
Abstract
The relative abundance of phosphoinositide (PI) species on the phagosome membrane fluctuates over the course of phagocytosis. PtdIns(3,4,5)P3 and PtdIns(3,4)P2 rapidly increase in the forming of the phagocytic cup, following which they disappear after sealing of the cup. In the present study, we monitored the clearance of these PI species using the enhanced green fluorescent protein-fused pleckstrin homology domain of Akt, a fluorescence probe that binds both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 in Raw 264.7 macrophages. The clearance of PIs was much faster when the phagocytosed particles were coated with IgG. The effect of IgG was not observed in the macrophages deficient in FcγRIIb, an inhibitory IgG receptor. To identify the lipid phosphatases responsible for the FcγRIIb-accelerated PI clearance, we prepared a panel of lipid phosphatase-deficient cells. The lack of a PI 5-phosphatase Src homology 2 domain-containing inositol-5-phosphatase (SHIP)1 or SHIP2 impaired the FcγRIIb-accelerated clearance of PIs. The lack of a PI 4-phosphatase Inpp4a also impaired the accelerated PIs clearance. In the FcγRIIb- and Inpp4a-deficient cells, acidification of the formed phagosome was slowed. These results suggested that FcγRIIb drives the sequential dephosphorylation system comprising SHIPs and Inpp4a, and accelerates phagosome acidification.
Introduction
Phagocytosis, an important process in innate and adaptive immune systems, refers to the uptake of large particles, including invading microorganisms and apoptotic ‘self’ cells, by specialized phagocytes. The process of phagocytosis involves specific cell-surface receptors. One group of receptors, including Man-Fuc receptors, dectin1 and TLR, interact directly with pathogens by recognizing conserved motifs on the microorganisms. Another set of receptors recognizes opsonins, i.e. serum components (e.g. Abs and complement components) that interact with the surface molecules of foreign particles. The clustering of opsonin receptors on phagocytes induces the extension of the membrane around the particle, resulting in the formation of a phagocytic cup. The newly formed phagosome then undergoes maturation, which is a series of fusion and fission events that culminates in the formation of the phagolysosome, the next step for microbial elimination. The maturation is also indispensable for the cross-presentation of exogenous ags in macrophages.
Phosphoinositide (PI) species play inevitable roles in the formation of phagosomes. 1 PtdIns(4,5)P2 increases locally in the phagocytic cup and promotes de novo actin polymerization to initiate particle ingestion.2,3 During the formation of the phagocytic cup, a transient and dramatic increase in PtdIns(3,4,5)P3 occurs. PtdIns(3,4,5)P3 recruits a pleckstrin homology (PH)-domain-containing effector myosin X, which drives pseudopod extension. 4 An additional role of PtdIns(3,4,5)P3 is the inactivation of Rac/Cdc42, which is necessary for the phagosome closure. 5 The sole pathway that produces PtdIns(3,4,5)P3 is the phosphorylation of PtdIns(4,5)P2 by class-I PI 3-kinases. 6 Thus, the phagocytic activity of murine macrophages is decreased by targeted deletion of the p85 adapter subunit of PI 3-kinases. 7 In Raw 264.7 macrophages, specific knockdown of the p110α catalytic subunit of PI 3-kinases impairs their phagocytic activity. 8 PtdIns(3,4,5)P3 on the phagocytic cup disappears immediately after sealing. Although the enzymes responsible for this elimination have not yet been specified, several PI phosphatases have been implicated in the phagocytic process. Cells lacking phosphatase and tensin homologue deleted on chromosome 10 (PTEN) that hydrolyzes PtdIns(3,4,5)P3, thereby producing PtdIns(4,5)P2, exhibit increased phagocytic activity.9,10 Src homology 2 (SH2) domain-containing inositol phosphatase (SHIP) 1 and SHIP2 (that hydrolyze PtdIns(3,4,5)P3 producing PtdIns(3,4)P2 are recruited to the phagocytic cup and inhibit phagocytosis.11–13 Furthermore, oculocerebrorenal syndrome of Lowe (OCRL), Inpp5b and Inpp5e, all of which possess in vitro 5-phosphatase activity toward PtdIns(3,4,5)P3,14,15 are reported to bind nascent phagosomes.13,16,17
In the present study, we examined the cellular localization of a fluorescent-tagged PH domain of Akt (Akt-PH) that binds both PtdIns(3,4,5)P3 and PtdIns(3,4)P2. Akt-PH accumulated on the phagocytic cup and disappeared rapidly after the formation of the phagosome. The disappearance was accelerated when the engulfed particles were opsonized with IgG. This effect of IgG was inhibited in the cells deficient in SHIP1 or SHIP2, but was unchanged in the cells deficient in OCRL, Inpp5b or Inpp5e. A similar inhibition of the IgG effect was observed in the cells deficient in a PtdIns(3,4)P2 4-phosphatase, Inpp4a or an inhibitory IgG receptor, FcγRIIb. Intriguingly, acidification of the phagosomes that contain IgG-opsonized particles was slowed in the cells deficient in Inpp4a or FcγRIIb. These results suggested that FcγRIIb drives the sequential dephosphorylation system comprising SHIPs and Inpp4a, and facilitates phagosome maturation by metabolizing PtdIns(3,4,5)P3 through PtdIns(3,4)P2 to PtdIns(3)P on the phagosome membrane.
Materials and methods
Materials
The sources of materials were as follows: zymosan, (FITC)-zymosan, TexRed-zymosan, anti-zymosan IgG and RPMI 1640 medium (Life Technologies, Carlsbad, CA, USA); protein assay kit (Bio-Rad, Foster City, CA, USA); anti-Inpp4a (#sc-12314) and anti-SHIP2 (sc-14502) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-PTEN (#9552) and anti-pAkt (#4058) (Cell Signaling Technology, Danvers, MA, USA); anti-SHIP1 (clone 32) (BD Transduction, San Jose, CA, USA).
Cells
Target sequence for shRNA.

Primer pairs for RT-PCR.

Opsonization of zymosan
Zymosan particles were sonicated for approximately 1 min. The particles were then opsonized with anti-zymosan IgG at 37°C for 60 min before use. The buffer used for the opsonization was EDTA-gelatin veronal buffer (GVB). The opsonized zymosan was washed thrice with GVB and finally suspended in incubation buffer. The zymosan preparation was sonicated for approximately 10 s immediately before use.
Monitoring PI dynamics along the course of phagocytosis
EGFP-Akt-PH was transfected with the Neon™ transfection system (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Approximately 24 h after the transfection, the cells were added with zymosan particles and placed on a BIOREVO BZ9000 microscope (Keyence, Osaka, Japan) equipped with a CFI Plan Apo VC60xH oil immersion lens, and phagocytosis was allowed to proceed at 37°C. The fluorescent images were collected every 1 min, and the intensity of the phagosome-associated fluorescence was analyzed using a BZ-II analysis system (Keyence).
Analysis of phagosome acidification
FITC-labeled zymosan or Texas Red-labeled zymosan was opsonized with IgG. Cells were added with the mixture of the fluorescent zymosan and placed on the microscope. Phagocytosis was allowed to proceed at 37°C. The fluorescent images were collected every 5 min, and the fluorescence intensity of FITC or Texas Red was analyzed.
Results
The PH domain of Akt (Akt-PH) binds both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 with comparable binding affinity.
20
In the experiment shown in Figure 1, Raw264.7 cells were transfected with the enhanced green fluorescent protein (EGFP)-fused Akt-PH, and the distribution of the fluorescence was monitored. When the cells were challenged with zymosan particles, the fluorescence appeared on the forming phagocytic cup (0–2 min), peaked after the cup closure (3–4 min), and then faded (Figure 1a, c). When the zymosan particles were coated with IgG (IgG-zymosan), the fluorescence was similarly detected on the cup, but disappeared rapidly after the cup closure (Figure 1b, c).
Accelerated clearance of PtdIns(3,4,5)P3/PtdIns(3,4)P2 from the phagosome membrane. Raw264.7 cells were transfected with EGFP-fused Akt-PH. (a, b) The cells were challenged with (a) zymosan or (b) IgG-coated zymosan, and the fluorescence was monitored every 1 min. (c) The intensity ratio of phagosome-associated fluorescence to total cellular fluorescence was determined. The combined results from three independent experiments (each contains three phagosomes) are shown as the means ± SEM.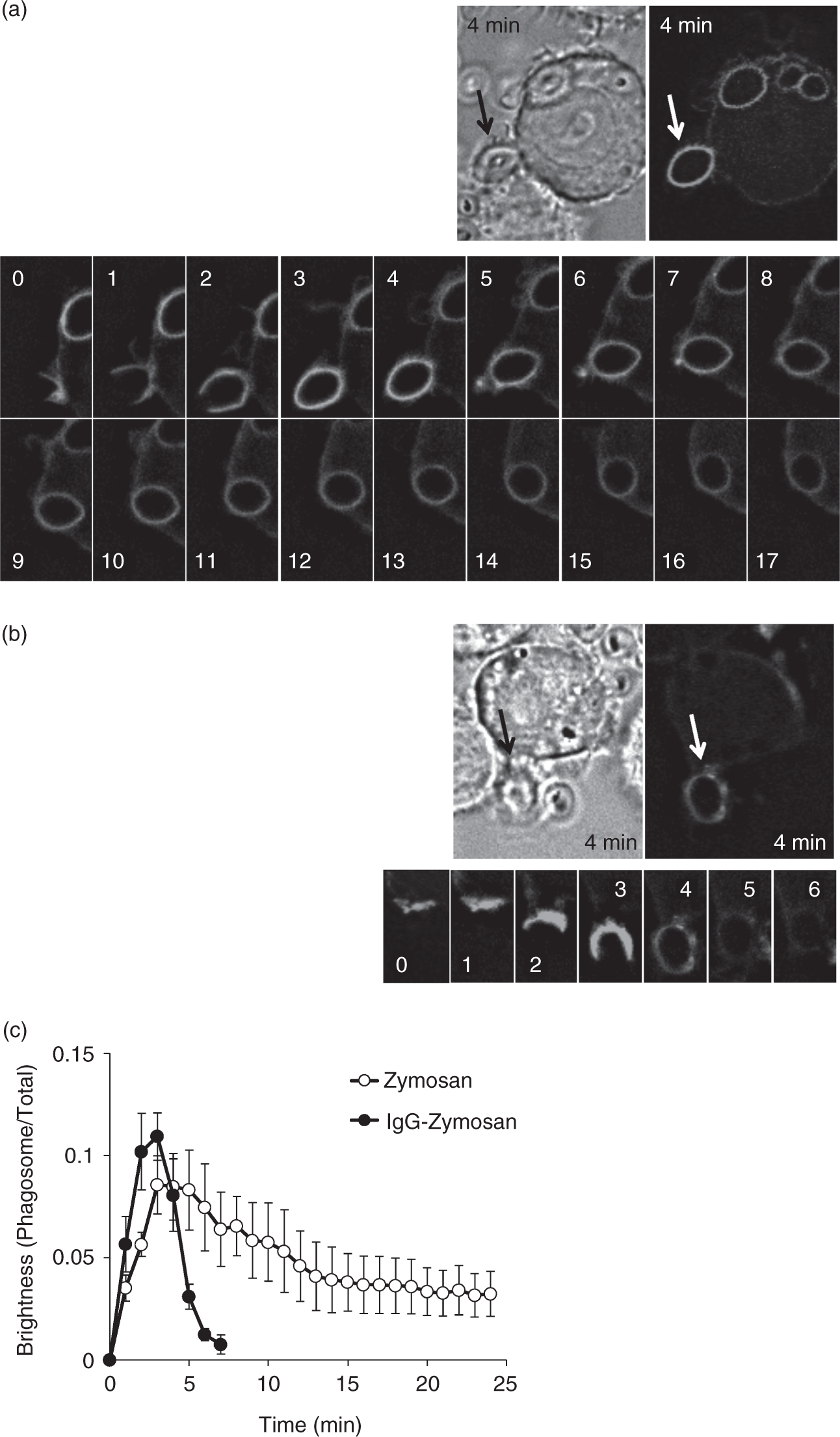
As shown in Figure 2, a panel of PI phosphatase-deficient Raw264.7 cells was prepared by use of specific short hairpin RNAs (shRNAs) (Figure S1) and examined for the change in the IgG-accelerated disappearance of the fluorescence. The lack of a PI 3-phosphatase PTEN abrogated the rate of fluorescence decay markedly (Figure 2a). The result indicated that a substantial amount of phagosomal PtdIns(3,4,5)P3 is hydrolyzed to PtdIns(4,5)P2. The lack of either one of the type-III PI 5-phosphatases, SHIP1 (Figure 2b) and SHIP2 (Figure 2c) also slowed the fluorescence decay. In contrast, the lack of Inpp5e (Figure 2e), Inpp5b (Figure 2f) or OCRL (Figure 2g), all of which possess the ability to hydrolyze PtdIns(3,4,5)P3 and are reported to bind nascent phagosomes,13–17 had little effect on the decay. Because SHIPs hydrolyze PtdIns(3,4,5)P3 but produce PtdIns(3,4)P2, the IgG-accelerated dissociation of Akt-PH [disappearance of both PtdIns(3,4,5)P3 and PtdIns(3,4)P2] suggests that PtdIns(3,4)P2 is further metabolized and effectively eliminated. The lack of Inpp4a, a PI 4-phosphatase that preferentially uses PtdIns(3,4)P2,
21
impaired the IgG-accelerated dissociation of Akt-PH (Figure 2d). Inpp4a is considered to be the sole PI 4-phosphatase that metabolizes PtdIns(3,4)P2 in Raw264.7 cells because we could not detect the mRNA expression of its isozyme, Inpp4b, in the cells. Thus, Inpp4a may be responsible for the elimination of PtdIns(3,4)P2, the product of SHIP1 and SHIP2, from the phagosome membrane.
The role of PI phosphatases in the clearance of PtdIns(3,4,5)P3/PtdIns(3,4)P2. The Raw264.7 cells deficient in (a) PTEN, (b) SHIP 1, (c) SHIP2, (d) Inpp4a, (e) Inpp5e, (f) Inpp5b or (g) OCRL were prepared by use of specific shRNA (see Figure S1). The deficient cells (closed symbols) and the vector control cells (open symbols) were transfected with EGFP-fused Akt-PH. The cells were challenged with IgG-coated zymosan, and the intensity ratio of phagosome-associated fluorescence to total cellular fluorescence was determined. The combined results from three independent experiments (each contains three phagosomes) are shown as the means ± SEM.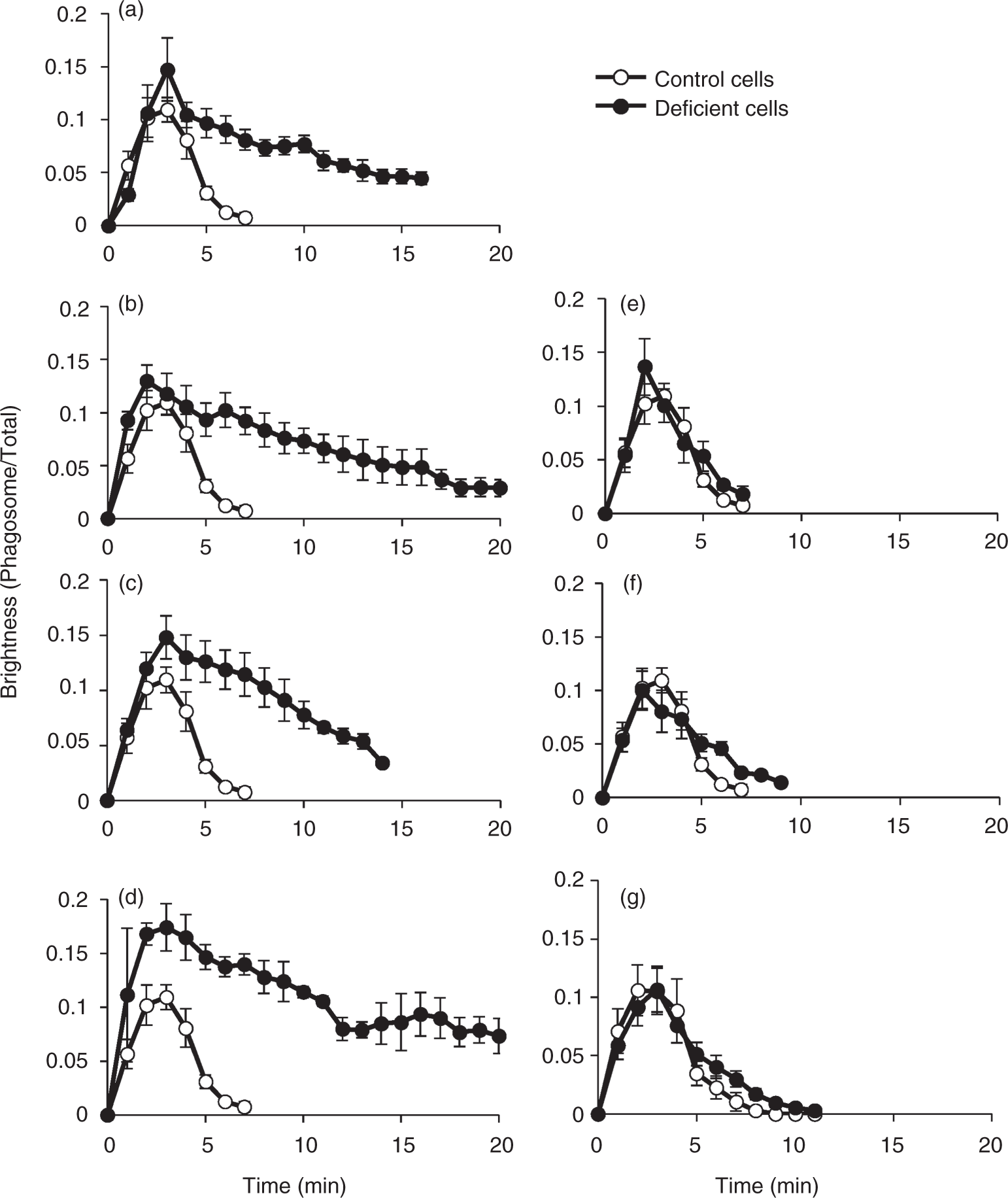
The relevance of the enzymatic activity of these phosphatases downstream of FcγR is shown in Figure S1, as augmentation of aggregated IgG-induced Akt phosphorylation, which depends on the cellular PtdIns(3,4,5)P3 and PtdIns(3,4)P2 level. The phosphorylation was obviously enhanced in the cells deficient in SHIP1, SHIP2 and Inpp4a (Figure S1A–C). In contrast, the enhancement of Akt phosphorylation was modest in shOCRL or shInpp5b cells (Figure S1E). Unexpectedly, the phosphorylation in shInpp5e cells was remarkably increased (Figure S1D), while the phagosomal accumulation of Akt-PH was comparable with the wild-type cells (Figure 2e). The increased Akt phosphorylation in shInpp5e cells may be limited to plasma membrane.
FcγRIIb is an inhibitory IgG receptor that possesses immune-receptor tyrosine-based inhibitory motif (ITIM) in its cytoplasmic tail. Because the phosphorylated ITIM binds SHIP1 and SHIP2,
22
it is likely that FcγRIIb mediates the action of IgG on the phagosome-associated fluorescence through the recruitment of these PI phosphatases. To test this hypothesis, we prepared two lines of FcγRIIb-deficient cells (clones 247 and 875; Figure 3a). The efficiency of the knockdown was confirmed by the marked increase in the aggregated IgG-induced phosphorylation of Akt, as determined by Western blotting of the whole cell lysates (Figure 3b for 247 clone). As expected, the dissociation of Akt-PH from the phagosome containing IgG-coated zymosan was markedly slowed in the FcγRIIb-deficient 247 cells (Figure 3c). When the engulfed zymosan was not opsonized with IgG, the FcγRIIb deficiency did not affect the dissociation of Akt-PH from the phagosome (Figure 3d). Similar results were obtained in the 875 cells (data not shown).
The role of FcγRIIb in the clearance of PtdIns(3,4,5)P3/PtdIns(3,4)P2. (a) FcγRIIb mRNA expression of two lines of Raw264.7 cells (clones 247 and 857). (b) Control cells and the 247 cells were stimulated with 30 µg/ml aggregated IgG. Whole-cell lysates were analyzed by Western blotting with the Ab against phosphorylated Akt (pSer473). (c, d) Control cells (open symbols) and the 247 cells (closed symbols) were transfected with EGFP-fused Akt-PH, and were challenged with (c) IgG-coated zymosan or (d) non-coated zymosan, and the intensity ratio of phagosome-associated fluorescence to total cellular fluorescence was determined. The combined results from three independent experiments (each contains three phagosomes) are shown as the means ± SEM.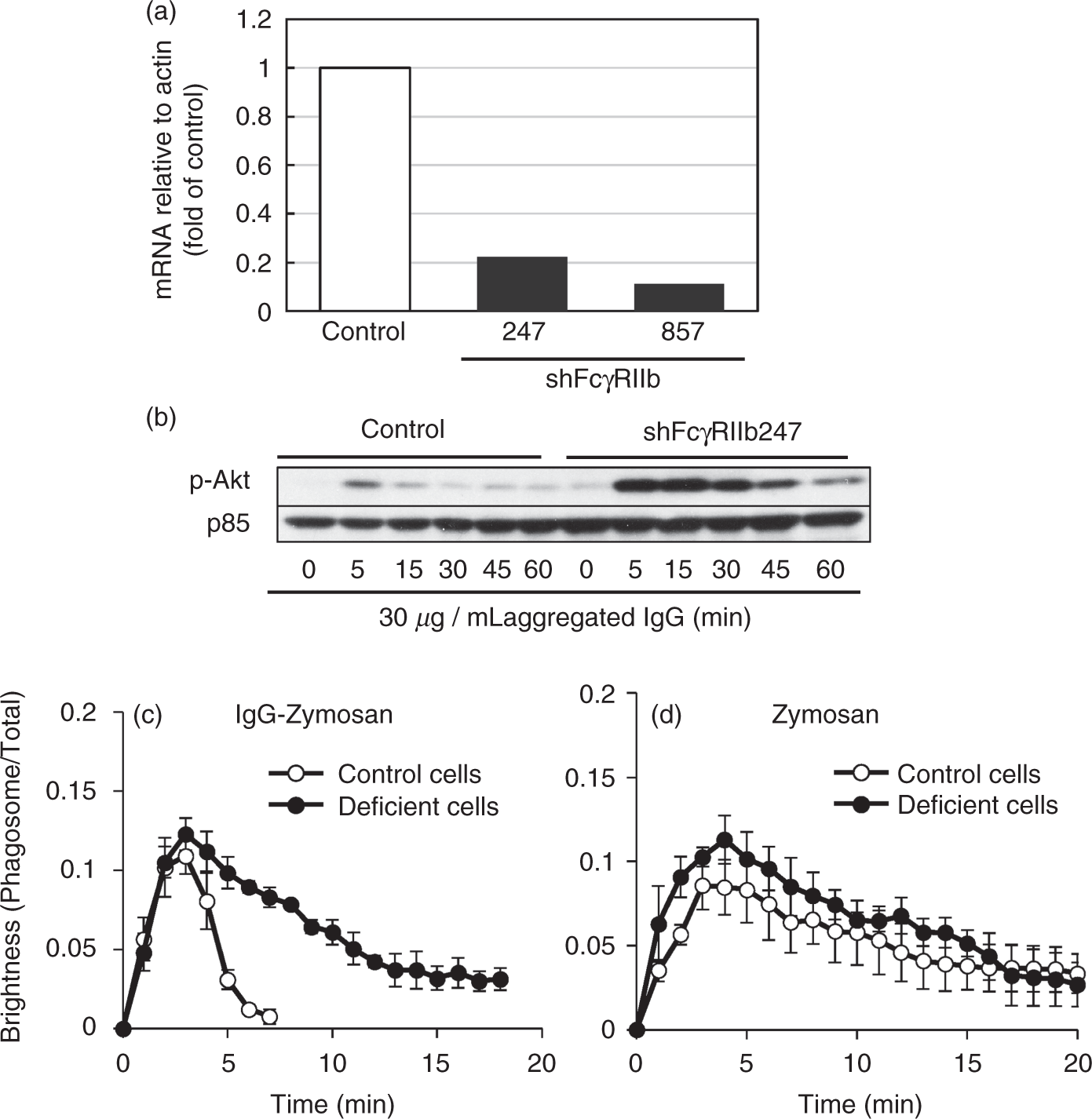
The above results suggested that FcγRIIb drove a sequential dephosphorylation system comprising SHIPs and Inpp4a that metabolizes PtdIns(3,4,5)P3 through PtdIns(3,4)P2 to PtdIns(3)P. Because PtdIns(3)P is known to facilitate phagosome maturation and acidification by recruiting a variety of proteins, including EEA1 (endosomal early antigen 1) and Hrs,
23
it is intriguing to consider that FcγRIIb-mediated acceleration of PtdIns(3,4,5)P3/PtdIns(3,4)P2 elimination accompanies the accelerated acidification of phagosome. In the experiment shown in Figure 4(a), FITC-labeled zymosan (green) or Texas Red-labeled zymosan (red) were coated with IgG. Raw264.7 cells were challenged with the mixture of these zymosan particles. In the figure, particle 1 is the IgG-coated FITC-zymosan that was not engulfed by the cells. The fluorescence intensity of the particle was not changed during the experimental period. Particle 2 is the IgG-coated FITC-zymosan that was engulfed and observed within the phagosome. The fluorescence of the particle decreased gradually. Because the FITC fluorescence is highly sensitive to acidic pH,
24
the result suggested that the phagosome was acidified during the period. In contrast, the fluorescence of the phagosomal TxR-zymosan (particle 3), the intensity of which is refractory to the pH change, was constant within the period. Interestingly, the decrease of the FITC fluorescence was slowed in the FcγRIIb-deficient cells (Figure 4b), indicating that phagosome acidification was delayed in the absence of FcγRIIb. The delayed acidification of phagosome was also observed in the Inpp4a-deficient cells (Figure 4c).
Slowed phagosome acidification in the FcγRIIb-deficient cells. (a) FITC-labeled (green) or Texas Red-labeled (red) zymosan were coated with IgG. Raw264.7 cells were challenged with the mixture of these zymosan particles, and their fluorescence was monitored every 5 min. Particle 1: the IgG-coated FITC-zymosan that was not engulfed during the experimental period. Particle 2: the engulfed IgG-coated FITC-zymosan. Particle 3: the engulfed IgG-coated TxR-zymosan. (b, c) Control cells (open symbols), the FcγRIIb-deficient cells (closed symbols in B) or the Inpp4a-deficient cells (closed symbols in C) were challenged with the IgG-coated FITC-zymosan, and the fluorescence of the engulfed zymosan was monitored every 5 min. The combined results from three independent experiments (each contains 10 phagosomes) are shown as the means ± SEM.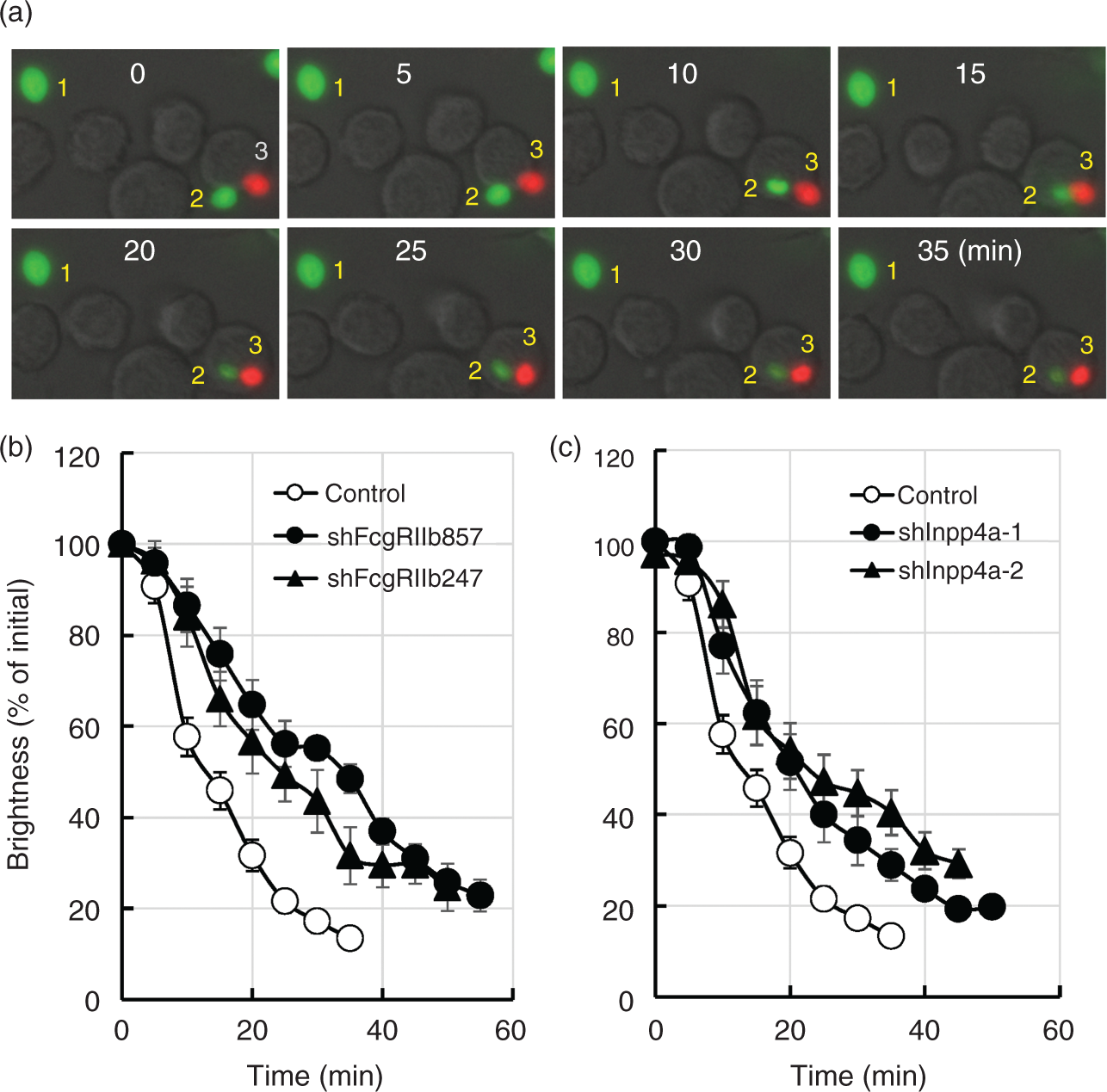
Discussion
The relative abundance of PI species on the phagosome membrane fluctuates along the course of phagocytosis. 1 The formation of a phagocytic cup accompanies a dramatic increase in PtdIns(3,4,5)P3. 25 After sealing of the cup, PtdIns(3,4,5)P3 disappears and the nascent phagosome acquires the microbicidal and degradative capability required for pathogen elimination by a process called phagosome maturation. 23 SHIP1, 11 SHIP2, 12 OCRL,14,15 Inpp5b 16 and Inpp5e13,17 possess the in vitro 5-phosphatase activity toward PtdIns(3,4,5)P3, and are reported to bind nascent phagosomes.
In the present study, we prepared a panel of PI phosphatase-deficient cells and examined the distribution of the transfected Akt-PH during the process of phagocytosis. We observed that the dissociation of this probe from the nascent phagosome was highly accelerated when the engulfed particles were opsonized with IgG (Figure 1). The specific knockdown of either SHIP1 or SHIP2 slowed the IgG-accelerated dissociation (Figure 2). Akt-PH binds both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 with comparable binding affinity. 20 Thus, the dissociation of this probe indicates that both these PI species are removed from the phagosome. Because SHIPs hydrolyze PtdIns(3,4,5)P3 but produce PtdIns(3,4)P2, the IgG-accelerated dissociation of Akt-PH suggests that PtdIns(3,4)P2 is further metabolized and effectively eliminated (Figure 2). Inpp4a is a PI 4-phosphatase that preferentially uses PtdIns(3,4)P2, 21 and is reportedly recruited to the phagosome membrane. 26 We observed that a specific knockdown of Inpp4a impaired the IgG-accelerated dissociation of Akt-PH (Figure 2). Inpp4a may efficiently remove the product of SHIPs, PtdIns(3,4)P2, from the phagosome that contains IgG-coated particles. SHIPs and Inpp4a are considered to constitute a sequential dephosphorylation system that metabolizes PtdIns(3,4,5)P3 through PtdIns(3,4)P2 to PtdIns(3)P. An additional intriguing finding of the present study is that a specific knockdown of FcγRIIb, an inhibitory receptor that attenuates FcγRIIa-mediated phagocytosis, 27 impaired the IgG-accelerated elimination of PtdIns(3,4,5)P3/PtdIns(3,4)P2 (Figure 3). This effect of FcγRIIb may occur through the recruitment of SHIP1 and SHIP2, because the phosphorylated ITIM of FcγRIIb is known to bind these 5-phosphatases. 22
In the striatum from Inpp4a–/– mice, internalization of cell surface N-methyl-d-aspartate-type glutamate receptors is decreased. 28 The fact indicates that Inpp4a is a positive regulator of clathrin-mediated endocytosis. In contrary, the uptake of large particles is increased in the macrophages from myeloid cell-specific Inpp4a-conditional knockout mice. 26 Thus, Inpp4a is a negative regulator of phagocytosis. In the Inpp4a–/– macrophages, IgG-induced phosphorylation of Akt is remarkably enhanced, 26 indicating the substantial role of this phosphatase in dephosphorylation of PtdIns(3,4)P2. It has also been reported that the conditional Inpp4a–/– mice cannot survive as long as the wild type after i.p. infection of Escherichia coli. 29 The killing activity toward E. coli may be dampened in these mice, as the phagosome acidification was down-regulated in Inpp4a-deficient macrophages (Figure 4).
PtdIns(3)P is known to facilitate phagosome maturation and acidification by recruiting a variety of proteins, including EEA1 and Hrs. 23 A major role of a class-III PI 3-kinase Vps34 in the synthesis of PtdIns(3)P has been demonstrated in both nascent phagosome and early endosome.30–32 However, it is reported that the enzyme cascade comprising the β-subtype of class-IA PI 3-kinase, PI 5-phosphatases and PI 4-phosphatases produces a substantial amount of PtdIns(3)P, which plays a role in transferrin uptake of HeLa cells. 33 The present study suggested that the activation of FcγRIIb accelerates the phagosomal PtdIns(3)P formation by driving the sequential dephosphorylation system as described above. In addition, we observed that IgG on the opsonized particles accelerates the phagosome acidification (Figure 4). FcγRIIa is known to stimulate phagocytosis by transmitting a signal through its cytoplasmic domain. In contrast, FcγRIIb attenuates the FcγRIIa-mediated phagocytosis in COS-1 model cells. 27 Recent studies have implicated FcγRIIb in immune tolerance and autoimmunity. 34 The present study suggested an additional role of the inhibitory receptor in the phagocytic process; once formed, the phagosomes that contain FcγRIIb–IgG complex are rapidly processed. During the maturation process, ER proteins are delivered to phagosome, where degraded peptides are loaded onto MHC classII. 35 It has been reported that a slow degradation of peptide antigen is crucial for regulated patterns of peptide degradation suitable for development of adaptive immune response. 35 Thus, it is intriguing to speculate that FcγRIIb transmits a signal that favors the rapid degradation of IgG-opsonized invaders, which need no longer be presented for development of adaptive immunity.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.