKβ and NF-κB signaling was associated with the loss in NF-κB-mediated negative regulation of caspase-1–dependent IL-1β processing and secretion. 41
We observed increased levels of pro-IL-1β mRNA and protein in p65Δmye inflammatory MΦs; however, the increased cytokine level was in the presence of reduced levels of inflammasome machinery. Importantly, the level of caspase-1 activity was sufficient to provide inflammasome activity to drive increased IL-1β cleavage and secretion. The observation of decreased levels of caspase-1 and increased IL-1β secretion in p65Δmye inflammatory MΦs further supports the concept of existence of additional pathways involved in altered IL-1β levels in myeloid cells following loss of IKKβ and NF-κB signaling independent of the negative regulation of inflammasome–dependent IL-1β processing and secretion. Our studies identified an alternative pathway whereby p65/NF-κB regulates IL-1β mRNA expression via transcriptional mechanism involving increased PU.1 and C/EBP-β interaction with the IL-1β and IL-6 promoters. There is emerging evidence of IKK-mediated signaling independent of NF-κB signaling, including regulation of MAPK pathways and TLR-induced IRF-5 signaling.61,62
One limitation of these analyses is that deletion of p65 in myeloid cells using the LysM-Cre system leads to deletion of p65 in common myeloid progenitor-derived cells, i.e. MΦs, neutrophils and DCs. Therefore, we cannot exclude the contribution of p65 signaling in neutrophils and DCs to the increased sera pro-inflammatory cytokines and mortality in vivo. Previous studies in LysMCreIKKβ mice revealed increased IL-1β release in both MΦs and neutrophils after endotoxin challenge. 41 However, MΦs were the major source of IL-1β under these conditions; IKKβ-deficient MΦs secrete 7–10-fold more IL-1β per cell after LPS stimulation than IKKβ-deficient neutrophils under the same conditions, and pharmacologic ablation of MΦs (Clondoranate) substantially decreases plasma IL-1β and improves endotoxic shock. 41 Consistent with this, we have demonstrated that IL-1β and IL-6 were elevated in serum and in p65Δmye peritoneal MΦs after U-LPS stimulation, indicating that MΦs and the cytokines that they produce are likely the factors contributing to endotoxic shock-induced death in p65Δmye mice.
In conclusion, our study shows that myeloid deletion of p65 aggravates LPS-induced shock via augmenting IL-1β and IL-6 production by LPS-stimulated MΦs via enhanced steady-state C/EBP-β and PU.1 interactions with the IL-1β and IL-6 promoters. Our findings highlight a previously undescribed role for p65 in the negative regulation of myeloid cell-derived pro-inflammatory cytokine production and LPS-induced shock and raise cautionary awareness with respect to the potential of employing NF-κB inhibitors for therapy of amplification of immune processes.
Footnotes
Acknowledgement
We thank Dr Joseph Qualls and members of the Division of Allergy and Immunology and Immunobiology, Cincinnati Children’s Hospital Medical Center, for critical review of the manuscript and insightful conversations. We would also like to thank Shawna Hottinger for editorial assistance and manuscript preparation.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH R01 AI 073553, AI 112626, DK 090119, Crohn s Colitis Foundation of America and Food Allergy Research Education Award (SPH). This project was also supported in part by PHS Grant P30 DK078392 of the Digestive Disease Research Core Center in Cincinnati.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.