
Abstract
Impaired resistance to Pseudomonas aeruginosa-induced pneumonia after cecal ligation and puncture (CLP), a mouse model for human polymicrobial sepsis, is associated with decreased IFN-γ, but increased IL-10, levels in the lung. We investigated the so far unknown mechanisms underlying this reduced IFN-γ synthesis in CLP mice. CD11b+ NK cells, but not T or NKT cells in the lung were impaired in IFN-γ synthesis upon challenge with Pseudomonas in vitro and in vivo after CLP. The inhibition of NK cells was independent of IL-10. IFN-γ synthesis of NK cells was only partly restored by addition of recombinant IL-12. Accessory cells including dendritic cells and alveolar macrophages were required for maximal IFN-γ secretion. But accessory cells of CLP mice suppressed the IFN-γ secretion from naive lung leukocytes. In turn, naive accessory cells were unable to restore the IFN-γ production from lung leukocytes of CLP mice. Thus, a disturbed interaction of accessory cells and NK cells is involved in the impaired IFN-γ release in response to Pseudomonas in the lung of CLP mice. Considering the importance of IFN-γ in the immune defense against bacteria the dysfunction of accessory cells and NK cells might contribute to the enhanced susceptibility to Pseudomonas after CLP.
Introduction
Sepsis is the host response to systemic, mostly, bacterial infection, and is associated with exaggerated inflammation that might cause organ damage. Despite effective control of inflammation on intensive care units, sepsis patients frequently acquire nosocomial life-threatening infections owing to the development of immunosuppression. 1 The pathomechanisms that cause sepsis-associated immunosuppression are only incompletely understood.
Pseudomonas aeruginosa is a common opportunistic Gram-negative pathogen and origin of pneumonia in sepsis patients. In healthy people, the efficient elimination of P. aeruginosa depends on the coordinated activity of innate immune cells. IFN-γ enhances the bactericidal activity of phagocytes and is associated with an effective clearance of P. aeruginosa.2–5 NK cells represent an important source of IFN-γ in diverse bacterial infections.6,7 NK cell-derived IFN-γ synthesis is triggered by IL-12, which is released from accessory cells like dendritic cells (DC) and macrophages upon contact with bacterial components (reviewed in Michel et al. 8 and Degli-Esposti and Smyth 9 ). Other cytokines, like IL-10, TGF-β or IL-4, inhibit the NK cell-derived IFN-γ production.10–13 Accordingly, pathologic responses to P. aeruginosa infection are frequently associated with the disturbance of the IL-12/IFN-γ axis. 14
Cecal ligation and puncture (CLP) is a common model for human polymicrobial sepsis. Corresponding to septic patients, mice that underwent CLP displayed an enhanced susceptibility to secondary infection with P. aeruginosa due to the development of immunosuppression. The impaired immune defense against P. aeruginosa-induced pneumonia after CLP is associated with reduced levels of IFN-γ, but enhanced amounts of IL-10, in the lung in comparison with sham mice that efficiently clear the infection. 15 The treatment of post-septic mice with naive DC improves survival of secondary P. aeruginosa infection and is paralleled by increased formation of IL-12, but reduced IL-10 synthesis, in the lung. 16 Moreover, after CLP the capacity of accessory cells in the lung to release IL-12 is strongly reduced.17,18 These findings point to a disturbed function of accessory cells in the lung. We have recently shown that polymicrobial sepsis induces a reprogramming of DC differentiation in the bone marrow that results in the generation of dysfunctional DC. 19 Such dysfunctional DC release enhanced levels of IL-10 and suppress the capacity of NK cells to release IFN-γ upon stimulation with P. aeruginosa in vitro. 19 Accordingly, upon adoptive transfer, DC generated from bone marrow of post-septic mice inhibit the clearance of P. aeruginosa in naive mice, and impair the formation of IFN-γ and IL-12 in the lung. 19 So far, it is unknown whether accessory cells similarly suppress NK cells during secondary infection with P. aeruginosa in post-septic mice in vivo.
In the present study, we investigated the modulation of NK cells in the lung of post-septic mice in response to secondary infection with P. aeruginosa and thereby focused on the role of the cytokine environment and accessory cells. CLP was used to induce polymicrobial sepsis, and secondary infection with P. aeruginosa was performed 4 d later.
Materials and methods
Mice
All mice used in this study were 7–12-wk-old female BALB/c mice purchased from Harlan Winkelmann (Borchen, Germany). The mice were housed in the laboratory animal facility of the University Hospital Essen under specific pathogen-free conditions and had access to standard rodent food and water ad libitum. The animal experiments were approved by the local ethics committee of the State Government of Nordrhein-Westfalen.
CLP
To induce polymicrobial sepsis in mice the CLP model was used as described previously.19,20 Briefly, after i.m. injection of 115 mg/kg ketamin and 13 mg/kg xylazin (both from CEVA Santé Animale, Dusseldorf, Germany) anesthesia, a midline laparotomy was performed, the cecum was exposed and a ligation was set at 50%. The cecum was punctured once with a 27-gauge needle and a small amount of content was extruded. Thereafter, the cecum was placed back into the abdominal cavity. Sham mice underwent the same procedure except for the ligation and puncture of the cecum. One milliliter of sterile saline was injected into the abdominal cavity for resuscitation before the incision was closed with two layers. This CLP model induced a moderate sepsis with 20% lethality within the first 24 h. Thereafter, none of the animals died. On d 4 after the operation, CLP mice did not show any signs of morbidity and displayed similar behavior to sham mice. All experiments dealing with the analyses of leukocyte phenotype and function were performed (with n = 3–4 sham and n = 4–5 CLP mice) and were independently repeated at least twice. In case of the determination of the bacterial load after secondary infection n = 5 mice per group were used.
P. aeruginosa infection
Secondary infection with P. aeruginosa was performed 4 d after CLP or sham operation, as described previously. 19 Briefly, P. aeruginosa (strain ATCC 27853) were grown on Tryptic–Soy–Agar plates (BD Trypticase Soy Agar; BD, Heidelberg, Germany) at 37℃, and then re-suspended and shaken for 1 h at 37℃ in tryptic soy broth (TSB) (Bacto, BD). The OD of the bacterial suspension at 550 nm was determined during the early logarithmic growth phase and the number of colony-forming units (CFU) was extrapolated from a standard growth curve. Bacteria were washed twice in RPMI medium (Biochrom, Berlin, Germany). Mice were anesthetized with diethyl ether and 2 × 108 CFU of P. aeruginosa bacteria were applied intranasally (i.n.) in a total volume of 20 µl. To address the ex vivo cytokine release, lung leukocytes were prepared 4 h after infection with P. aeruginosa, as described below. In order to determine the bacterial load, lungs were homogenized 24 h after infection with P. aeruginosa, as reported previously. 19 Serial dilutions of homogenates were cultured on TSB agar plates and colonies were counted after 18 h.
Isolation and culture of lung leukocytes
Single-cell suspensions of lungs were prepared after perfusion of pulmonary vessels with sterile PBS and subsequent digestion of the tissue in 2 mg/ml Collagenase P solution (Roche Diagnostics, Penzberg, Germany) for 45 mins at 37℃. After meshing through a 30-µm cell strainer, erythrocytes were lysed using a standard lysis buffer (0.15 M NH4Cl, 10 mM KHCO3 and 0.1 mM Na2EDTA; all Sigma Aldrich, Taufkirchen, Germany). Cell viability was usually > 95% as determined by trypan blue exclusion. Cells were cultured in very low endotoxin medium (VLE RRMI1640 medium; Biochrom) with stable glutamine supplemented with 10% FCS (Biochrom), 10 mM HEPES (Biochrom), 0.06 mg/ml penicillin, 0.02 mg/ml gentamicin, 0.05 mM β-mercaptoethanol (all Sigma Aldrich) and 3 ng/ml rmIL-2 (R&D Systems, Wiesbaden, Germany), which was used to improve the survival of NK cells. All cell cultures were set up at least in triplicates. Lung leukocytes were cultured for 18 h at a density of 8 × 105 cells per well (96-well round bottom-plates). In some experiments, lung cells were stimulated with 2.5 or 5 × 106 CFU inactivated P. aeruginosa bacteria per well (kindly provided by Y. Zhang) with 0.5 ng/ml recombinant murine IL-12 (R&D Systems) or with 10 µg/ml neutralizing Abs against IL-10 (Clone JES052A5; R&D Systems 21 ), as indicated in the figure legends. For some experiments, lung leukocytes were depleted of DX5+ NK/ T (NKT) cells or CD11c+ cells using anti-CD49b (DX5) or anti-CD11c Abs-coupled MicroBeads (both Miltenyi, Bergisch Gladbach, Germany), respectively, according to the manufacturer’s instructions. Separation of adherent cells was achieved by incubation of total lung leukocytes in tissue culture plates for 2 h at 37℃. Non-adherent cells were harvested and cultured as described above. After 18 h, supernatants of leukocyte cultures were used to determine the content of IFN-γ, IL-10 (DuoSet; R&D Systems) or IL-12p70 (Ready-SET-Go; eBioscience, San Diego, CA, USA) by means of ELISA. Where indicated, leukocytes were analyzed for intracellular IFN-γ expression by flow cytometry (see below).
Flow cytometry
After blocking non-specific binding using 5 µg/ml Fc Block (BD Biosciences, Heidelberg, Germany) single cell suspensions of the lung were stained with fluorochrome-labeled Abs against CD3 (clone 145-2C11), CD11c (clone HL3), CD11b (clone M1/70), CD49b (clone DX5), CD69 (clone H1.2F3), Ly6G (clone 1A8), IL-12 receptor β1 (clone 114), IFN-γ receptor 1 (clone 2E2) or IFN-γ (clone XMG1.2). All Abs were obtained from BD Biosciences or eBioscience. For detection of intracellular IFN-γ cells were incubated with 0.66 µl/ml monensin (GolgiStop; BD Biosciences) for the last 4 h of the culture. After surface staining with Abs against CD3, CD11b and CD49b, lung cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences) and stained for intracellular IFN-γ. For all stainings appropriate isotype controls were used. Cells were analyzed by flow cytometry using a FACSCalibur and Cell QuestPro software (both from BD Biosciences). The threshold for positive staining was set where < 2% of the cells in the isotype control stained false-positively.
Statistics
Data of the bacterial load of individual mice were illustrated as a scatter dot plot showing the median. Differences were analyzed by the non-parametric Mann-Whitney U-test. Remaining data were expressed as mean + SD and were tested for statistical difference using Student’s t-test or by one-way ANOVA, followed by Newman–Keuls multiple comparison test, as indicated in the figure legends. Statistical analyses were performed using GraphPad Prism 5.0 and a P-value < 0.05 was considered significant.
Results
Impaired IFN-γ release from lung leukocytes after polymicrobial sepsis
First, we investigated the impact of polymicrobial sepsis on the cytokine secretion pattern of leukocytes in the lung after secondary infection with P. aeruginosa. Therefore, mice were i.n. infected with P. aeruginosa 4 d after sham or CLP operation. The bacterial load in the lung was determined 24 h later. As expected, the lungs of CLP mice contained >104-fold more bacteria than lungs of sham mice (Figure 1A). In a previous study, we showed that no bacteria were detectable in lungs of sham or CLP mice in the absence of secondary infection with P. aeruginosa.
19
To address the ex vivo cytokine release after secondary infection, lung leukocytes from CLP and sham mice were isolated 4 h after secondary infection with P. aeruginosa and were cultured in medium only. Lung leukocytes from sham mice released high levels of IFN-γ and low amounts of IL-10 (Figure 1B). In contrast, lung leukocytes from CLP mice secreted low levels of IFN-γ, but enhanced levels of IL-10 (Figure 1B).
Bacterial load and cytokine release from leukocytes in the lung after opportunistic Pseudomonas infection. Four d after CLP or sham operation, mice were infected i.n. with 2 × 108 CFU P. aeruginosa bacteria (Pa). (A) Twenty-four h later the bacterial load in the lung was determined. The graph shows the data of individual mice from one of three independent experiments (n = 5 mice per group). The horizontal lines indicate the median. Statistical difference was analyzed by the non-parametric Mann–Whitney U-test. (B) Four h after Pa infection lung leukocytes were prepared and cultured in medium only to address the ex vivo cytokine response. The levels of IFN-γ and IL-10 in the supernatants were determined. The graph shows the mean + SD of triplicate cultures of cells from individual mice (n = 3–4 mice per group). Statistical differences were analyzed by unpaired Student's t-test. For ex vivo cytokine release from lung leukocytes of mice after sham or CLP operation, but without secondary infection, see ‘medium’ values shown in (C). (C) Four d after CLP or sham operation lung leukocytes were pooled per group (n = 3 mice per group) and cultured with or without inactivated Pa as indicated. The concentration of IL-12p70, IFN-γ and IL-10 in the supernatants was determined. The data represent the mean + SD of triplicate cultures of one representative experiment of five. Statistical differences were analyzed by one-way ANOVA, followed by Newman–Keuls multiple comparison test. *P < 0.05; **P < 0.01; ***P < 0.001; #P < 0.001 versus medium.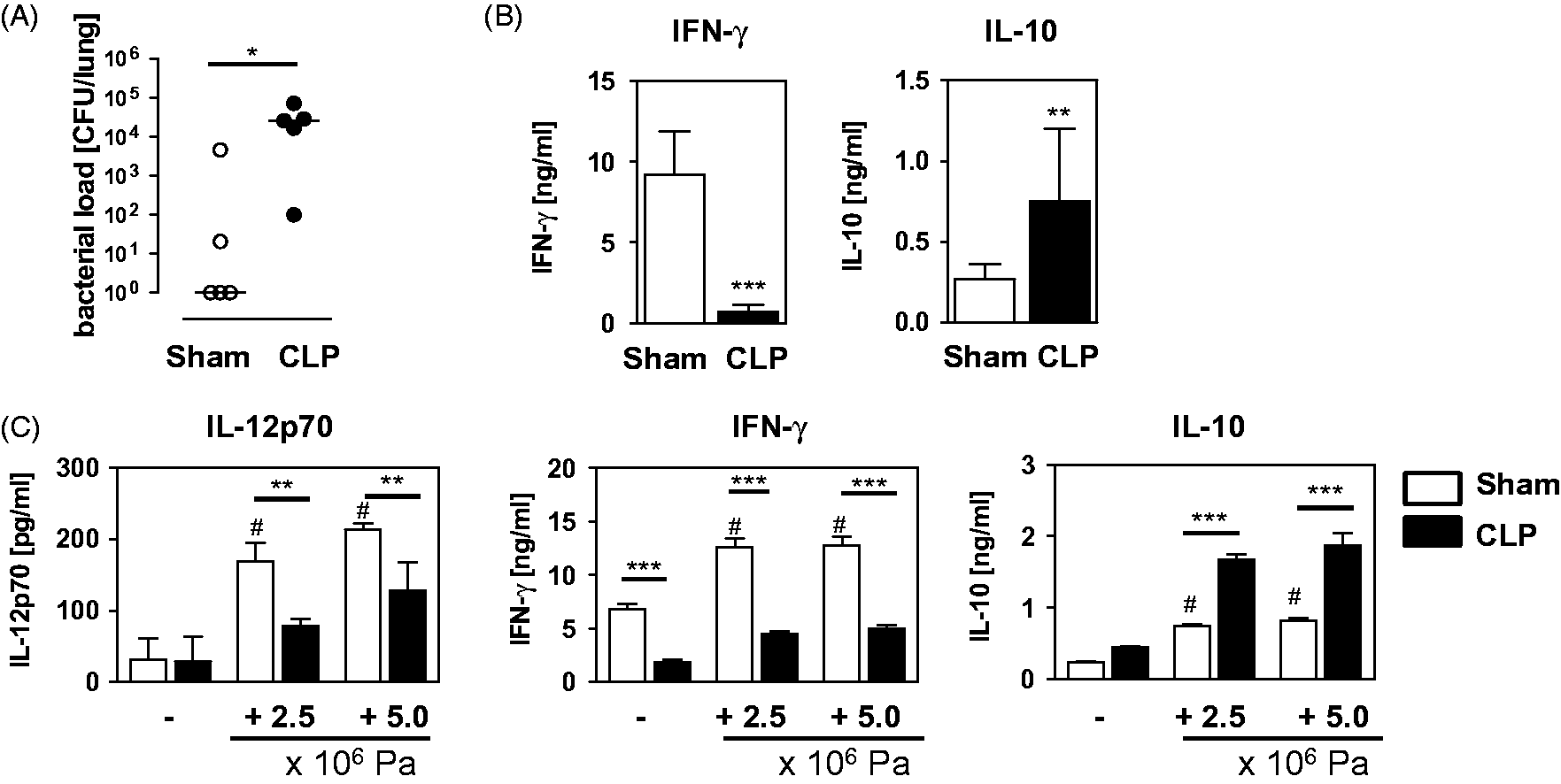
To determine whether the diverse cytokine response of lung leukocytes to P. aeruginosa was already established during sepsis, lung leukocytes were isolated 4 d after sham or CLP operation and were stimulated with inactivated P. aeruginosa bacteria in vitro. After sham operation, lung leukocytes clearly released IL-12 and IFN-γ, and, to a lesser extent, IL-10 upon stimulation with P. aeruginosa (Figure 1C). In contrast, lung leukocytes from CLP mice secreted significantly less IL-12 and IFN-γ, but strongly increased levels of IL-10 in comparison with cells from sham mice (Figure 1C). Thus, as a consequence of polymicrobial sepsis, leukocytes in the lung are impaired in the P. aeruginosa-induced production of IL-12 and IFN-γ.
Cellular composition of lung leukocytes after CLP and after secondary P. aeruginosa infection
The bias of lung leukocytes from CLP mice towards decreased IL-12/IFN-γ synthesis in response to P. aeruginosa might be associated with an altered leukocyte composition. To address this issue, lung leukocytes were isolated 4 d after sham or CLP operation or 4 h after subsequent secondary i.n. infection with P. aeruginosa. The absolute number of DC and alveolar macrophages, as well as of CD11b+ NK cells that are characteristic sources of IL-12 and IFN-γ, respectively, in bacterial infections was determined by flow cytometry. Likewise, data on granulocytes were acquired. The gating strategy of DC, alveolar macrophages, CD11b+ NK cells and granulocytes is shown in Figure 2A. The absolute number of granulocytes in the lung was clearly increased after CLP (Figure 2B). Secondary infection with P. aeruginosa led to a strong influx of granulocytes into the lung of both sham and CLP mice (Figure 2B). After CLP, the absolute number of alveolar macrophages and CD11b+ NK cells was reduced by 65% and 50%, respectively (Figure 2D, E), whereas the number of DC did not change significantly (Figure 2C). Upon secondary infection with P. aeruginosa, the number of DC increased in CLP mice (Figure 2C). While the number of alveolar macrophages decreased in sham mice after secondary infection with P. aeruginosa it remained constant in the lungs of CLP mice (Figure 2D). However, the number of NK cells in the lung did not vary in either group after secondary infection with P. aeruginosa (Figure 2E). Thus, in CLP mice secondary infection with P. aeruginosa triggers the expansion of the granulocyte and DC population, whereas the number of CD11b+ NK cells does not change and, consequently, remains reduced.
Cellular composition of lung leukocytes after CLP and after opportunistic Pseudomonas infection. Four d after CLP or sham operation, lung leukocytes were either prepared directly or 4 h after an additional intranasal infection with 2 × 108 CFU P. aeruginosa bacteria (Pa). Innate immune cells were quantified by flow cytometry. (A) Gating strategy of Ly6G+ granulocytes, CD11c+CD11blow alveolar macrophages (AM; region I), CD11cintCD11b+ dendritic cells (DC; region II) and CD3-DX5+CD11b+ NK cells. Representative dot plots from sham mice are shown. Dot plots from CLP mice showed a similar pattern, but the size of the populations differed. Absolute number of (B) granulocytes, (C) DC, (D) alveolar macrophages and (E) CD11b+ NK cells per lung. Numbers indicate the percentage of the gated population among total leukocytes. Data show mean + SD of 3–4 mice per group from three independent experiments. Statistical differences were analyzed by one-way ANOVA, followed by Newman–Keuls multiple comparison test. *P < 0.05; **P < 0.01; ***P < 0.001. FSC: forward side scatter.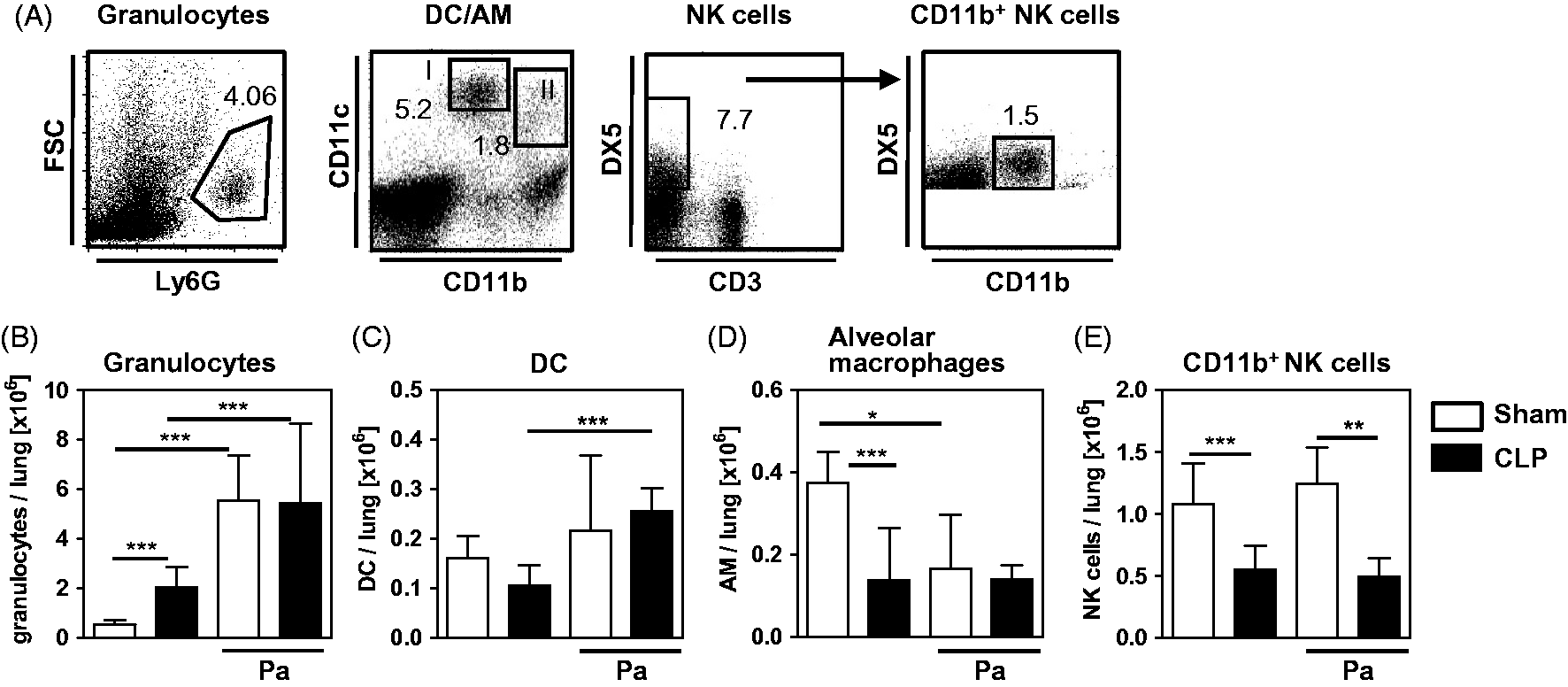
NK cells are impaired in IFN-γ secretion after CLP
Next, the cell population that is the main source of IFN-γ in response to P. aeruginosa among total lung leukocytes was identified. Therefore, lung leukocytes were prepared 4 d after sham or CLP operation, and stimulated with inactivated P. aeruginosa bacteria in vitro. Intracellular IFN-γ in T cells, NKT cells and CD11b+ NK cells was determined. Pseudomonas aeruginosa did not induce the production of IFN-γ in CD3+DX5- T cells from sham or CLP mice (Supplementary Figure 1A). Likewise, NKT cells of both groups did not respond to P. aeruginosa with enhanced IFN-γ synthesis (Supplementary Figure 1B). In contrast, CD11b+ NK cells from sham mice increased the expression of IFN-γ after stimulation with P. aeruginosa (Figure 3A, B). The percentage of NK cells from CLP mice that produced IFN-γ was significantly reduced in comparison with NK cells from sham mice both in the absence and presence of P. aeruginosa. (Figure 3A, B). In contrast, CD11b+ NK cells from CLP mice expressed significantly enhanced levels of the activation marker CD69 than cells from sham mice (Figure 3C, D). To further prove that NK cells are a major source of IFN-γ after stimulation with P. aeruginosa lung leukocytes from naive mice were depleted of DX5+ cells and the release of IFN-γ into the supernatant was determined after stimulation with P. aeruginosa. Depleted lung leukocytes contained < 0.05% CD11b+ NK cells (Figure 3E). Accordingly, depletion of DX5+ cells clearly reduced the release of IFN-γ from unstimulated and stimulated leukocytes (Figure 3F).
Pseudomonas-induced IFN-γ secretion from NK cells in vitro and in vivo. (A, B) Four d after CLP or sham operation, lung leukocytes were pooled from each group and were cultured with or without inactivated P. aeruginosa bacteria (Pa), and the production of IFN-γ in CD11b+ NK cells was determined by intracellular staining and flow cytometry. (A) Gating strategy of DX5+CD11b+ NK cells and representative dot plots of the IFN-γ expression. Isotype control Abs were used to determine the threshold of positive staining (Iso). (B) Summary of the percentage of IFN-γ+ cells among CD11b+ NK of four independent experiments with pooled cells from 3–4 mice per group. Data shows the mean + SD. Statistical differences were tested using one-way ANOVA followed by Newman–Keuls multiple comparison test. (C, D) Four d after CLP or sham operation, the expression of CD69 was determined on gated CD11b+ NK cells. Representative dot plots of one sham and one CLP mouse are shown (C). (D) Data of five independent experiments with 3–4 mice per group are summarized as a bar graph showing the mean + SD. Statistical differences were tested using paired Student’s t-test. (E, F) Lung leukocytes from naive mice were depleted of NK cells. (E) Representative dot plot showing the percentage of CD11b+ NK cells among lung leukocytes before and after NK cell depletion. (F) Total lung leukocytes and cells depleted of NK cells (DX5-depl.) were incubated with or without inactivated P. aeruginosa bacteria (Pa) and the amount of IFN-γ in the supernatants was determined. The graph shows the mean + SD of triplicate cultures of one representative out of two experiments. Statistical differences were tested using one-way ANOVA, followed by Newman–Keuls multiple comparison test. (G) Four d after CLP or sham operation, mice were infected i.n. with 2 × 108 CFU P. aeruginosa bacteria. Four h later, lung leukocytes were prepared and the ex vivo IFN-γ production was determined using intracellular staining as described in (A). The bar graph shows the mean + SD of 3–4 mice per group. Statistical difference was determined using unpaired Student's t-test. Numbers in dot plots indicate the percentage of gated or positive cells. *P < 0.05; **P < 0.01; ***P < 0.001. ns: not significant.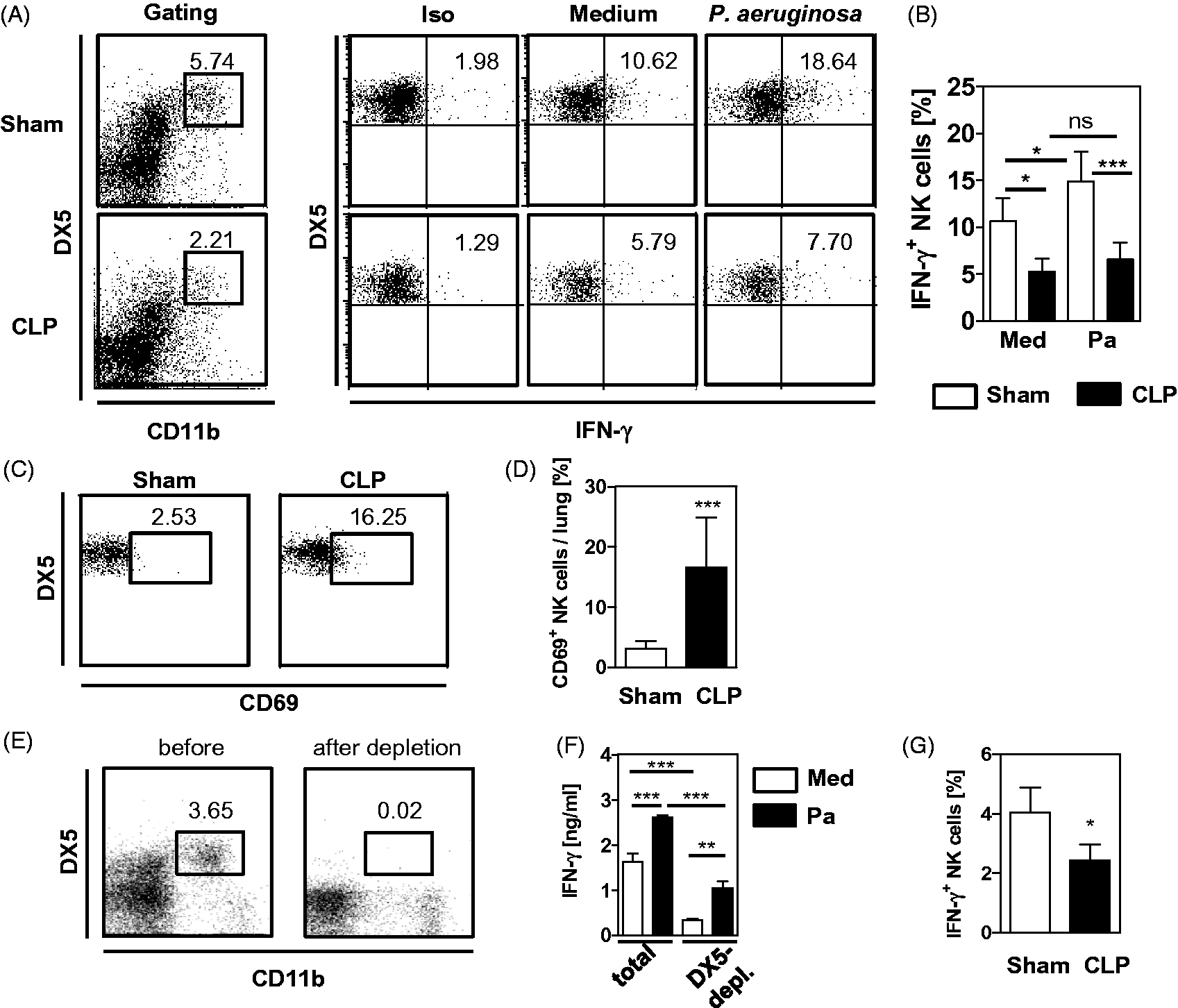
Further, we investigated whether the suppression of NK cells from post-septic mice likewise occurred during secondary infection with P. aeruginosa in vivo. Therefore, lung leukocytes were prepared 4 h after secondary infection of sham and CLP mice, and ex vivo intracellular IFN-γ was determined in CD11b+ NK cells. Similarly to the stimulation of lung leukocytes in vitro, NK cells from CLP mice were inferior to NK cells from sham mice in IFN-γ synthesis (Figure 3G). Taken together, although they display signs of activation mirrored by an enhanced expression of CD69, NK from CLP mice are impaired in IFN-γ synthesis upon challenge of leukocytes with P. aeruginosa in vitro and in vivo.
Role of the cytokine environment in the impaired IFN-γ synthesis of NK cells after CLP
Upon stimulation with P. aeruginosa, lung leukocytes from CLP mice released increased levels of IL-10, but only low amounts of IL-12 in comparison with leukocytes from sham mice. To examine whether blocking of IL-10 or supplementation with recombinant IL-12 might restore the IFN-γ secretion from lung leukocytes after CLP, stimulation with P. aeruginosa was performed in the presence or absence of neutralizing Abs against IL-10 or of additional IL-12. Blocking of IL-10 had no effect on IFN-γ release from leukocytes of CLP mice (Figure 4A). Leukocytes from sham mice vigorously responded with increased IFN-γ secretion to the IL-12 supplementation during stimulation with P. aeruginosa (Figure 4B). Lung leukocytes from CLP mice increased secretion of IFN-γ in the presence of additional IL-12, but remained inferior to leukocytes form sham mice (Figure 4B). Supplementation with IL-12 did not change the IL-10 formation of leukocytes from both groups (Figure 4B). Accordingly, stimulation of leukocytes with IL-12 alone strongly increased the percentage of NK cells that were positive for IFN-γ from sham mice, but was much less effective on NK cells from CLP mice (Figure 4C). T cells and NKT cells did not respond to IL-12 with the secretion of IFN-γ (data not shown). Moreover, the expression of the IFN-γ receptor and IL-12 receptor on NK cells and/or accessory cells was determined. After CLP, the expression of the IFN-γ receptor on NK cells, alveolar macrophages, and DC did not change significantly (Figure 4D). Likewise, the expression of the IL-12 receptor on CD11b+ NK cells did not change significantly after CLP (Figure 4E). In summary, NK cells in the lung from CLP mice respond to IL-12 with an enhanced secretion of IFN-γ, but, by far, do not reach the activity of NK cells from sham mice. Neutralization of IL-10 does not restore the IFN-γ secretion from lung leukocytes of CLP mice.
Influence of IL-10 and IL-12 on the Pseudomonas-induced secretion of IFN-γ. Four d after CLP (n = 4) or sham (n = 5) operation, lung leukocytes were prepared. (A, B) Cells were pooled per group and were cultured in the presence of inactivated P. aeruginosa bacteria. (A) IL-10 was blocked by neutralizing Abs (αIL-10) or (B) recombinant IL-12 was added. The level of IFN-γ and IL-10 in the supernatants was determined. Data show the mean + SD of triplicate cultures of one representative experiment out of two experiments. (C) Four d after CLP or sham operation, pooled lung cells were incubated with or without recombinant IL-12 and intracellular IFN-γ expression in CD11b+ NK cells was analyzed by flow cytometry. Representative dot plots are shown. Numbers indicate the percentage of positive cells. Statistical differences were determined by one-way ANOVA, followed by Newman–Keuls multiple comparison test. (D) Lung leukocytes from individual mice were analyzed for the percentage of CD11b+ NK cells, alveolar macrophages (AM) and DC expressing the receptor for IFN-γ. (E) Percentage CD11b+ NK cells that express the receptor for IL-12. Gating for (D) and (E) was performed as shown in Figure 2A. Data show the mean + SD of individual mice and were tested for statistical significance using unpaired Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.001. ns: not significant.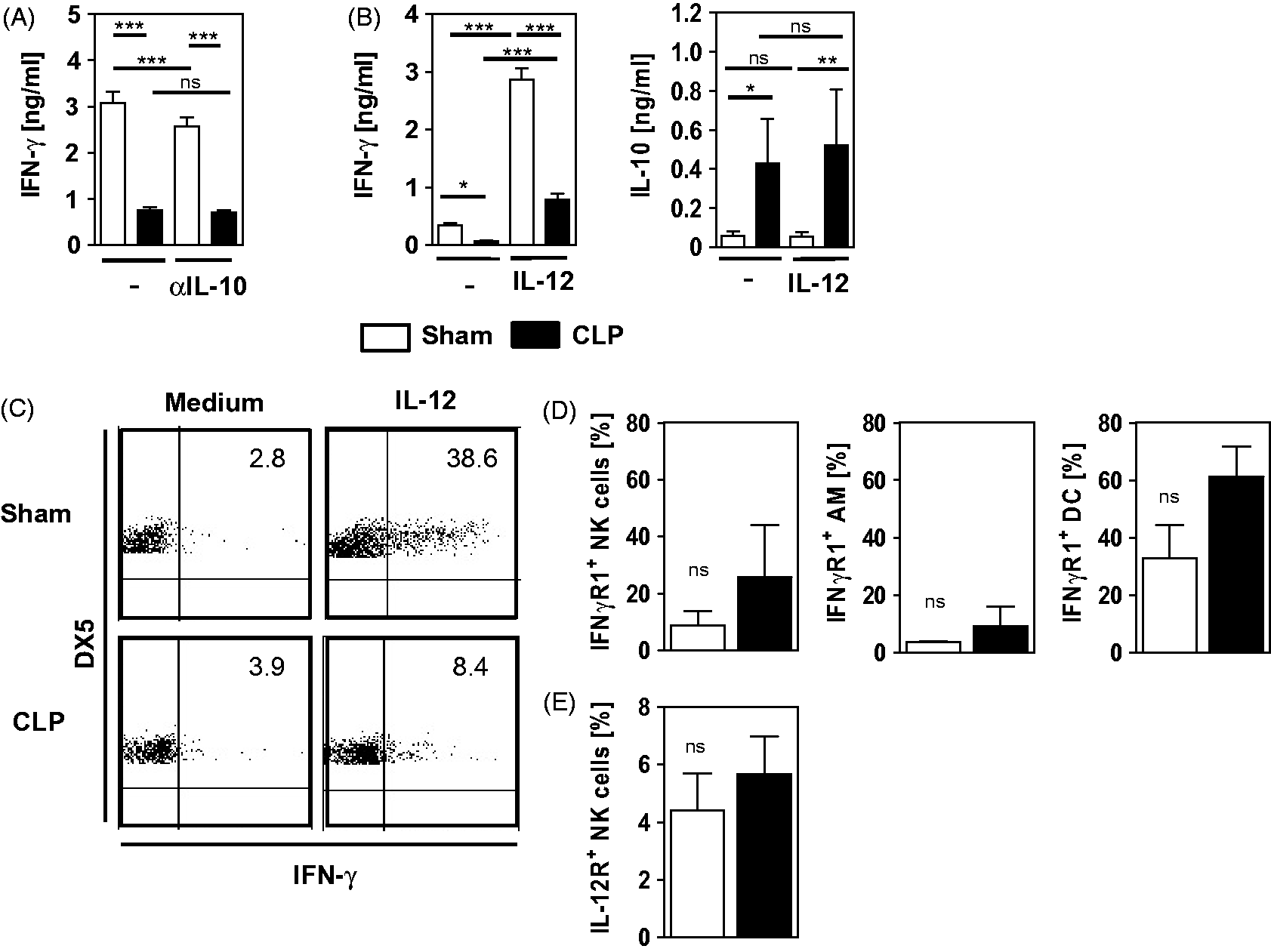
Involvement of accessory cells in the impaired IFN-γ synthesis of NK cells after CLP
Next, we investigated whether accessory cells like DC and alveolar macrophages might contribute to the NK cell suppression after CLP. Therefore, lung leukocytes from sham and CLP mice were depleted of CD11c+ cells that represent DC and alveolar macrophages. Depleted cells contained < 1.5% CD11c+ cells (Figure 5A). Total and CD11c-depleted lung cells were stimulated with P. aeruginosa. CD11c-depleted leukocytes from sham mice released significantly less IFN-γ than total leukocytes (Figure 5B). Moreover, CD11c+ cells were isolated from lung leukocytes of sham and CLP mice, as well as from naive mice. The purity of CD11c+ cells was at least 83% (Figure 5A). Auto-fluorescent alveolar macrophages constituted the major part of CD11c+ cells (Figure 5A). CD11c+ cells from sham and CLP mice were added to naive CD11c-depleted lung leukocytes. These co-cultures were stimulated with P. aeruginosa and the release of IFN-γ was quantified. In the presence of CD11c+ cells from CLP mice naive leukocytes released significantly less IFN-γ than in the presence of CD11c+ cells from sham mice (Figure 5C).
Impact of accessory cells on the IFN-γ release from lung leukocytes. Four d after CLP (n = 4) or sham (n = 5) operation, lung leukocytes were prepared, pooled per group and, in part, depleted of CD11c+ cells. (A) Gating scheme and representative dot plots that show the depletion and enrichment of CD11c+ cells from leukocytes of CLP, sham and naive mice. (B) Total or depleted (CD11c-) lung cells were incubated with inactivated P. aeruginosa bacteria. (C) Enriched CD11c+ cells from CLP and sham mice were added to CD11c-depleted leukocytes from naive mice and were incubated with inactivated P. aeruginosa bacteria. (D) Enriched CD11c+ cells from lungs of naive mice were added to CD11c-depleted lung leukocytes of CLP and sham mice, and were incubated with inactivated P. aeruginosa bacteria. Bar graphs show the amount of IFN-γ in the supernatant as mean + SD of triplicate cultures of one representative experiment out of two. Statistical differences were determined by one-way ANOVA, followed by Newman–Keuls multiple comparison test. (E, F) Naive lung cells were depleted of macrophages through plastic adherence. (E) Dot plots show the percentage of CD11c+ auto-fluorescent macrophages and CD11cint non-auto-fluorescent DC among total and non-adherent naive leukocytes. (F) Percentage of CD11b+ NK cells before and after the plastic adherence step. (G) Total and non-adherent cells were cultured in the presence of inactivated P. aeruginosa bacteria and the amount of IFN-γ in the supernatant was determined. Data shows the mean + SD of triplicate cultures of one representative experiment out of two. Statistical differences were tested using unpaired Student's t-test. *P < 0.05; **P < 0.01; ***P < 0.001.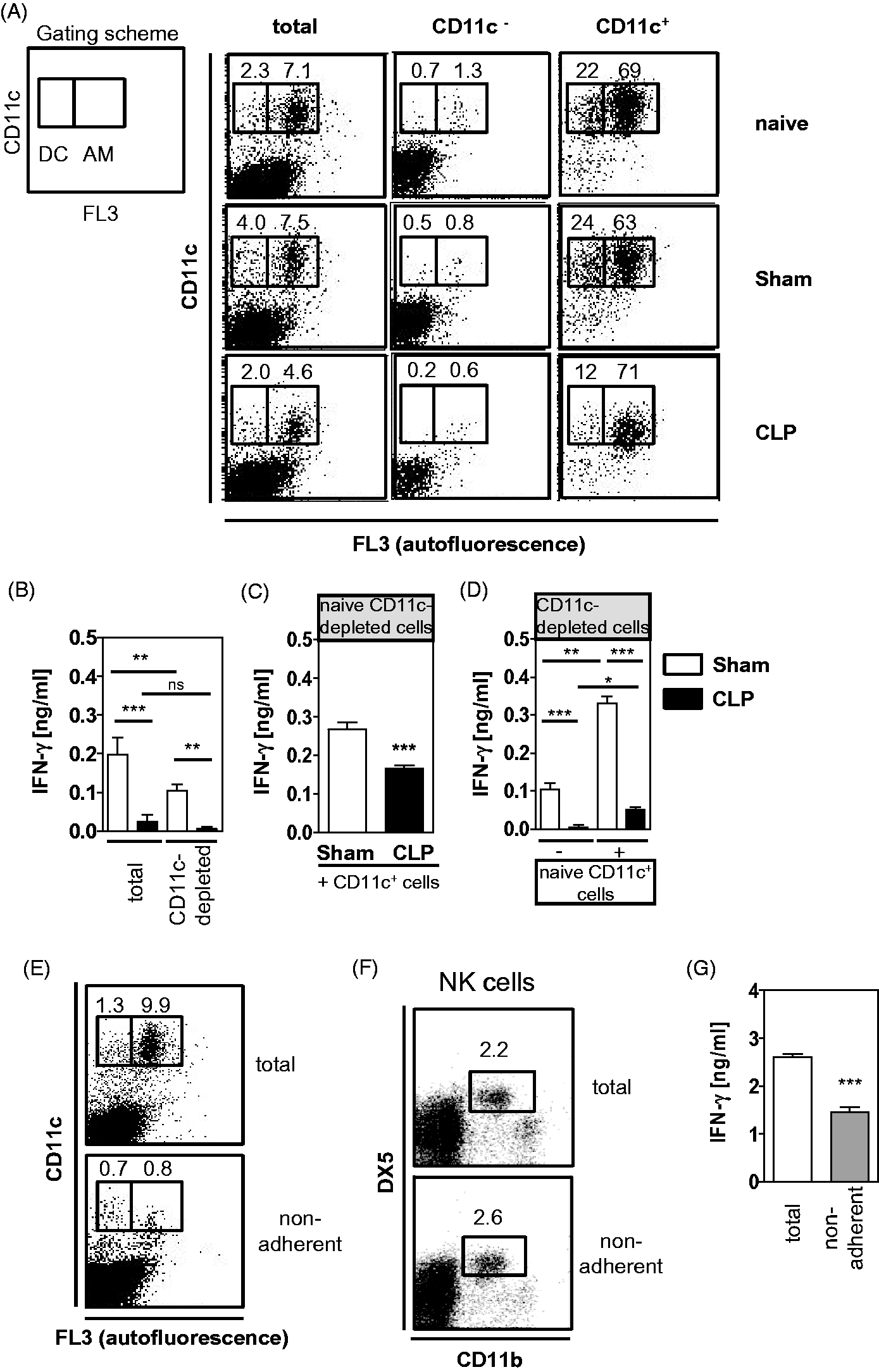
To examine whether, in turn, naive accessory cells might restore the IFN-γ secretion from lung leukocytes after CLP, CD11c-depleted lung leukocytes from sham and CLP mice were co-cultured with CD11c+ cells from naive mice. Addition of naive CD11c+ cells to CD11c-depleted leukocytes from sham mice strongly increased the release of IFN-γ (Figure 5D). In contrast, addition of naive CD11c+ cells only marginally increased the IFN-γ release from CD11c-depleted leukocytes from CLP mice (Figure 5D).
Moreover, to specify their contribution to the IFN-γ secretion from lung leukocytes macrophages were removed from naive total leukocytes through plastic adherence prior to stimulation with P. aeruginosa. Non-adherent cells contained < 1% alveolar macrophages (Figure 5E). The adherence step did not influence the percentage of CD11b+ NK cells (Figure 5F). Total leukocytes released twofold more IFN-γ than non-adherent cells (Figure 5G). Taken together, accessory cells from lungs of CLP mice suppress the capacity of naive leukocytes to release IFN-γ. But naive accessory cells do not restore the capacity of lung leukocytes from CLP mice to secrete IFN-γ.
Discussion
Previous reports have shown that the impaired clearance of P. aeruginosa in the lung of post-septic mice is associated with a shift of the cytokine expression pattern towards decreased IFN-γ, but increased IL-10 levels, in lung and plasma.15,16 Insufficient IFN-γ synthesis in bacterial infections is frequently associated with impaired bacterial clearance and, accordingly, is considered to contribute to the enhanced susceptibility to secondary infections during sepsis. 15 So far, homogenates of total lung or bronchoalveolar lavage fluids from septic mice were the basis of such cytokine analyses. The limitation of tissue homogenates or lavage fluids is that they do not provide information on the cellular source of the cytokines. In addition to diverse leukocyte populations, non-immune cells, like airway epithelial cells, may also release pro- and anti-inflammatory mediators.22,23
Using isolated lung leukocytes we now provide evidence that lung leukocytes from post-septic mice secrete decreased IFN-γ, but increased IL-10, levels after secondary infection with P. aeruginosa ex vivo (Figure 1B) and thus mirror the known cytokine pattern of total organ preparations. This finding suggests that alterations in the leukocyte function contributed to the suppressed IFN-γ formation upon secondary P. aeruginosa infection in the lung of post-septic mice. Lung leukocytes of CLP mice similarly responded to P. aeruginosa in vitro (Figure 1C), as did lung leukocytes after secondary infection in vivo (Figure 1B). We conclude that the suppressed capacity of lung leukocytes for IFN-γ secretion was already established after CLP and did not solely depend on leukocytes, including neutrophils and DC that were recruited into the lung upon secondary infection in vivo (Figure 2B, C).
In line with a previous report, we identified CD11b+ NK cells as the main source of IFN-γ in response to P. aeruginosa among total lung leukocytes (Figure 3B). T cells and NKT cells that secrete IFN-γ in other infectious diseases did not produce IFN-γ upon stimulation of the leukocytes with P. aeruginosa (Supplementary Figure 1). We show here for the first time that the capacity of NK cells in the lung to produce IFN-γ in response to P. aeruginosa is clearly reduced in vitro (Figure 3B) and ex vivo (Figure 3G) as a consequence of polymicrobial sepsis. Interestingly, Murphey et al. 15 reported that the percentage of IFN-γ+ NK cells in the spleen of post-septic mice after secondary infection with P. aeruginosa is increased, while the mean cytokine production per NK cell is reduced. This observation partly contradicts our study, but might be explained by the usage of different routes for infection in both studies. Murphey et al. 15 applied Pseudomonas bacteria i.v. that might result in an altered responsiveness of NK cells or might target, in particular, splenic NK cells, whereas we used the i.n. route, which addresses primarily the lung before the bacteria disseminate into the spleen. Moreover, differences in the function and maturation state of NK cells in the spleen and in the lung have been described, and might contribute to the diverse extent of IFN-γ production by NK cells during secondary infection after polymicrobial sepsis. 24 In line with this assumption is the finding that splenic NK cells from septic mice do not express increased levels of the activation marker CD69 in contrast to NK cells in the lung as we show here (Figure 3D). 25
Accessory cells, like DC and macrophages, or monocytes stimulate NK cell-derived IFN-γ secretion through soluble mediators like IL-12 and cell contact-dependent mechanisms.8,26 Accordingly, depletion of accessory cells, including DC and macrophages from lung leukocytes, reduced the formation of P. aeruginosa-induced IFN-γ (Figure 5B, F), indicating the relevance of accessory cells in the cytokine response to P. aeruginosa. CD11c-depleted lung leukocytes were not completely impaired in IFN-γ secretion (Figure 5B). This observation might be explained by the presence of few remaining CD11c+ cells after the depletion step and/or by CD11c- accessory cells, like interstitial macrophages, that might also induce the synthesis of IFN-γ (Figure 5A). Moreover, NK cells as the main source of IFN-γ might be directly stimulated by P. aeruginosa independently of accessory cells as they express TLR and NKp44, which may be triggered by P. aeruginosa-derived molecules.25,27 During polymicrobial sepsis, alveolar macrophages and DC are impaired in the secretion of IL-12 and therefore might be incompetent in NK cell stimulation upon secondary infection with P. aeruginosa.17,18 However, attempts to detect IL-12 in the supernatants from isolated CD11c+ cells after exposure to P. aeruginosa in vitro have failed so far (data not shown). We have recently shown that the differentiation of DC from bone marrow is modulated during and after sepsis leading to the formation of dysfunctional DC that suppress the IFN-γ release from naive NK cells. 19 In line with these previous findings, we show here for the first time that accessory cells, including DC from lungs of post-septic mice, suppressed the P. aeruginosa-induced IFN-γ secretion from naive lung leukocytes (Figure 5C). Thus, we hypothesize that the suppression of NK cell-derived IFN-γ synthesis in the lung of post-septic mice is at least partly mediated by dysfunctional accessory cells.
So far, potential underlying mechanisms of a disturbed cross-talk of accessory cells and NK cells in the lung of post-septic mice are unknown. The finding that addition of recombinant IL-12 was able to increase the synthesis of IFN-γ in NK cells from CLP mice implies that an insufficient level of IL-12 contributed to the reduced activation of NK cells in post-septic mice (Figure 4C). However, IL-12 did not completely restore the IFN-γ production from NK cells of CLP mice (Figure 4C). It is unlikely that a reduced expression of the IL-12 receptor was responsible for the reduced responsiveness of NK cells to IL-12 after CLP as NK cells from sham and CLP mice did not differ significantly in the expression of this receptor (Figure 4E). We have previously shown that splenic DC reduce their expression of the IFN-γ receptor during sepsis and become insensitive to stimulation with IFN-γ. 21 Regarding the cross-talk of accessory cells and NK cells, IFN-γ plays an important role as it provides a positive feedback on accessory cells and further drives the IL-12 production and subsequent NK cell stimulation. However, alveolar macrophages and DC in the lung from CLP mice did not display a diminished expression of the IFN-γ receptor (Figure 4D). Therefore, it is unlikely that a reduced expression of the IFN-γ receptor contributed to the disturbed cross-talk between NK cells and accessory cells after CLP.
Moreover, the extent of NK cell-derived IFN-γ production depends on the balance between stimulatory and inhibitory signals in the microenvironment, like IL-12 and IL-10, respectively. Lung leukocytes of CLP mice released enhanced amounts of IL-10 (Figure 1B, C) upon stimulation with Pseudomonas in vitro and in vivo, which might inhibit NK cell activation. However, neutralization of IL-10 did not affect the IFN-γ production from leukocytes of CLP mice (Figure 4A) and argues against the relevance of IL-10 in P. aeruginosa-induced IFN-γ production. In addition, the enhanced synthesis of IFN-γ after addition of recombinant IL-12 to leukocytes from CLP mice did not change the production of IL-10 (Figure 4B). These findings suggest that the formation of P. aeruginosa-induced IL-10 and IFN-γ in the lung of post-septic mice was independently regulated. Considering the relevance of IFN-γ in efficient elimination of Pseudomonas the IL-10-independent formation of IFN-γ might explain why the neutralization of IL-10 prior to secondary infection with Pseudomonas does not improve the outcome. 28 However, we cannot exclude that other cytokines like TGF-β or IL-4 contributed to the suppressed stimulation of NK cells in the lung of post-septic mice in terms of IFN-γ production.11–13
Given that insufficient signals from accessory cells were responsible for the suppressed activity of NK cells during sepsis we expected a restoration of the IFN-γ levels when accessory cells in lung leukocytes from CLP mice were replaced by accessory cells from naive mice. However, the release of IFN-γ from CD11c-depleted leukocytes of CLP mice only slightly increased upon addition of naive accessory cells and, thus, remained inferior to the cytokine release of leukocytes from sham mice (Figure 5D). This finding suggests that the impaired capacity of lung leukocytes to produce IFN-γ was not only the consequence of a reduced activity of accessory cells, but additionally originated in a direct modulation of NK cells during sepsis. Souza-Fonseca-Guimaraes et al. 25 have recently shown that isolated splenic NK cells from septic mice are impaired in their IFN-γ secretion upon direct, accessory cell-independent stimulation with TLR agonists and cytokines. This sepsis-induced NK cell tolerance requires the presence of regulatory T cells during the development of sepsis. 25 As NK cells in the spleen and in the lung display diverse functions as mentioned above, it remains to be elucidated whether the unresponsiveness of NK cells in the lung that we show here similarly depended on the influence of regulatory T cells during the onset of sepsis. 24 Moreover, IL-10 and TGF-β seem to be required for the development of NK cell tolerance or unresponsiveness, but, once established, NK cell unresponsiveness was maintained independently from IL-10, as discussed above (Figure 4A).25,29 NK cells in the spleen of septic mice express reduced levels of the receptor for IL-18 that acts synergistically with IL-12 on NK cells to release IFN-γ. 30 The reduced IL-18 receptor expression might render NK cells in the septic host less sensitive for stimulatory signals. Furthermore, modulation of the IL-12 receptor signaling and subsequent activation of STAT4, for example by suppressor of cytokine signaling, or by the formation of inhibitory microRNA might be involved in the unresponsiveness of NK cells in the lung during sepsis.31–33 However, so far there exists no data on intracellular signaling pathways in NK cells during sepsis.
The suppressed capacity of NK cells to release IFN-γ (Figure 3B) seems to contradict the state of activation that is mirrored by the increased expression of CD69 (Figure 3D). The secretion of IFN-γ from activated NK cells is a transient process. Thereafter, NK cells acquire a state termed “exhaustion” that is characterized by a reduced sensitivity to IL-12 and impaired IFN-γ synthesis. 34 NK cells are activated during the early phase of sepsis and migrate into the peritoneal cavity where they promote hyperinflammation through activation of myeloid cells. 35 Activation of NK cells has also been observed in the blood of septic patients. 36 We observed that NK cells in the lung of CLP mice, in addition to the enhanced expression of CD69, weakly responded to IL-12 (Figure 4C) and therefore resembled the above-mentioned ‘exhausted’ NK cells. Thus, we assume that NK cell exhaustion after previous activation might contribute to the development of unresponsiveness that we observed for NK cells in the lung of post-septic mice.
In summary, the present study shows that NK cells in the lungs of post-septic mice are impaired in the production of IFN-γ upon secondary infection with P. aeruginosa in vitro and in vivo. The unresponsiveness of NK cells seemed to be mediated by a disturbed interaction with accessory cells and by a reduced sensitivity to IL-12 possibly as consequence of previous stimulation. The inhibition of NK cell function during polymicrobial sepsis might be responsible for the increased susceptibility of the septic host to opportunistic infections, for example with P. aeruginosa.
Footnotes
Acknowledgements
We are grateful to Yang Zhang for providing inactivated P. aeruginosa bacteria, and to Michaela Bak and Marion Frisch for excellent technical assistance. We cordially thank Wiebke Hansen for critically reading the manuscript.
Funding
The study was supported by the Deutsche Forschungsgemeinschaft (DFG, GRK1045 and FL-391/3-1 to S.B.F.).
Conflict of interest
The authors do not have any potential conflicts of interest to declare.