
Abstract
Phosphatidylinositol 3-kinase (PI3K)/Akt signaling has been implicated in the anti-inflammatory response in a mouse model of endotoxemia and sepsis. The present study focused on the role of inositol polyphosphate-4-phosphatase type I (Inpp4a), which dephosphorylates PtdIns(3,4)P2 to PtdIns(3)P, in bacterial infections. We prepared myeloid cell-specific Inpp4a-conditional knockout mice. Macrophages from these mice showed increased Akt phosphorylation and reduced production of inflammatory cytokines in response to LPS or Escherichia coli in vitro. The Inpp4a knockout mice survived for a shorter time than wild type mice after i.p. infection with E. coli, with less production of inflammatory cytokines. Additionally, E. coli clearance from blood and lung was significantly impaired in the knockout mice. A likely mechanism is that the Inpp4a-catalyzed dephosphorylation of PtdIns(3,4)P2 down-regulates Akt pathways, which, in turn, increases the production of inflammatory mediators. This mechanism at least fits the decreased E. coli clearance and short survival in the Inpp4a knockout mice.
Introduction
Bacterial infection is a common cause of sepsis-associated organ failure and results in high morbidity and mortality rates worldwide.1,2 Despite the availability of antibiotics, the mortality rate from sepsis remains high.3,4 The inflammatory response during infection requires tight regulation to achieve effective pathogen removal without overwhelming inflammation. 5 Thus, it is important to understand the mechanisms that limit inflammation, maintain homeostasis and promote survival in the septic patients. Mouse models of sepsis have led to controversial results. Numerous studies suggest that a decreased production of inflammatory cytokines leads to ameliorating tissue injury and recovery rates from sepsis.5,6 Conversely, other studies indicate that an augmented inflammation response exacerbates the clearance of bacteria but improves host survival.7,8
Two types of inositol polyphosphate-4-phosphatases, type I (Inpp4a) and type II (Inpp4b), have been identified in mammals.
9
Inpp4a and Inpp4b use PtdIns(3,4)P2 as a preferential substrate and produce PtdIns(3)P as an end product.
9
Inpp4a has been found on early and recycling endosomes.
9
The RNAi-based knockdown of this isozyme reduces the endocytosis of transferrin in HeLa cells.
10
The role of Inpp4a as a positive regulator of clathrin-mediated endocytosis is supported by the reduced internalization of cell surface N-methyl-
The PI3K/Akt signaling pathway regulates the decreased production of pro-inflammatory and the increased production of anti-inflammatory cytokines.13,14 Previous reports using genetically modified mice and/or pharmacological reagents revealed that the PI3K/Akt pathway provides beneficial anti-inflammatory properties in a mouse model of endotoxemia and sepsis.15–18 In this context, myeloid-specific deletion of PTEN (phosphatase and tensin homolog deleted on chromosome 10), which antagonizes the PI3K/Akt pathway by hydrolyzing PtdIns(3,4,5)P3 to produce PtdIns(4,5)P2, was reported to augment phagocytosis and attenuate inflammatory cytokine production. 19 As a result, PTEN knockout bring mice fewer adverse events and longer survival in response to bacterial infections. 19 Inpp4a is predicted to antagonize the PI3K/Akt pathway through the dephosphorylation of PtdIns(3,4)P2 to produce PtdIns(3)P. However, the involvement of Inpp4a in innate immunity has not been established. Here, we investigated the role of Inpp4a during bacterial infection. We prepared myeloid-specific Inpp4a knockout mice because the homozygotes showed 100% mortality by the fourth week of age. The myeloid Inpp4 knockout mice lived normal; however, the myeloid Inpp4a-knockout became susceptible to E. coli loading into peritoneal cavity. We interpreted the results to mean that decreased inflammatory cytokine production resulted in the impaired bacterial clearance.
Materials and methods
Reagents
LPS (E. coli serotype 0111:B4) was purchased from Sigma (St. Louis, MO, USA). The rabbit m Ab against pAkt (Ser473) was purchased from Cell Signaling (Danvers, MA, USA). The m Abs for mouse Ly6G and F4/80 were purchased from Biolegend (San Diego, CA, USA) and labeled with fluorescein and HiLyte Fluor 647, respectively, using Labeling Kits (Dojindo, Kumamoto, Japan). The protein assay kit was purchased from Bio-Rad (Hercules, CA, USA). The ELISA kits were purchased from (PeproTech, Rocky Hill, NJ, USA).
Preparation of myeloid-specific Inpp4a knockout mice
The conditional targeting vector was constructed to delete a genomic fragment containing the 23rd coding exon of the mouse Inpp4a gene using homologous recombination (Figure 1). Exon 23 encodes the phosphatase domain essential for Inpp4a-mediated dephosphorylation of PtdIns(3,4)P2. Two loxP sequences were introduced into intron 22 and one sequence was introduced into intron 23. The LacZ-PGK-Neor cassette was inserted in an antisense orientation to Inpp4a transcription between the two lox sites in intron 22. The linearized construct was electroporated into 1 × 107 E14K mouse embryonic stem (ES) cells.
20
ES cell colonies resistant to G418 (0.3 mg/ml; Life Technologies, Carlsbad, CA, USA) were screened for homologous recombination by standard PCR. Correctly targeted ES cells were injected into C57BL/6J (CLEA Japan, Tokyo, Japan) blastocysts to generate chimeric mice. Chimeric male mice were crossed with C57BL/6J females, and offspring that successfully transmitted the mutation to the germ line were crossed with MeuCre40 transgenic mice to generate Inpp4aflox/+ mice.
21
An oligo primer (5′- AGGATAAGAGGATTGAGCACATCG-3′) common to the Inpp4a+ and Inpp4aflox alleles, a primer specific for the Inpp4a+ allele (5′- CCAAACTCCAGGGAAGGAACTG-3′) and a primer specific for the Inpp4aflox allele (5′-TATTGACAAGGAGGAGGGCACCAC-3′) were used to detect the 422-bp and 546-bp product for the Inpp4a+ and Inpp4aflox alleles, respectively. Methods to produce Inpp4aneo/+ were previously described.
11
To delete Inpp4a in macrophages, we crossed Inpp4aneo/+ mice with CD11bCre Tg mice that express Cre recombinase under the control of the CD11b promoter.
22
The resulting CD11bCreInpp4aneo/+ mice were crossed with Inpp4aflox/flox mice to generate CD11bCreInpp4aneo/flox mice. All experimental protocols were reviewed and approved by the Akita University Institutional Committee for Animal Studies (a-1-2611).
Conditional disruption of the mouse Inpp4a gene. (a) The genomic structure of the wild type mouse Inpp4a gene surrounding exon 23, which encodes the phosphatase domain. (b) The targeting vector consisted of the Neor gene flanked by two loxP sequences (black arrowheads) and a third loxP sequence flanking exon 23. (c) The recombined allele contained three loxP sequences and the Neor gene. (d) The floxed allele resulting from the Cre-mediated deletion of Neor. (e) The deleted allele resulting from the Cre-mediated deletion of exon 23.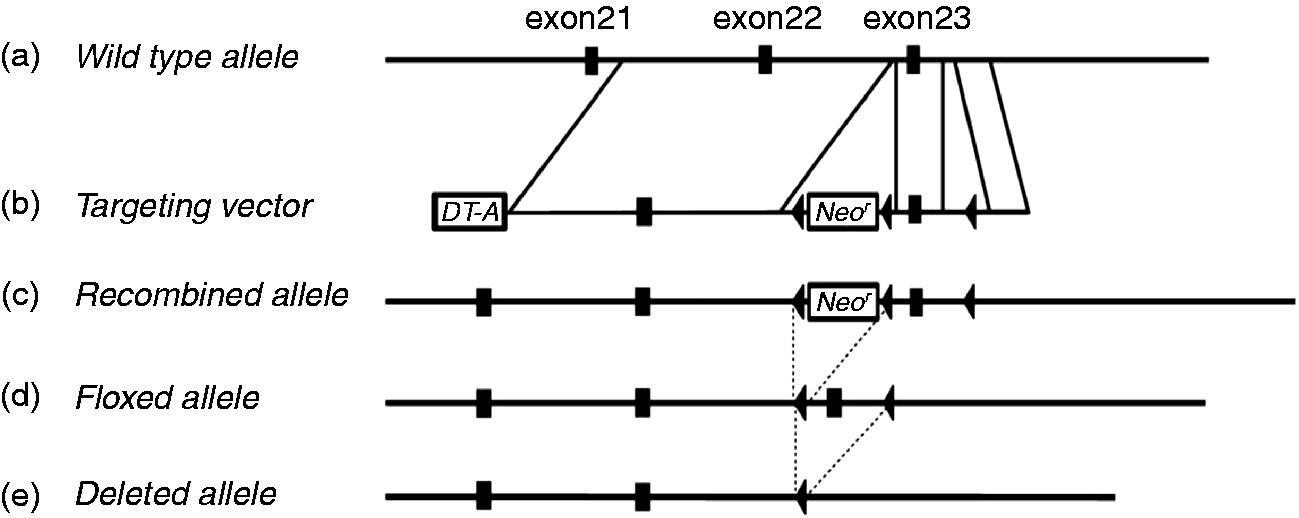
Cell preparation and culture
Peritoneal macrophages were harvested from mice. Briefly, mice were injected i.p. with 2–3 ml 3% thioglycollate broth. After 3 d, the peritoneal exudate cells were harvested by washing the peritoneal cavity with ice-cold PBS. The cells were seeded at approximately 1 × 106 cells/well in 12-well plates in RPMI 1640 medium supplemented with 10% FCS and incubated in humidified 5% CO2 at 37℃ for 1–2 h. Non-adherent cells were washed with PBS, and the attached cells were designated as macrophages. The cells were incubated with 0.1 µM LPS or 108 CFU/ml E. coli for 6 h. The culture supernatant was harvested to determine cytokine production.
Western blotting
Cells in the 12-well plates were washed with PBS and lysed in 100 µl lysis buffer containing 25 mM Tris-HCl (pH 7.4), 0.5% Nonidet P-40, 150 mM NaCl, 1 mM sodium orthovanadate (Na3VO4), 1 mM EDTA, 0.1% BSA, 20 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, 2 µM leupeptin, 20 µM p-amidinophenylmethylsulfonyl fluoride and 1 mM dithiothreitol. The cell lysates were centrifuged at 20,000 g for 10 min. The supernatants were collected, and the protein concentration was determined using the Bio-Rad assay kit. Total cell lysates (100 µg protein) were mixed with 10 µl of 5× sample buffer [62.5 mM Tris (pH 6.8), 1% SDS, 10% glycerol, 5% 2-mercaptoethanol and 0.02% bromophenol blue] and heated at 100℃ for 5 min. The proteins were separated by SDS-PAGE and transferred electrophoretically onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). The membrane was blocked with 5% skimmed milk and incubated with the appropriate Abs. Ab binding was detected using a chemiluminescent substrate (Perkin-Elmer, Waltham, MA, USA).
ELISA
Cytokine levels were determined using ELISAs, according to the manufacturer's instructions.
Mouse model of E. coli peritonitis
Mice were injected i.p. with 1 × 109 or 2 × 108 CFU E. coli (DH5α) and killed 6 h or 16 h post-injection to collect peritoneal lavage fluid (PLF) or blood and lung, respectively. PLF supernatants were collected for the ELISA cytokine measurements. The pelleted cells were stained with fluorescently labeled anti-Ly6G and anti-F4/80 to determine the numbers of neutrophils and monocytes by flow cytometry using the guava easyCyte system (Merck Millipore, Darmstadt, Germany). CFU counts of bacteria were determined by plating serial dilutions of PLF, blood or lung homogenates on LB agar plates. Plates were incubated at 37℃ for 16–20 h, and the colonies were counted the following day. For the survival study, mice were treated with 1 × 109 CFU E. coli, and their health status was assessed every 3 h.
Statistical analysis
All data are expressed as the mean ± SEM. The survival data were plotted as Kaplan–Meier survival curves and analyzed using the log-rank test.
Results
Increased Akt activity in macrophages from myeloid-specific Inpp4a knockout mice (Inpp4aCD11b/KO)
Inpp4a was almost completely abolished in peritoneal macrophages from Inpp4aCD11b/KO mice (Figure 2a). The basal level of Akt phosphorylation was increased in Inpp4a-deficient cells (Figure 2a, b). Akt phosphorylation after exposure to LPS or E. coli was also augmented in these cells (Figure 2a, b).
Increased Akt phosphorylation in macrophages from Inpp4aCD11b/KO mice. Thioglycollate broth-elicited macrophages from wild type (WT) and Inpp4aCD11b/KO (KO) mice were stimulated with 0.1 µg/ml LPS or 108 E. coli for 6 h. (a) The total cell lysates were analyzed by Western blotting using specific Abs. (b) The phospho-Akt (pAkt) blots from (a) were analyzed using NIH ImageJ and normalized to the density of p85 regulatory subunit of class I PI3K (p85). The results are expressed relative to the control value of LPS-stimulated WT cells. The combined results from three separate experiments are shown as the mean ± SEM. *P < 0.05.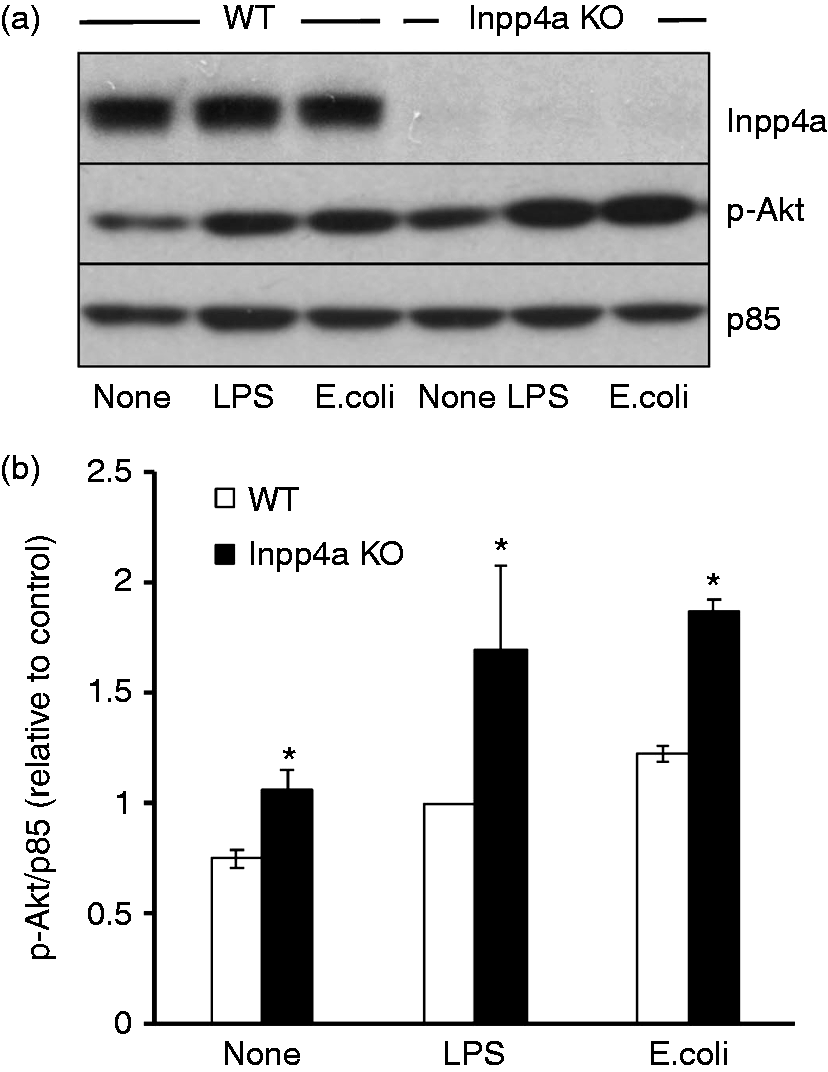
Decreased inflammatory cytokine production in macrophages deficient in Inpp4a in vitro
The production of IL-12/IL23 p40 and TNF-α in response to TLR4 stimulation with LPS or E. coli was decreased in peritoneal macrophages from Inpp4aCD11b/KO mice (Figure 3a, b). These results are in agreement with previous reports suggesting that the PI3K/Akt signaling pathway down-regulates the TLR-mediated production of pro-inflammatory cytokines.23,24
Decreased pro-inflammatory cytokine production in macrophages from Inpp4aCD11b/KO mice in vitro. Thioglycollate broth-elicited macrophages from wild type (WT) and Inpp4aCD11b/KO (KO) mice were stimulated with 0.1 µg/ml LPS or 108 E. coli/106 macrophages for 6 h. The cytokine concentration in the supernatants was determined by ELISA. The results are expressed as relative to the control value of LPS-stimulated WT cells. Data are presented as the mean ± SEM of five mice/group. *P < 0.05.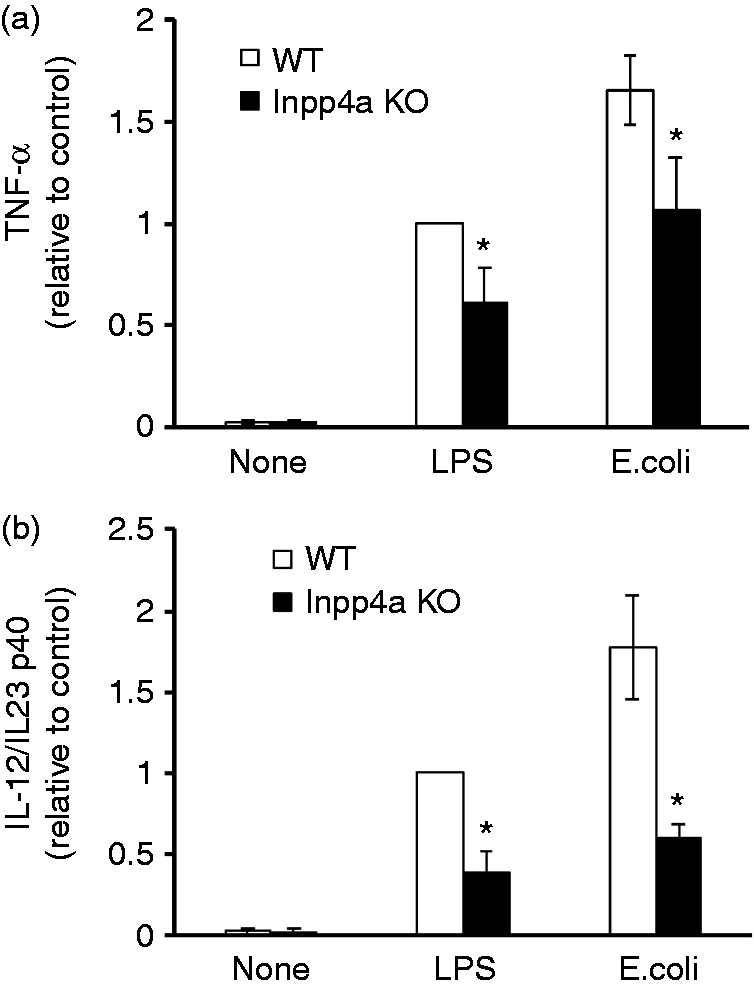
Impaired survival of Inpp4aCD11b/KO mice after E. coli infection
Excessive inflammation is a major cause of high mortality from sepsis.
6
However, some evidence suggests that the decreased production of pro-inflammatory cytokines increases mortality due to impaired bacterial clearance.7,8 Wild type and Inpp4aCD11b/KO mice were given an i.p. injection of 1 × 109 CFU E. coli (DH5α), and their survival was monitored. The knockout mice showed a significantly decreased survival rate after the E. coli injection (Figure 4).
Impaired survival of Inpp4aCD11b/KO mice after an i.p. injection of E. coli. Inpp4aCD11b/KO and wild type mice were injected with DH5α (1 × 109 CFU) and monitored over 60 h. Survival data were analyzed by a log-rank test to compare Kaplan–Meier survival curves. The P-value is depicted in the graph. n = 7 mice/group.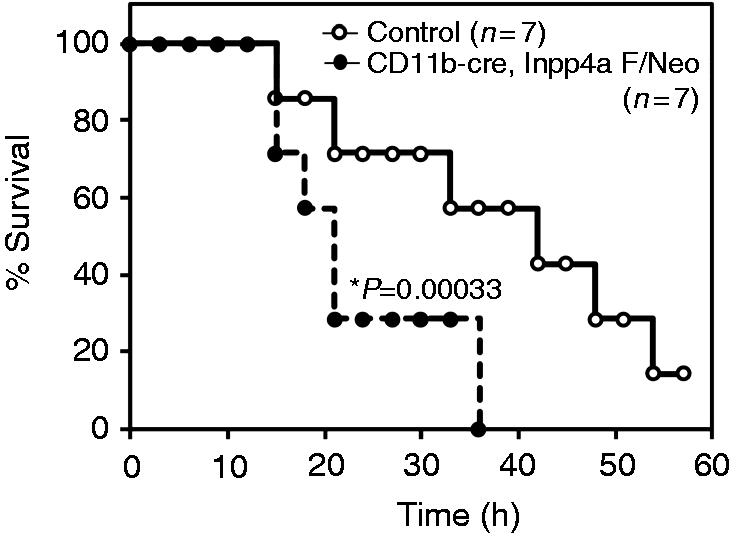
Impaired E. coli clearance from the blood and lung of Inpp4aCD11b/KO mice
E. coli in the blood was significantly increased in myeloid-specific Inpp4a-deficient mice at 6 h after i.p. injection (Figure 5a). The level of E. coli in lung homogenates was unchanged 6 h after the injection (data not shown) but was significantly increased after 16 h (Figure 5b). The majority of the knockout mice died 12–20 h after the injection (Figure 4); thus, an increase in lung bacteria and resultant pneumonia may have been the cause of death. We hypothesize that the decreased production of pro-inflammatory cytokines by macrophages was responsible for the decreased clearance of E. coli in Inpp4aCD11b/KO mice (Figure 3). Interestingly, E. coli clearance from the peritoneal fluid showed a non-significant trend toward a decrease in the Inpp4a knockout mice (Supplementary Figure 2).
Impaired clearance of E. coli from the blood and lung of Inpp4aCD11b/KO mice. Inpp4aCD11b/KO and wild type (WT) mice were injected with DH5α (1 × 109 CFU). Bacterial CFU counts were determined in (a) blood and (b) lung homogenates. Data are presented as the mean ± SEM of five mice/group. *P < 0.05; **p>0.01. KO: knockout.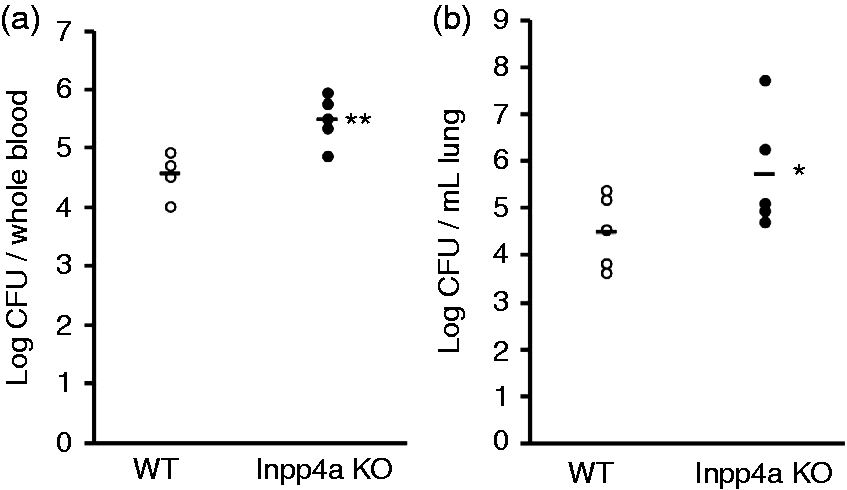
Decreased cytokine accumulation in the peritoneal fluid of Inpp4aCD11b/KO mice after an i.p. injection of E. coli
The peritoneal fluids were harvested 6 h after the injection of E. coli, and the levels of cytokines were determined. The accumulation of IL-1β, IL-6, IL-12/IL-23 p40 and TNF-α were decreased in Inpp4aCD11b/KO mice (Figure 6). The CC-chemokine MCP-1 was also decreased (Figure 6).
Decreased cytokine accumulation in the peritoneal fluid of Inpp4aCD11b/KO mice after an i.p. injection of E. coli. Inpp4aCD11b/KO and wild type (WT) mice were injected with DH5α (1 × 109 CFU). The peritoneal exudates were harvested with PBS in a total volume of 2 ml. Cytokine levels were determined by ELISA. Data are presented as the mean ± SEM of five mice/group. *P < 0.05. KO: knockout.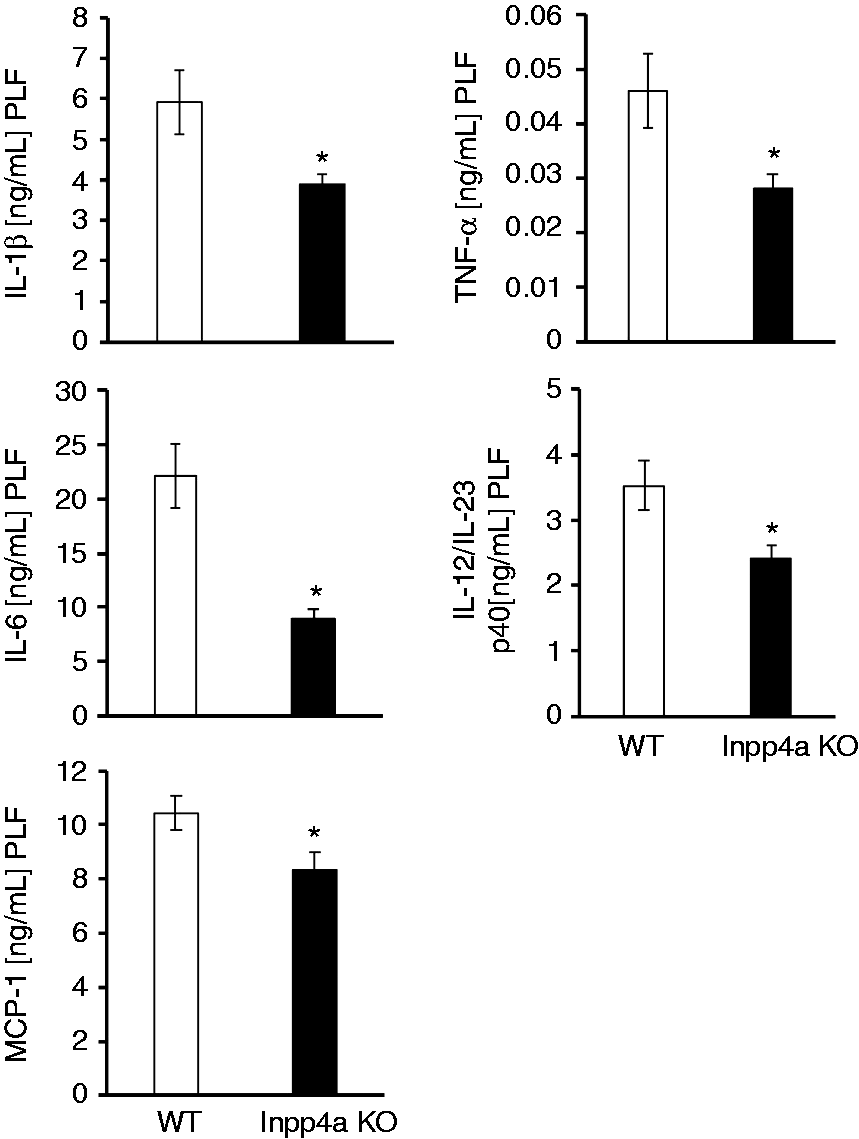
Decreased neutrophil influx into peritoneal cavity of Inpp4aCD11b/KO mice after E. coli injection
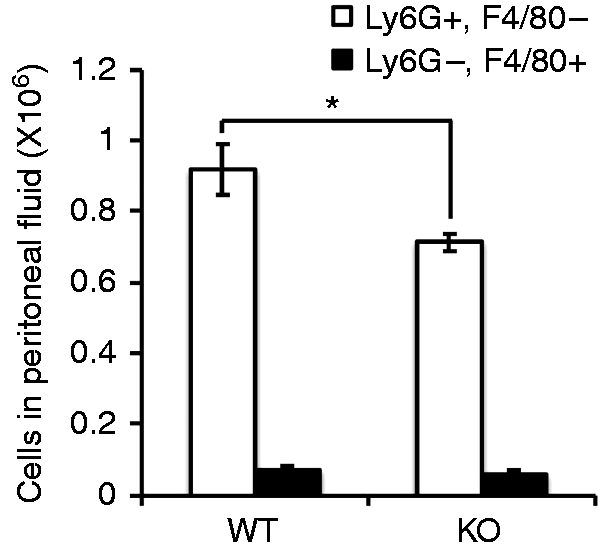
The numbers of neutrophils and monocytes as determined by Ly6G and F4/80, respectively, were unchanged in the blood and bone marrow of Inpp4aCD11b/KO mice (Figure 7). However, the neutrophil influx into the peritoneal cavity after E. coli injection was decreased (Figure 8). MCP-1 regulates neutrophil recruitment after E. coli infection.25,26 Thus, we can speculate that the dampened influx of neutrophils in the Inpp4a knockout mice (Figure 8, Supplementary Figure 3) is caused by the decreased production of MCP-1 and pro-inflammatory cytokines (Figure 6). Conceivably, this effect partly explains the impaired clearance of E. coli in myeloid-specific Inpp4a-deficient mice.
The number of neutrophils and monocytes in the blood and bone marrow of wild type (WT) and Inpp4aCD11b/KO mice. Blood and bone marrow cells were obtained from untreated mice and stained with a fluorescein-labeled anti-Ly6G Ab and a HiLyte Fluor 647-labeled anti-F4/80 Ab. The numbers of neutrophils (Ly6Ghigh, F4/80low) and monocytes (Ly6Glow, F4/80high) were determined by flow cytometry. Data are presented as the mean ± SEM of three mice/group. KO: knockout. Decreased neutrophil influx into the peritoneal cavity of Inpp4aCD11b/KO mice after E. coli injection. The peritoneal lavage fluids were collected 6 h after infection. The cells in the lavage fluids were stained with a fluorescein-labeled anti-Ly6G Ab and a HiLyte Fluor 647-labeled anti-F4/80 Ab. The numbers of neutrophils and monocytes were determined by flow cytometry. Data are presented as the mean ± SEM of four mice/group. *P < 0.05. WT: wild type; KO: knockout.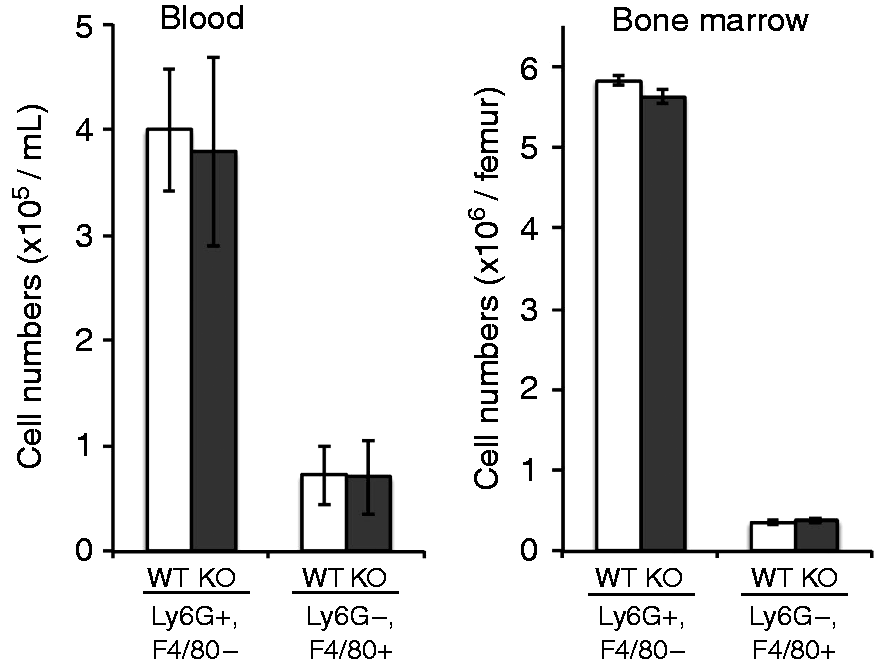
Discussion
The role of PI3K/Akt in innate immunity is controversial. We, and others, have previously shown that TLR-mediated inflammatory responses in macrophages are negatively regulated by the PI3K pathway.15,26,27 Similar results were reported in dendritic cells and human PBMCs.23,24 In contrast, the PI3K pathway was reported to regulate positively the TLR-mediated activation of NF-kB and the production of inflammatory cytokines in neutrophils and TLR-expressing 293T cells.28,29 The process of phagocytosis, another important innate immune response, is also dependent on PI3K activity.30,31 The activation of the PI3K/Akt pathway through down-regulation of PtdIns(3,4,5)P3 phosphatases, such as SHIPs or PTEN, augments the phagocytic activity of macrophages. 32 Furthermore, myeloid-specific PTEN knockout mice showed improved survival after murine pneumococcal pneumonia as a result of enhanced phagocytosis and the decreased production of inflammatory cytokines. 19
In the present study, we observed that macrophages from Inpp4aCD11b/KO mice showed increased Akt activation upon stimulation with LPS or E. coli (Figure 2). In agreement with previous publications, 13 this augmented Akt activity was accompanied by a decrease in the production of inflammatory cytokines (Figure 3). This in vitro result partly explains the decreased accumulation of inflammatory cytokines in the peritoneal fluid of the Inpp4aCD11b/KO mice after an i.p. injection of E. coli (Figure 6). In these mice, the neutrophil influx into the peritoneal cavity was decreased (Figure 8, Supplementary Figure 3), probably owing to the decreased accumulation of MCP-1 and inflammatory cytokines (Figure 6). Thus, Inpp4a plays a role in the bacteria clearance through the negative regulation of the Akt/PI3K pathway.
The phagocytosis of pathogens by macrophages is one of the most important initial responses against bacterial invasion. 33 Defects in phagocytic function lead to impaired survival in septic patients. 34 In a mouse model of sepsis, TREM-2–/– mice showed early augmented inflammation, accelerated resolution and improved survival against an LPS challenge. 35 However, upon infection with E. coli, the beneficial phenotype of these mice is counteracted by reduced phagocytosis. 35 Different phagocytic activities may explain why Inpp4a knockout mice but not PTEN knockout mice exhibit an impaired survival rate from sepsis. Phagocytic activity against E. coli is reportedly increased in macrophages from PTEN knockout mice, 19 while the activity was not changed in macrophages from Inpp4a knockout mice (Supplementary Figure 1). We have observed that Inpp4a-deficient Raw264.7 macrophages engulf less E. coli than wild type macrophages (data not shown). In contrast, these cells engulf IgG-coated erythrocytes far better than wild type cells, 12 suggesting that the role of Inpp4a in endocytosis depends on the target species. The enhanced phagocytosis of IgG-coated erythrocytes in Inpp4a-deficient cells is accompanied by the increased accumulation of PtdIns(3,4,5)P3 and PtdIns(3,4)P2 around the nascent phagocytic cup. 12 The excessive PtdIns(3,4)P2 accumulation around the endosome was reported to delay endosome maturation. 36 Thus, phagosome maturation and the degradation of E. coli may be delayed in the macrophages from Inpp4aCD11b/KO mice. In agreement with this speculation, phagosome acidification at an early time point (10 min) was significantly lower in Inpp4a-deficient macrophages (Supplementary Figure 4). Further examination of this hypothesis is required.
Ticam1 (TRIF)–/– neonates are highly susceptible to Gram-negative sepsis. 7 The dampened clearance of E. coli results in high mortality in these mice. The concentrations of inflammatory cytokines in the peritoneal exudate of these mice are significantly lower compared with wild type mice 6–12 h after E. coli injection. However, 24 h after administration, the levels of inflammatory cytokines are higher than with the wild type, owing to the poor clearance of E. coli. 7 In the present study, we observed that inflammatory cytokines in the peritoneal fluid of Inpp4aCD11b/KO mice were significantly lower 6 h post-infection (Figure 6). The concentrations were higher 16 h after administration, although the difference was not statistically significant (Supplementary Figure 5). This fluctuation is similar to the data obtained from Ticam1–/– mice and may partly explain the impaired survival of Inpp4aCD11b/KO mice. As Akt/PI3K negatively regulates TICAM-1/TRIF-mediated production of inflammatory cytokines, 37 it is very likely that TICAM-1/TRIF-mediated activation of inflammasome, 38 which is important early control of bacterial infection, is also attenuated in Inpp4aCD11b/KO mice. In summary, the present study shows the role of Inpp4a in the homeostatic regulation of pro- and anti-inflammatory responses during sepsis. These results confirm the critical role of phosphoinositide metabolism in innate immunity in vivo.
Supplemental Material
Supplemental material for Myeloid cell-specific inositol polyphosphate-4-phosphatase type I knockout mice impair bacteria clearance in a murine peritonitis model
Supplemental Material for Myeloid cell-specific inositol polyphosphate-4-phosphatase type I knockout mice impair bacteria clearance in a murine peritonitis model by Shin Morioka, Kiyomi Nigorikawa, Junko Sasaki, Kaoru Hazeki, Yoshihiro Kasuu, Takehiko Sasaki and Osamu Hazeki in Innate Immunity
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a Grant-in-Aid (Kakenhi) (grant numbers 23590078 to O Hazeki) from the Japan Society for the Promotion of Science.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.