
Abstract
Sepsis is a serious clinical condition of excessive systemic immune response to microbial infection. The pro-inflammatory stage of sepsis is generally launched by innate cells such as macrophages. They release inflammatory cytokines, activate other immune cells and cause severe tissue/organ damage. In this study, we have revealed that recombinant Trichinella spiralis (TS) excretory–secretory protein (rTsP53) exhibited anti-inflammatory properties and rescued mice from LPS-induced endotoxemia, which is a common model for sepsis study, potentially through the induction of M2 macrophages. rTsP53 treatment significantly decreased inflammatory cytokines (IL-6, IFN-γ and TNF-α) and increased IL-4, IL-10, IL-13 and TGF-β secretion, both in circulation and in tissues. rTsP53 also induced the activation and infiltration of F4/80+CD163+ macrophages to inflammatory tissues, increased M2 macrophage-related Arg1 and Fizz1 expression, and decreased M1 macrophage-related iNOS expression. PCR array showed that rTsP53 activated several genes that involve the survival of macrophages and also anti-inflammatory genes such as SOCS3. Together, our results show that rTsP53 activates M2 macrophages, which has strong anti-inflammatory potential to prevent LPS-induced lethal sepsis.
Introduction
Sepsis is a serious clinical condition of excessive systemic immune response to microbial infection. Despite substantial advances in medical science and therapeutic agents, sepsis remains one of the most important health problems worldwide. This is partially because currently no reliable host or pathogen biomarkers exist with which to prevent the progress of sepsis. 1 In Western countries, the incidence of sepsis in intensive care units ranges from 10% to 73%; mortality is about 20–30%.2,3 The progression of sepsis can be roughly categorized into two distinct but concomitant stages, termed systemic inflammatory response syndrome (SIRS) and compensatory anti-inflammatory response syndrome. 4 SIRS is initiated by innate immune cells such as macrophages, which release inflammatory cytokines upon the detection of pathogens or activation by cytokines, to mobilize host immune system for further combat. This stage is also referred to as the cytokine storm and is thought to be responsible for lethal collateral organ damage during the early stage of sepsis. Modulating septic inflammatory responses is therefore the focus of current clinical therapies, including selective neutralization of inflammatory mediators by Abs, the administration of cytokines that modulate immune responses, the administration of anticoagulant molecules, and so on. 5
The activation of macrophages is generally categorized into at least two activation states, based on function. The activation of macrophages (classical activation) was at first described as enhanced microbicidal activity to BCG and Listeria in the 1960s; 6 later, it was linked to IFN-γ and LPS exposure.7,8 Upon activation, macrophages express iNOS and make nitrogen and oxygen intermediates, acquiring strong cytotoxicity. They also increase the secretion of inflammatory cytokines such as IL-1β, IL-12, IL-23 and TNF-α. By consequence, M1 macrophages are the key mediators of inflammation and Th1 responses, and are also the major tissue destroyer in inflammatory diseases. 9 In contrast, Th2 cytokines IL-4 and IL-13 have been shown to induce alternative activation of macrophages (M2 macrophages). They selectively induce the expression of Man receptor CD206 and the secretion of the suppressive cytokines IL-10 and TGF-β, without dampening (albeit restricting) the expression of MHC II molecules.8,10 M2 macrophages are less efficient APCs, secrete low levels of IL-12 and high levels of suppressive cytokines; consequently, they inhibit inflammation. They also promote cell division through the production of polyamines, and, as a result, are considered as tissue repairing/remodeling cells. 9 Several other stimuli have also been shown to induce the M2-like phonotype; thus, sub-groups of M2 macrophages are classified based on the stimuli, termed M2a (activated by Th2 cytokines), M2b (immunocomplex or TLR ligand) and M2c (IL-10 or glucocorticoids) macrophages. 11 Currently, no reliable markers can distinguish M1 and M2 macrophages; however, gene expression-based studies suggest that iNOS is strongly associated with M1 macrophages, while arginase 1 (Arg1), Fizz1 (also known as RELMα) and Ym1/2 are more associated with M2 macrophages. 12 It has to be noted that the activation states of macrophages do not represent end points of macrophage differentiation. The M1/M2 macrophage is highly flexible and reversible both in vivo and in vitro. 9 Repolarizing macrophages is therefore a potential therapeutic strategy for diseases. 13
Trichinella spiralis is a nematode parasite that infects the gastrointestinal tract. The parasite modulates the host immune system through muscle larvae excretory–secretory products (ES protein), suppress immune responses and sometimes remain asymptomatic during infection. 14 Our previous study has demonstrated that recombinant T. spiralis 53-kDa protein (rTsP53) exhibited a protective effect on experimental ulcerative colitis, potentially through the induction of Th2 responses and M2 macrophages. 15 Although rTsP53 possesses a strong immune-modulating property, whether it is protective with regard to sepsis has not yet been addressed. In the present study, we investigated the anti-inflammatory property of rTsP53 in LPS-induced endotoxemia in mice model and the underlying signaling pathways of rTsP53 regulating M1/M2 macrophage polarization.
Materials and methods
Extraction, purification and identification of rTsP53
The methods of purification and identification of rTsP53 have been described elsewhere. 15 Briefly, total RNA was extracted from T. spiralis larvae using TRIzol (Life Technologies, Carlsbad, CA, USA) and reverse transcribed to cDNA by using SuperScript III Reverse Transcriptase kit according to the manufacturer’s instructions (ThermoFisher Scientific, Waltham, MA, USA). The PCR product of TsP53 was digested by restriction enzymes, inserted into pET-28a (+) plasmids and transformed into Escherichia coli. The recombinant TsP53 was further induced by 1.0 mM IPTG at 28℃, then extracted and purified with BugBuster Ni-NTA His-Bind Purification Kit (EMD Millipore, Billerica, MA, USA). Endotoxin was removed using Pierce High Capacity Endotoxin Removal Resin (ThermoFisher Scientific), resulting in levels < 0.1 endotoxin units/ml as indicated by the Pierce LAL Chromogenic Endotoxin Quantitation Kit (ThermoFisher Scientific), following the manufacturer’s protocol. The concentration of rTsP53 was determined by a Bradford assay. The expression and purity of rTsP53 were analyzed by 12.0% SDS-PAGE and identified by Western blotting using the serum of rTsP53-immunized rats.
Animal endotoxemia model
Male BALB/c mice aged 8–10 wk were purchased from the experimental animal center of Sun Yat-sen University. Experiments followed the protocols approved by the Animal Ethics Committee of Sun Yat-sen University. Mice were housed in ventilated cages with wire net floors in a controlled room and were fed with a normal laboratory diet. Mice were deprived of food 24 h prior to the induction of endotoxemia but were allowed to access tap water. To induce endotoxemia, mice were injected i.p. with 15 mg/kg LPS [from E. coli 055:B5 ultrapure (Invivogen, San Diego, CA, USA) and was prepared according to the manufacturer’s instructions].
Survival rate monitoring
One-hour post-endotoxemia induction, mice were left untreated or injected i.p. with PBS, 1 mg/kg, 5 mg/kg or 10 mg/kg rTsP53 (n = 20 per group). Survival rates of mice were monitored for 72 h. Survival curves are representative of the day on which mice were found dead. GraphPad Prism software (GraphPad Inc., La Jolla, CA, USA) was used to generate the Kaplan–Meier curves and to calculate statistical significance.
Blood and tissue sample preparation
One h post-endotoxemia induction, mice were left untreated or injected i.p. with PBS, 1 mg/kg, 5 mg/kg or 10 mg/kg rTsP53. Surviving mice were sacrificed 12, 24, 48 and 72 h later (n > 5 for each group at each time point). Whole-blood samples were collected and allowed to clot for 30 min. Clots were removed by centrifuge and sera were used for ELISA. Cytokines were determined by platinum ELISA kit (eBioscience, San Diego, CA, USA) according to manufacturer’s instructions. Lung and liver tissues were surgically removed, homogenized by using a DEPC water-treated syringe and treated with Trizol (100 mg tissue/ml) for at least 5 min. RNAs were extracted by TRIzol according to the manufacturer’s protocol for the PCR analysis.
Hematoxylin and eosin staining
One post-endotoxemia induction, mice were injected i.p. with PBS, 1 mg/kg, 5 mg/kg or 10 mg/kg rTsP53. Surviving mice were sacrificed 72 h later (n > 5 for each group) and liver and lung tissues were surgically removed, fixed in 10% neutral-buffered formalin, embedded in paraffin, and sectioned and stained with hematoxylin and eosin. Slides were then examined under a microscope for pathological analysis.
Real-time RT-PCR
Primers used for quantitative RT-PCR in this study.

Immunohistochemistry staining
One post-endotoxemia induction, mice were injected i.p. with PBS or 5 mg/kg rTsP53. Surviving mice were sacrificed 72 h later (n > 5 for each group), liver and lung tissues were fixed in buffered 4% formalin solution (pH 7.4) and embedded in paraffin. All slides were dewaxed and rehydrated using xylene and graded alcohols, followed by incubation in 3% H2O2 solution for 10 min to quench the endogenous peroxidase activity. The tissue section was then heated by a microwave oven to repair the Ag. Non-specific binding was blocked with normal goat serum. The slides were incubated with the diluted (1:50) primary Abs [rat anti-mouse F4/80 Ab (Abcam, Cambridge, MA, USA); rabbit anti-mouse HLA-DR Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or rabbit anti-mouse CD163 Ab (Santa Cruz Biotechnologies)] at 4℃ for 16 h. The biotinylated secondary IgG Ab [goat anti-rat Cy3-conjugated Ab (Abcam); goat anti-rabbit FITC-conjugated Ab (Abcam)] was then added to the tissue section and incubated at 37℃ for 30 min. Next, DAPI staining was applied to the nucleus. Finally, the tissue sections were dried and sealed by a fluorescence decay resistance agent with a cover slide. PBS was applied as a negative control.
Peritoneal macrophage preparation and flow cytometry assay
One h post-endotoxemia induction, mice were injected i.p. with PBS or 5 mg/kg rTsP53. Surviving mice were sacrificed 48 h later (n > 5 for each group), peritoneal exudate cells were obtained by lavage of the peritoneal cavity. The fluid lavage was centrifuged (250 g, 5 min, 4℃). Cells were adjusted to 2 × 106 cells/ml with DMEM medium supplemented with 10% FBS. Cells were cultured for 4 h at 37℃ in 5% CO2. Non-adherent cells were removed and fresh medium was added in, and cells were cultured for another 6 h. Macrophages were then detached by pipetting the culture medium across wells. Single-cell suspensions were incubated with anti-mouse CD16/32 mAb (BioLegend, San Diego, CA, USA) and stained with allophycocyanin/cyanine7 (APC/Cy7) conjugated anti-mouse F4/80 mAb; FITC-conjugated anti-mouse CD206 mAb; phycoerythrin-conjugated anti-mouse/human CD11b mAb (all from BioLegend) and FITC-conjugated rabbit anti-mouse CD163 Ab (Biosun, Beijing, China). Dead cells were excluded by propidium iodide staining. Samples were run by FACS Calibur flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA) and were analyzed by FACSDiva 6.1.3 software (BD Biosciences); single cells were gated during analysis.
Bone marrow-derived macrophage preparation
Bone marrow-derived macrophages (BMDM) were obtained as previously described, with minor modifications. 16 In brief, bone marrow cells were grown in DMEM containing 30% L929 cell-conditioned medium and 10% heat-inactivated FBS, incubated at 37℃ in 5% CO2 for 96 h. Non-adherent cells were then removed and fresh medium added for another 24 h. These cells were considered as M0 cells and were harvested with a cell scraper and washed with ice-cold culture medium. Cells were then cultured with a different concentration of rTsP53 as indicated. Twenty-four h post-culture, supernatants were harvested for ELISA assay with eBioscience’s platinum ELISA kit. Total RNAs of cultured cells were extracted by TRIzol, according to the manufacturer’s protocol.
PCR array
PCR array was performed by the Mouse Signal Transduction Pathway Finder PCR Array (Qiagen), according to the manufacturer’s instructions. For BMDM, cells were co-incubated with 5 µg/ml rTsP53 for 24 h and total RNA was extracted with TRIzol. For peritoneal macrophages, mice were injected with LPS and 5 mg/kg rTsP53 1 h later, as described. Forty-eight h post-LPS injection, surviving mice were sacrificed (n > 5). Peritoneal macrophages were prepared as described and total RNA was extracted by TRIzol. RNA was then converted to first-strand cDNA with PrimeScript II 1st Strand cDNA Synthesis Kit (TaKaRa). PCR array data were calculated by the comparative cycle threshold method, normalized against multiple housekeeping genes and expressed as the mean fold change in rTsP53-treated samples relative to the controlled samples.
Statistical analysis
All data are expressed as mean ± SEM. The difference in survival rate between each group was analyzed by Kaplan–Meier survival function analysis and the significance was determined by the log-rank test. Statistical changes in inflammatory factors and gene expression were analyzed by ANOVA with Dunnett’s test as a post-hoc procedure to compare multiple treatment conditions with controls where appropriate. Differences were considered significant when P < 0.05. All analyses were performed using IBM SPSS Version 20 (SPSS Statistics V20; IBM, Armonk, NY, USA).
Results
Effect of rTsP53 on survival rate and organ damage of LPS-induced endotoxemia in mice
We first established systemic inflammation by injecting BALB/c mice i.p. with 15 mg/kg LPS. As shown in Figure 1, this dose of LPS produced severe responses with a very low survival rate (20%). To evaluate the therapeutic effect of rTsP53, we injected i.p. different doses of rTsP53 1 h post-LPS injection. One injection of rTsP53 showed a very clear therapeutic effect and rescued mice from lethal endotoxemia in a dose-dependent manner. To our surprise, as low a dose as 1 mg/kg rTsP53 displayed a mild therapeutic effect, although this was not significant (40%). The survival rate increased to 80% at a dose of 5 mg/kg, while a dose of 10 mg/kg prevented all mice from lethal sepsis (100%). These data clearly demonstrate the therapeutic effect of rTsP53 in LPS-induced septic inflammation.
rTsP53 prevented lethality in mice with sepsis. Kaplan–Meier plot of mouse survival after different treatments for sepsis. BALB/c mice were injected i.p. with 15 mg/kg LPS to induce sepsis and separated into five groups (n = 20 each): untreated, PBS, 1 mg/kg rTsP53, 5 mg/kg rTsP53 or 10 mg/kg rTsP53. Significance of survival analyses was performed by log-rank test (*P < 0.05, **P < 0.001).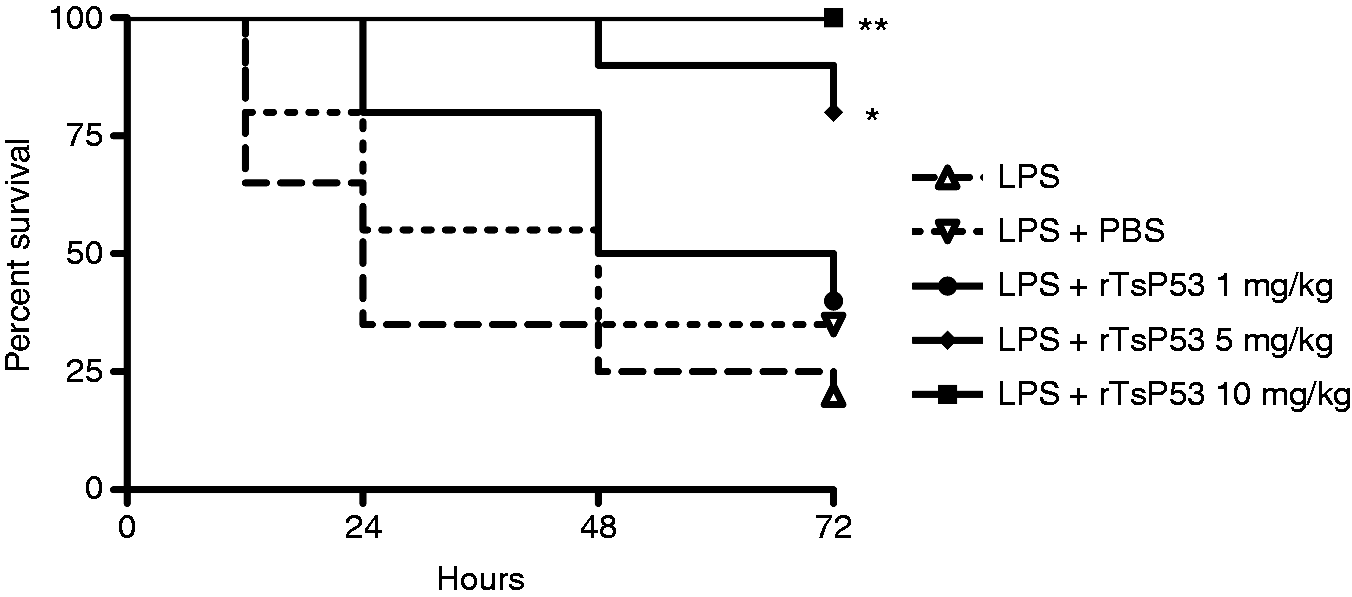
rTsP53 attenuated serum Th1 cytokines, and increased Th2 and regulatory cytokines in LPS-induced endotoxemia
One characteristic of sepsis is an increased circulating level of pro-inflammatory cytokines. To investigate whether rTsP53 rescued mice through the inhibition of systemic inflammation and cytokine storm, we examined the serum cytokine pattern of mice with or without rTsP53 treatment. We observed that as early as 12 h post-LPS injection, inflammatory cytokines (IFN-γ, IL-6 and TNF-α) were increased in septic mice, while Th2 cytokines (IL-4 and IL-13) and regulatory cytokines (IL-10 and TGF-β) remained low, and this pattern remained similar throughout the entire experimental period (until 72 h post-LPS; Figure 2 and data not shown). The injection of rTsP53 significantly decreased inflammatory cytokines (Figure 2a–c), and increased Th2 and regulatory cytokines in a dose-dependent manner (Figure 2d–g). At a dose of 10 mg/kg, all inflammatory cytokines dropped to about half to one-third of the cytokines detected in septic mice (Figure 2a–c), while Th2 and suppressive cytokines increased 3–6-fold more than those of septic mice (Figure 2d–g). These data indicate that rTsP53 rescued mice from lethal sepsis through the induction of suppressive cytokines and the inhibition of systemic inflammatory cytokines.
rTsP53-attenuated serum Th1 cytokines, increased Th2 and regulatory cytokines in LPS-induced septic mice. BALB/c mice were injected i.p. with LPS and PBS or rTsP53, as described in the ‘Materials and methods’. Naïve mice were used as control (–). Forty-eight h post-LPS injection, sera were collected (n > 5 for each group) and (a) IFN-γ, (b) IL-6, (c) TNF-α, (d) IL-4, (e) IL-13, (f) IL-10 and (g) TGF-β were analyzed by ELISA. Experiments were repeated three times with similar results (**P < 0.01 vs. LPS + PBS control).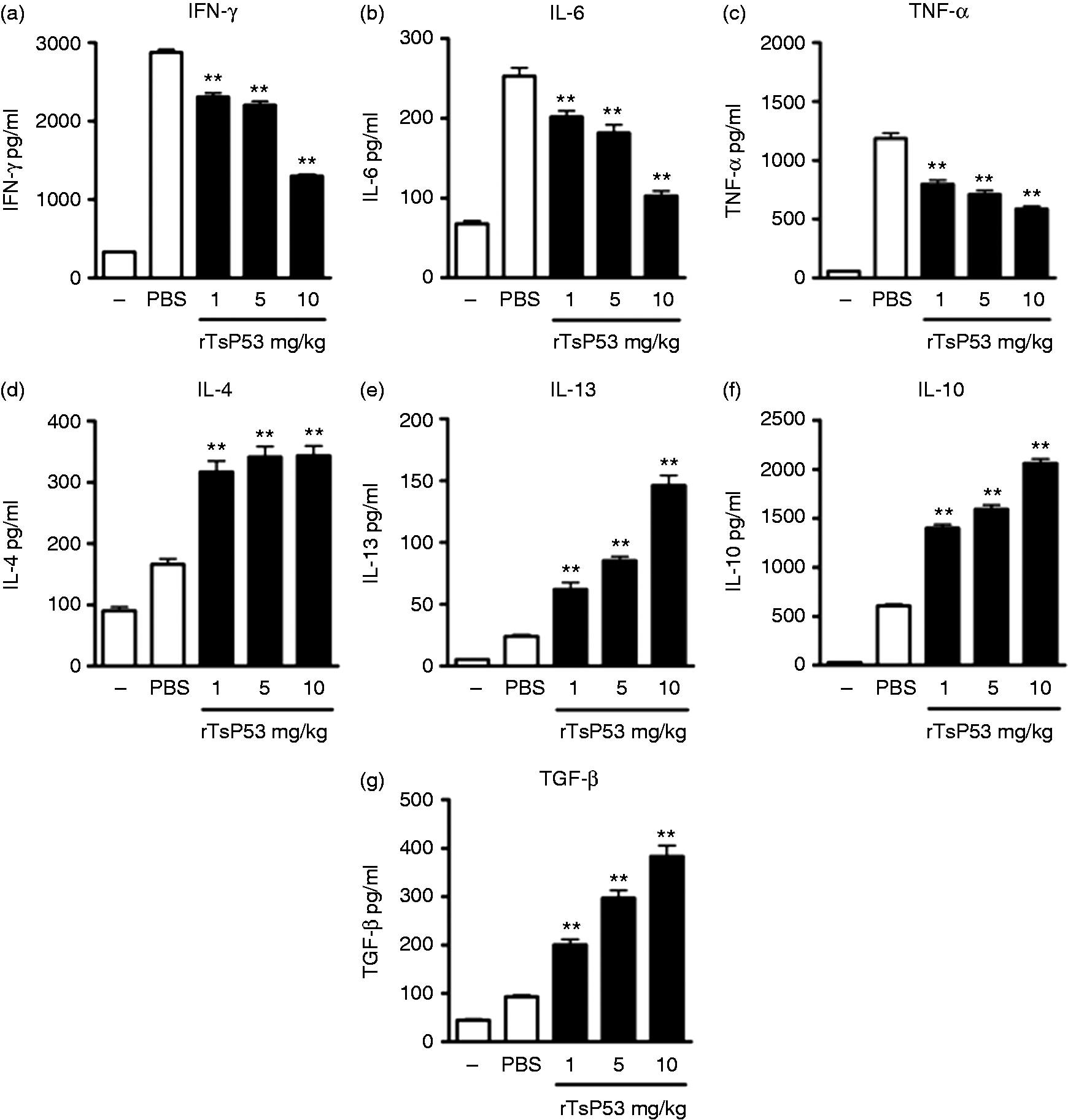
rTsP53 prevented tissue destruction of LPS-induced septic mice and attenuated tissue inflammation
As rTsP53 decreased systemic inflammatory cytokines, we next examined whether rTsP53 prevented tissue destruction caused by sepsis. We therefore performed liver and lung tissue pathological analysis after the induction of sepsis. Hematoxylin and eosin staining showed obvious pathologic damage in the liver and in the lung tissue of mice 72 h post-LPS injection (Figure 3; LPS + PBS vs. normal). A high amount of degeneration and necrosis was found in hepatic tissue of mice with endotoxemia, such as enlargement (Figure 3E), vacuolization or dikaryon (Figure 3D) in the cell nucleus, as well as congestion in the hepatic venous system (Figure 3C). Pulmonary slice also showed congestion, exudation, leukocyte infiltration (Figure 3I), and severe destruction of the alveolus structure. After rTsP53 intervention, significant pathological improvement could be observed in the liver and lung tissue, especially for a dose of 10 mg/kg. At this dose, all tissue structure was preserved, indicating that rTsP53 could remarkably prevent LPS-induced multiple organ damage.
rTsP53 prevented tissue destruction from LPS induced inflammation. BALB mice were injected i.p. with LPS and rTsP53, as described in the ‘Materials and methods’ (n > 5 for each group). Seventy-two h post-LPS injection, liver and lung tissues were collected for hematoxylin and eosin staining. Black arrow indicated pathological modification of tissue including dikaryon in cell nucleus (D), congestion (C), enlargement of cell nucleus (E) and necrosis of hepatocyte (N). White arrow indicates the congestion, exudation and leukocyte infiltration in alveolus (I).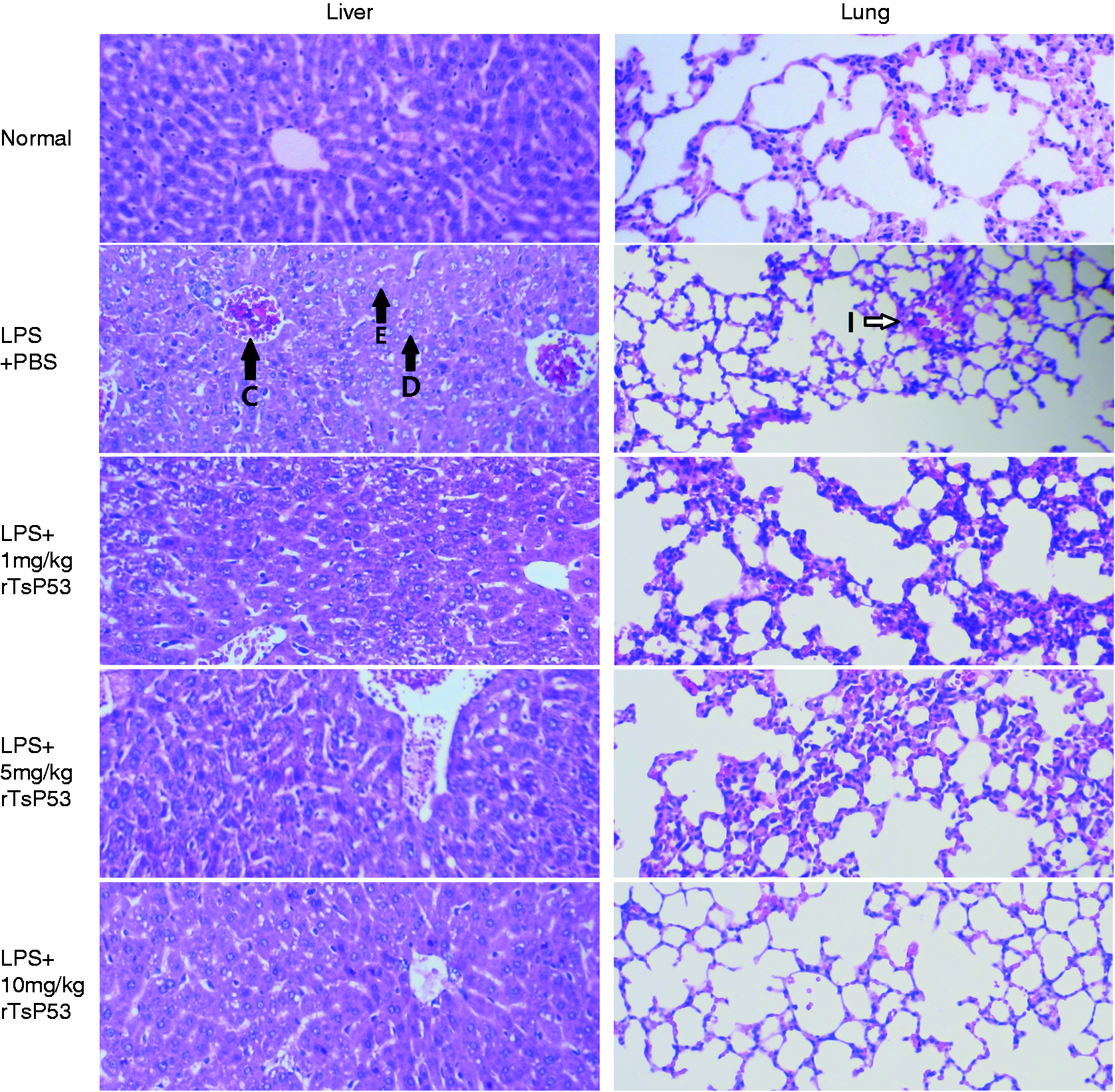
Using quantitative real-time PCR, we further examined the tissue cytokine pattern induced by rTsP53. We found that tissue cytokine expression profile was similar to that in peripheral sera (Figure 4a–d). Compared with LPS-treated mice, the mRNA levels of IL-6 and TNF-α were down-regulated in both liver and lung tissue of rTsP53-treated mice (Figure 4a, c), while TGF-β expression was significantly up-regulated (Figure 4b, d).
rTsP53 attenuated the inflammatory cytokines and increased M2 macrophages in tissues. BALB/c mice were injected i.p. with LPS and PBS or rTsP53, as described in the ‘Materials and methods’; naïve mice were used as control (–). Forty-eight h post-LPS injection, mRNAs of liver and lung tissues were collected (n > 5 for each group) and the expression of (a,c) IL-6, TNF-α, iNOS, (b,d) IL-10, TGF-β, Fizz1 and Arg1 were analyzed by RT-PCR and normalized to GAPDH expression. Experiments were repeated at least three times with similar results. (ns: not significant; *P < 0.05; **P < 0.01).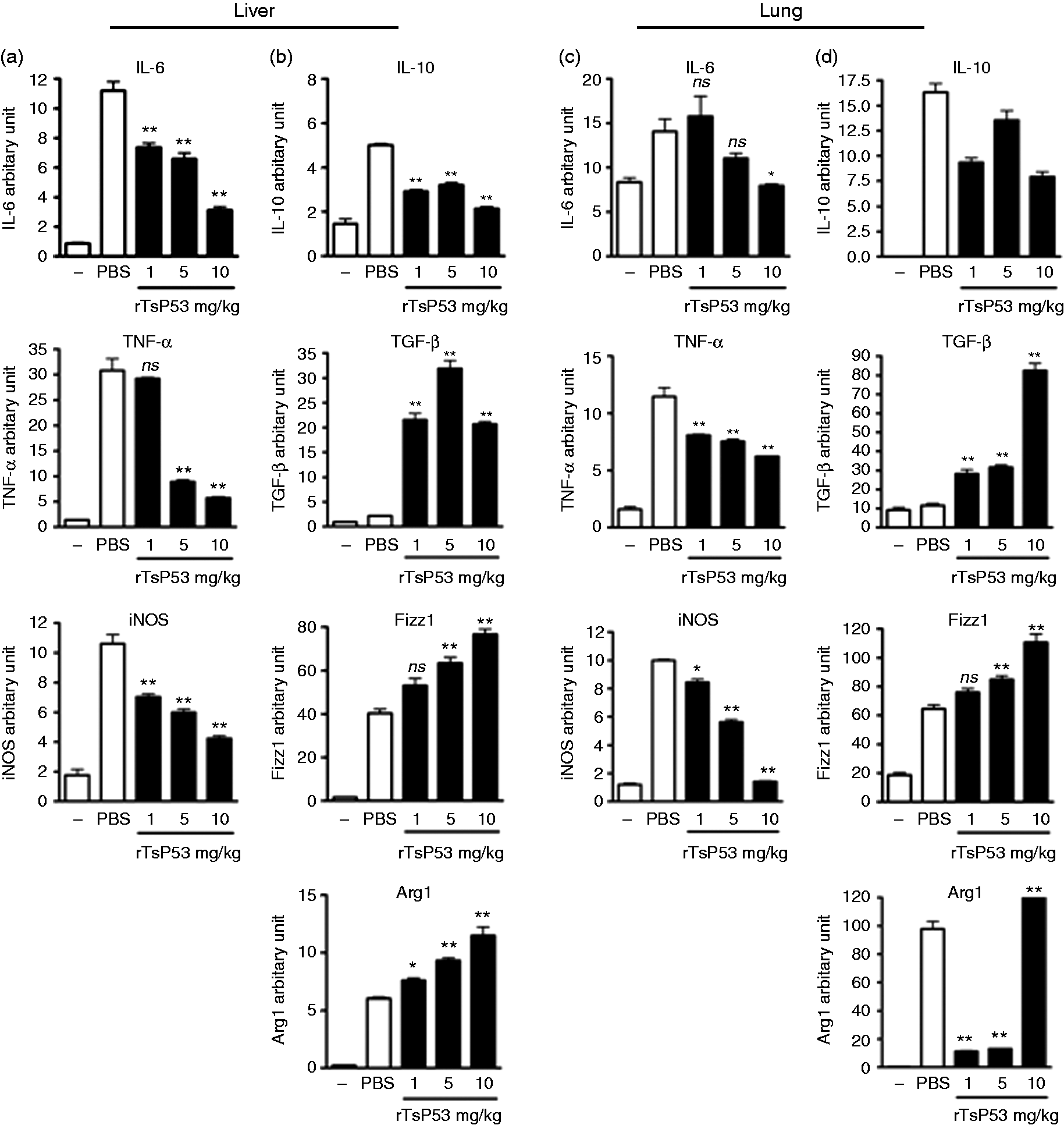
We have previously shown that rTsP53 potentially induces the activation of M2 macrophages and inhibits Th1 responses; 15 we therefore examined weather rTsP53 induced M2 macrophages in septic mice. As shown in Figure 4, rTsP53 treatment significantly down-regulated the expression of M1 macrophage-related iNOS in both liver and lung tissue, while the expression of M2 macrophage-related Arg1 and Fizz1 was increased. Taken together, these data strongly suggested that rTsP53 activated M2 macrophages and, by consequence, prevented severe tissue inflammation and destruction.
rTsP53 induced the activation and infiltration of M2 macrophages to inflammatory tissues
As the M2 macrophage-related gene was increased in rTsP53-treated tissues, we further performed a double immunohistochemistry tissue staining of liver and lung tissue 72 h post-LPS injection, to confirm the infiltration of M2 macrophages. As M2 macrophages express restricted levels of the MHC II molecule and higher levels of CD163,
17
we therefore chose these two molecules to distinguish between M1 and M2 macrophages. As shown in Figure 5, F4/80+MHC-II+ M1 macrophages were found in both lung and liver tissue of septic mice (Figure 5a), while F4/80+CD163+ M2 macrophages were rarely seen in the septic tissues (Figure 5b). Upon rTsP53 treatment, F4/80+MHC-II+ macrophages could still be seen in the tissues, while a substantial increase in F4/80+CD163+ staining was now perceived in tissues. These data clearly showed that rTsP53 induced activation and infiltration of M2 macrophages to inflammatory sites.
rTsP53 induced the activation and infiltration of M2 macrophage in tissues. BALB mice were injected i.p. with LPS and rTsP53, as described in the ‘Materials and methods’ (n > 5 for each group). Seventy-two h post-LPS injection, lung and liver tissues were collected for immunohistochemical staining. Representative histology is shown. Tissues were stained with (a) F4/80 (red) plus MHC-II (green) and DAPI (blue), or (b) F4/80 (red), CD163 (green) and DAPI (blue).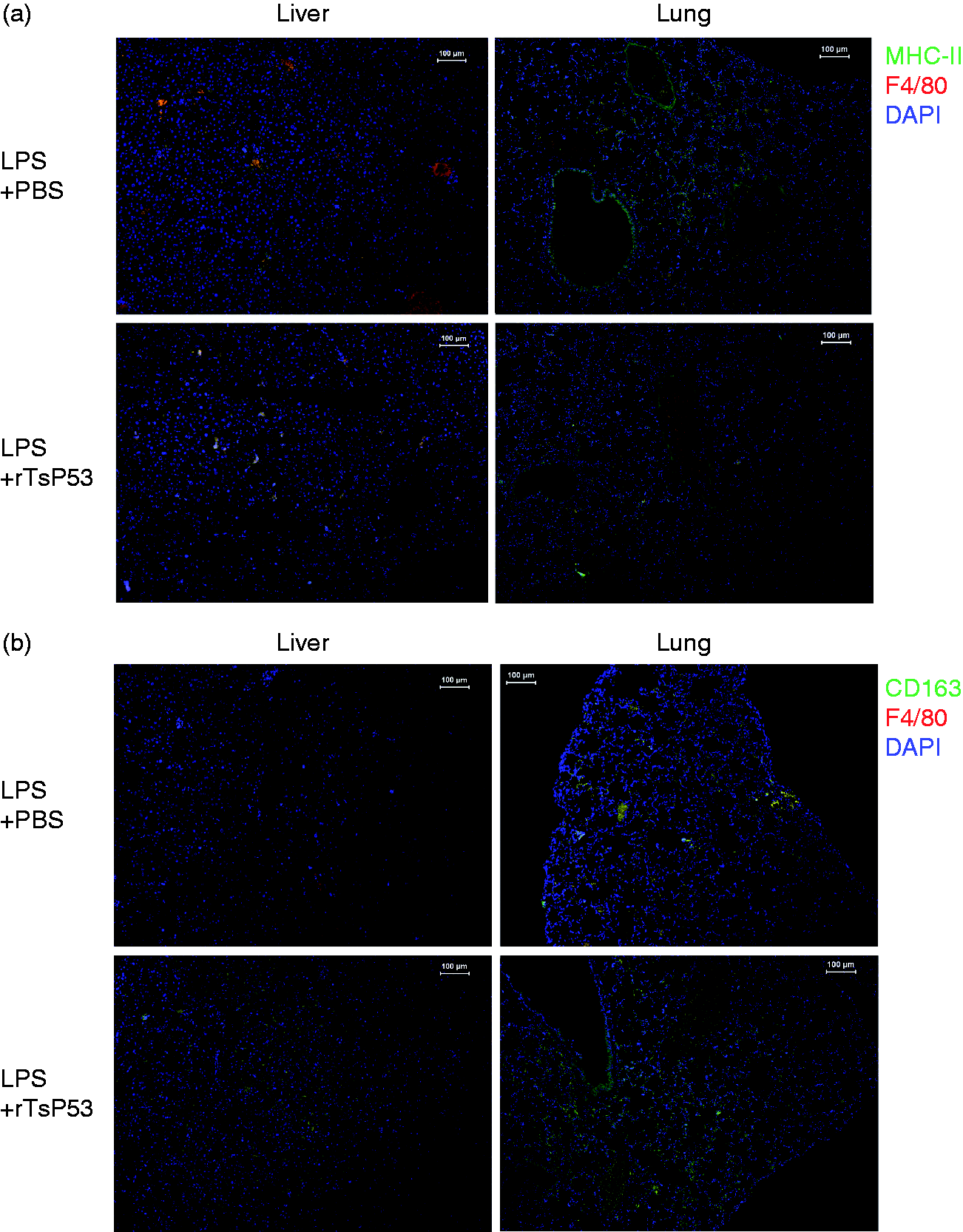
Flow cytometric and genetic analysis of rTsP53 activated M2 macrophages
Flow cytometry analysis of peritoneal macrophages showed very similar results. Forty-eight h post-sepsis induction, we compared the peritoneal macrophage activation states between mice that were treated or not with rTsP53. As shown in Figure 6, upon 5 mg/kg rTsP53 treatment, the percentage of M2 macrophage increased dramatically (identified by CD163+ and CD206+), while the percentage of F4/80+CCR7+ M1 macrophages decreased significantly.
rTsP53 activated peritoneal macrophages to M2 macrophages. BALB mice were injected i.p. with LPS and treated with 5 mg/kg rTsP53 or PBS. Forty-eight h post-LPS injection, peritoneal macrophages were analyzed by flow cytometry for the expression of CD206, CD163 and CCR7. Representative dot plots are shown. Numbers in the plots represent the mean ± SEM of the results of at least three mice.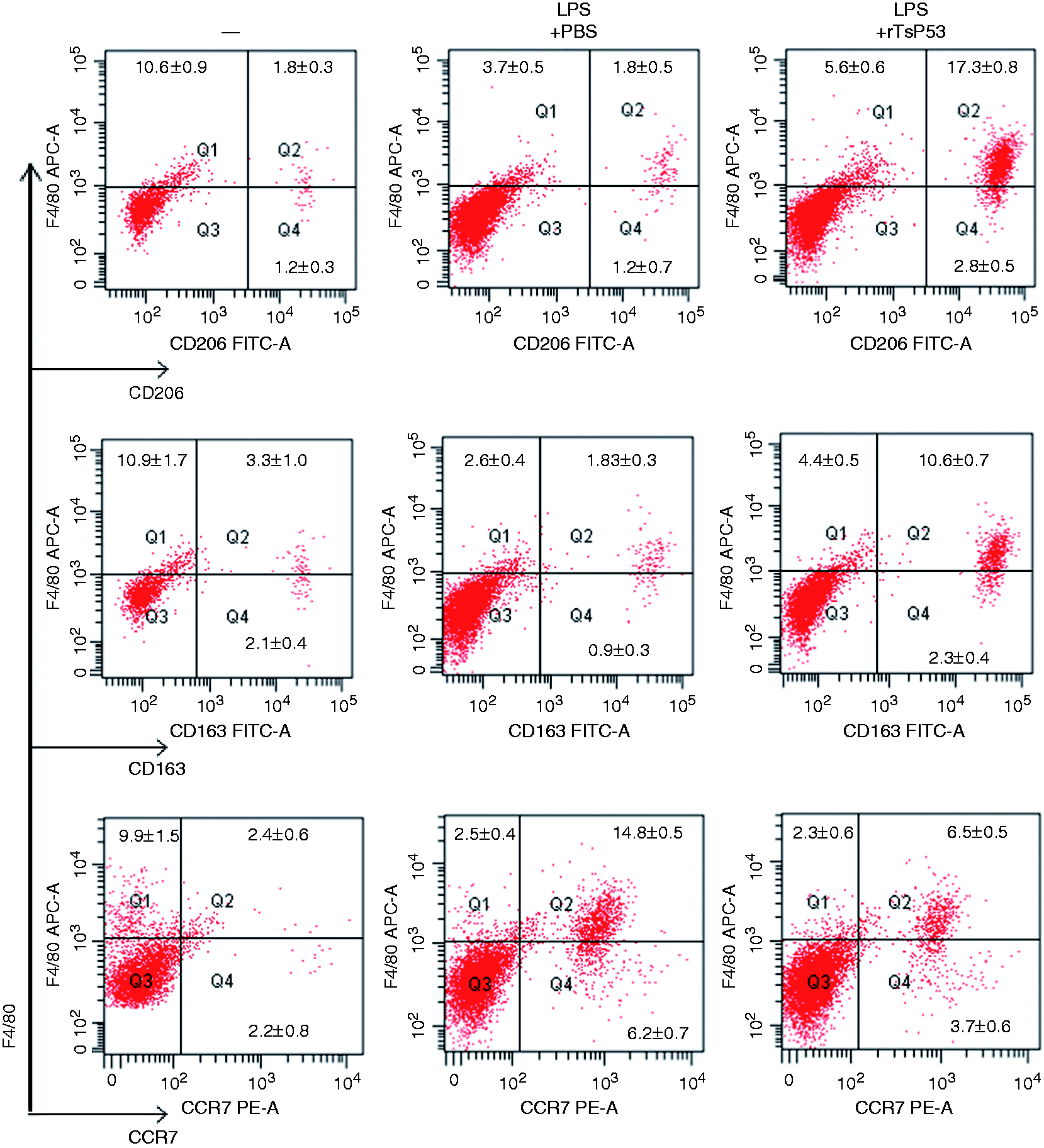
PCR array analysis of rTsP53-treated peritoneal macrophages.
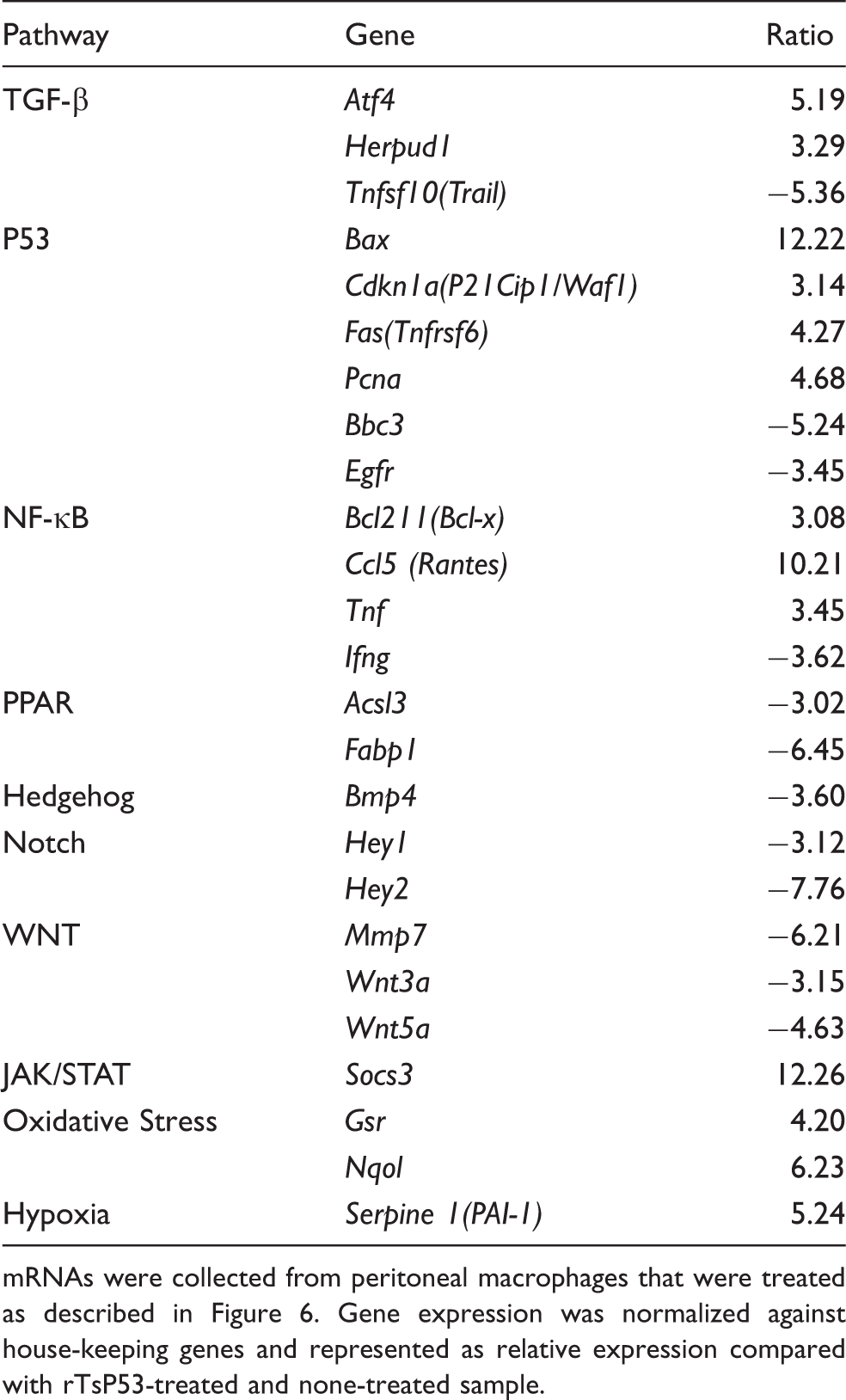
mRNAs were collected from peritoneal macrophages that were treated as described in Figure 6. Gene expression was normalized against house-keeping genes and represented as relative expression compared with rTsP53-treated and none-treated sample.
rTsP53 polarized BMDM to M2 phenotype and induced gene expression
To further investigate the impact of rTsP53 on un-stimulated macrophages, BMDMs were extracted and cultured in the presence or absence of rTsP53. We have found that treatment of rTsP53 inhibited the production of IL-6 and TNF-α (Figure 7a, b) and increased the production of IL-4 and IL-13 in a dose-dependent manner (Figure 7d, e). The mRNA level of iNOS was clearly down-regulated after rTsP53 exposure in a dose-dependent manner (Figure 7c), while Fizz1 and Arg1 expression was up-regulated (Figure 7f, g).
rTsP53 polarized bone-marrow derived macrophages to the M2 phenotype. In vitro (a) TNF-α, (b) IL-6, (d) IL-4 and (e) IL-13 from rTsP53-treated BMDM culture were analyzed by ELISA at 48 h. The expression of (c) iNOS, (f) Fizz1 and (g) Arg1 was analyzed by RT-PCR. Data are representative of at least three independent experiments (ns: not significant; *P < 0.05; **P < 0.01).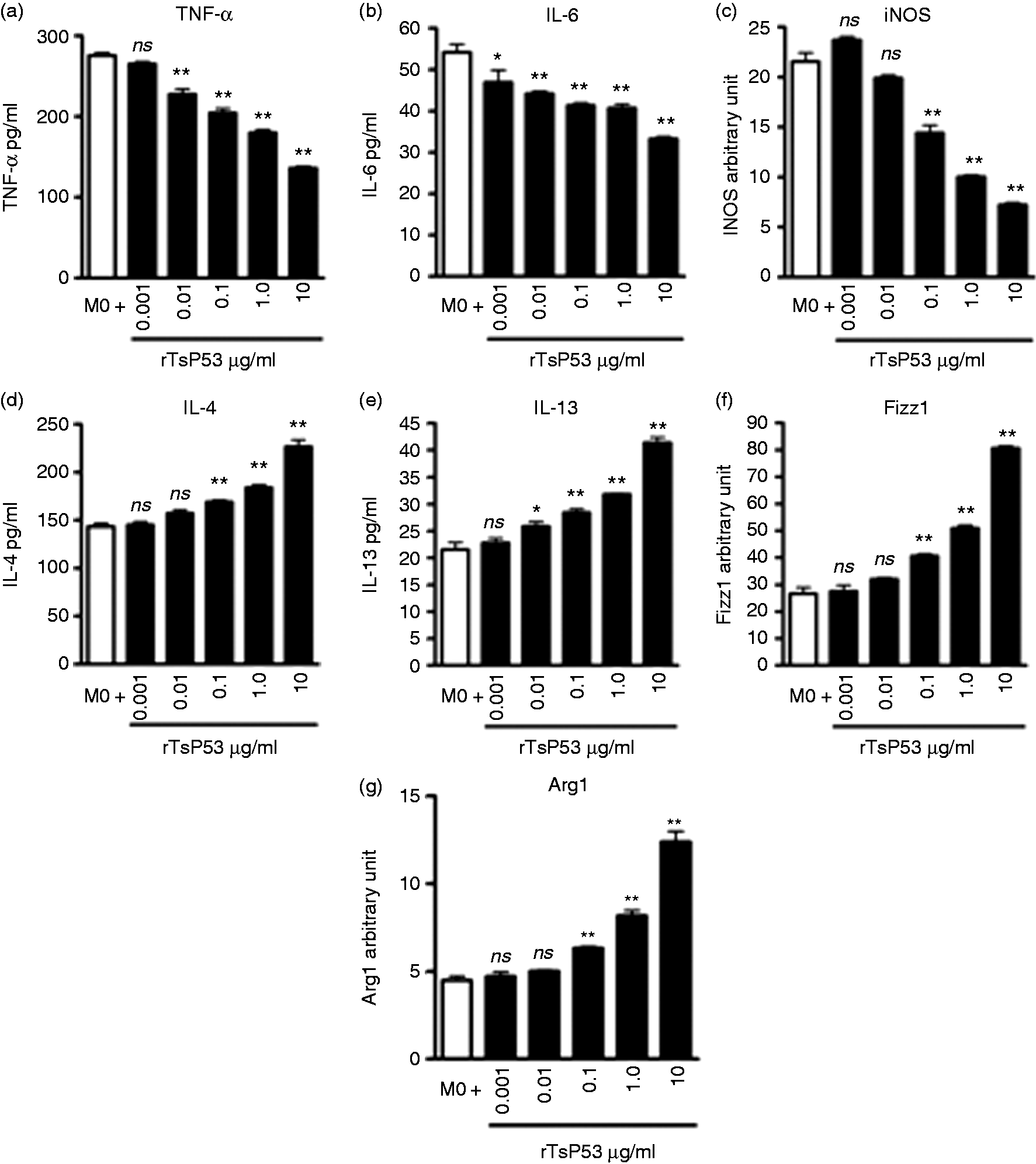
PCR array analysis of rTsP53-treated BMDM.
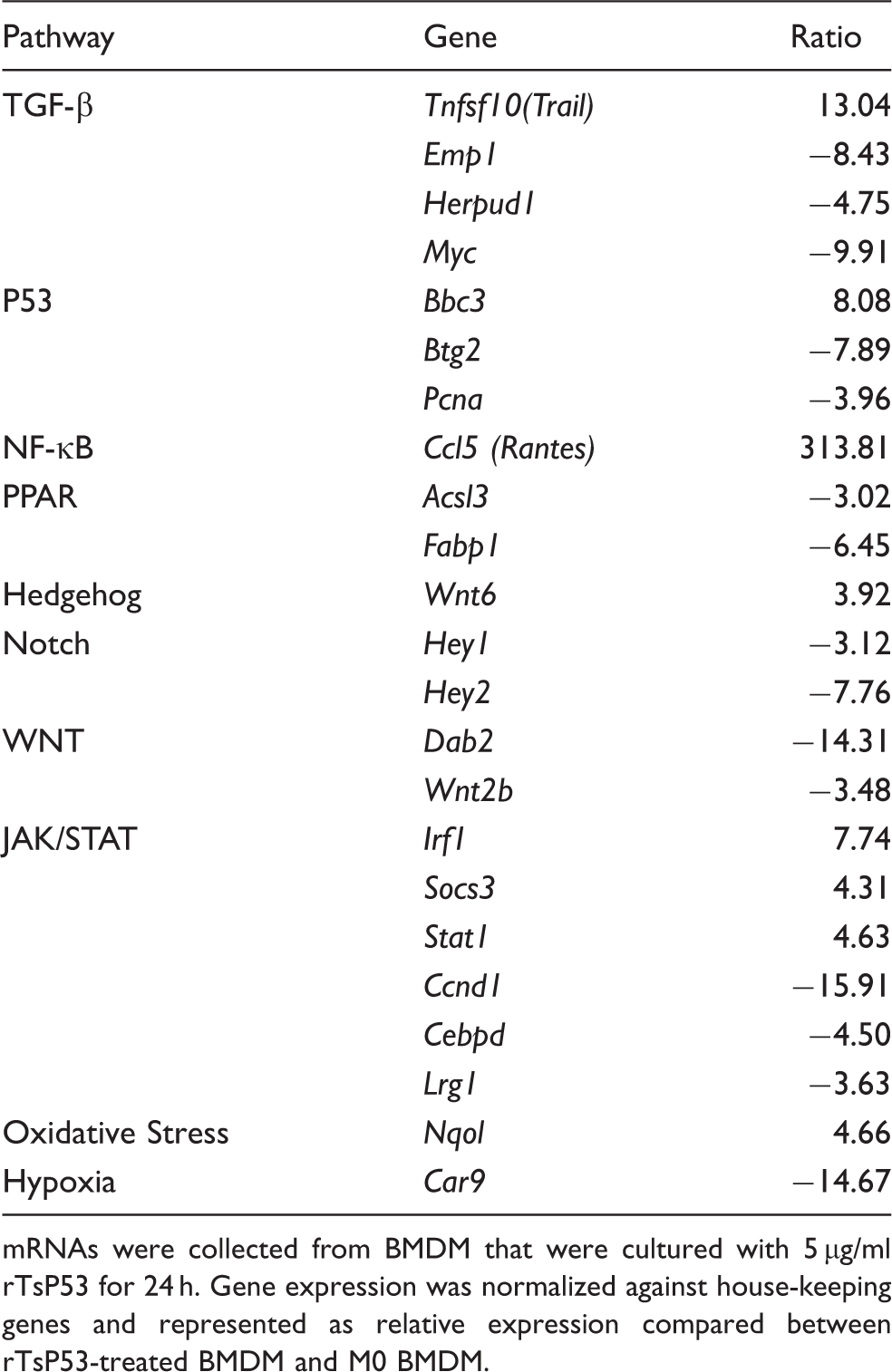
mRNAs were collected from BMDM that were cultured with 5 µg/ml rTsP53 for 24 h. Gene expression was normalized against house-keeping genes and represented as relative expression compared between rTsP53-treated BMDM and M0 BMDM.
Discussion
The data presented herein indicate that rTsP53 regulates immune responses and possesses protective activity in vivo. The present study demonstrated that rTsP53 changed the macrophage activation states via the decrease of pro-inflammatory cytokines (IL-6 and TNF-α), and via an induction of anti-inflammatory cytokines (IL-10 and TGF-β). Moreover, rTsP53 improved LPS-induced multiple organs damage via activating JAK/STAT and oxidative stress signal pathways, leading to enhanced M2 polarization in vivo and survival of animals.
Sepsis is generally caused by pathogens and PAMPs such as LPS. The severe form of sepsis includes severe shock and multiple organ failure, and is often lethal. Although treatment has improved in the last decade, the incidence and the mortality of sepsis remain high even in modern countries.2,3 The complex inflammatory responses observed in sepsis involve dynamic equilibrium amongst a number of opposite molecular mechanisms. 18 A previous study has shown that rTsP53 could effectively attenuate the tissue damage during colonic inflammation, showing its ability to prevent colitis. 15 In the present study, we demonstrated that rTsP53 could also play a significantly protective role in an LPS-induced septic mice model. There was a decrease of systemic inflammatory cytokines, significant pathological improvement in the liver and lung tissue of endotoxemia mice after rTsP53 intervention, including reduced multiple degeneration and necrosis in hepatic cells, congestion in hepatic venous system and exudative destruction of lung tissue, indicating that rTsP53 could remarkably improve LPS-induced multiple organ damage. The most remarkable point is that rTsP53 could clearly improve the survival rate of endotoxemia BALB/c mice, and therefore has clinical therapeutic potential for early-stage sepsis. Recently, another study has demonstrated that secretory product could protect mice from sepsis, potentially through mannose receptor CD206 and through the blocking of the MyD88 signal pathway. 19 In line with that study, we further demonstrate that the anti-inflammatory property of rTsP53 is through the induction of M2 macrophages.
Current theory holds that dysregulation in cytokine networking is considered to play a fundamental role in mortality and morbidity in sepsis. Macrophages become activated in response to signals present in damaged organs, or associated with pathogens, which can acquire phenotypes with distinct function under different microenvironments. M1 macrophages refer to classically activated pro-inflammatory macrophages, while M2 macrophages stand for alternatively activated macrophages. Generally, M1 macrophages express high levels of IL-1β, IL-6, IL-12, IL-23, TNF-α and iNOS, and produce low levels of IL-10. In contrast, M2 macrophages induce high levels of IL-10 and low levels of IL-12 and IL-23, and up-regulate the expression of scavenger receptor CD163, Man receptors CD206 and Fizz1.17,20 As early-stage sepsis is the result of systemic inflammatory responses, it is generally considered as an M1-associated disease. 21 This is supported by our data, which demonstrate that LPS increases both systemic and local IL-6 and TNF-α level, increases CCR7+ macrophages and infiltrates MHC-II+ M1 macrophages in tissues. It has to be noted that as soon as 24 h post-LPS treatment, the increases in inflammatory cytokines and tissue damage were observed, and we therefore excluded the involvement of adaptive immune responses. The inflammation and tissue damage were therefore, in principle, caused by innate responses. The treatment of rTsP53 suppressed the elevation of pro-inflammatory cytokines, while the expression of anti-inflammatory cytokines was boosted. Moreover, we also observed that CD206+ and CD163+ macrophages were increased and infiltrated to inflammatory tissues upon rTsP53 treatment, accompanied by a decrease in tissue damage. However, abundant MHC-II+ macrophages could still be observed in liver and lung tissue after rTsP53 treatment. It might be that M2 macrophages expressed restricted but still substantial class II molecules, and therefore the staining did not discriminate M1 macrophages specifically. 8 Currently, no reliable surface marker distinguishes between M1 and M2 macrophages; thus, we examined the expression of more representative markers such as iNOS and Fizz1. We have found that iNOS was significantly down-regulated in rTsP53-present mice, while Fizz1 was up-regulated. Taken together, these data suggest that rTsP53 stimulated M2 polarization, increased Th2-like and regulatory cytokines, and attenuated Th1 cytokines tissue inflammation.
To establish novel therapies for sepsis, it is important to understand the mechanisms that regulate macrophage activation. Although it is clear that rTsP53 activates macrophage and induces the secretion of suppressive cytokines, the signaling transduction pathways involved are not yet established. It has been suggested that rTsP53 could activate STAT6 in M2 macrophages. 22 We performed a Mouse Signal Transduction Pathway Finder PCR Array assay in peritoneal macrophages and BMDM to reveal more potential mechanisms. We found that there were increased expressions of several genes upon rTsP53 treatment, such as Bax, Ccl5 and Socs3. BAX is a pro-apoptotic protein of the Bcl-2 family. It has been shown that bacteria could induce the apoptosis of macrophages through the activation of BAX. 23 It is interesting that the same pattern was not observed in BMDM cultured with rTsP53. One explanation is that, as has been suggested previously, the increase in M2 macrophages is the consequence of the decrease in M1 macrophages. 24 Therefore, it is possible that mixed M1 and M2 macrophages were obtained from endotoxemic mice, while treatment of rTsP53 induced the apoptosis of M1 macrophages. As BMDM were not stimulated by LPS and did not fully induce M1 activation, the apoptosis (and the expression of Bax) was not observed under the treatment of rTsP53. Further experiment will be needed to address this hypothesis. At the same time, we observed an increase in CCL5 in both peritoneal macrophage and BMDM that were treated with rTsP53. CCL5 is a chemokine that recruits blood mononuclear cell through interaction with CCR1, CCR3 and CCR5. However, it has also been shown to promote the survival of macrophages under certain circumstances. 25 Therefore, rTsP53 could support the survival of M2 macrophage through the induction of CCL5. Another interesting remark is that both peritoneal macrophages and BMDM increase the expression of SOCS3 upon treatment with rTsP53. SOCS3 is an important feedback inhibitor of the JAK/STAT signaling pathway, 26 while the JAK/STAT pathway is the most critical pathway for the decision of M1/M2 activation under the stimulation of different cytokines. 27 For example, SOCS3 has been shown to bind directly to JAK1, JAK2 and TYK2, serving as a noncompetitive tyrosine kinase inhibitor, 28 and play important roles in the prevention of neuroinflammation and suppression of Th1 and Th17 cells. 29 Most importantly, it has been shown that the deficiency of SOCS3 promotes M1 macrophage activation and inflammation, 30 while the delivery of SOCS3 through liposomes could protect mice from lethal endotoxic shock. 31 Our preliminary data have also shown that the expression of CD206 is perturbed in SOCS3 KO macrophages upon treatment with rTsP53 (manuscript in preparation). Taken together, these data clearly demonstrate that rTsP53 prevents inflammation through the induction of SOCS3 in M2 macrophages.
In summary, we have shown that rTsP53 functions as a modulator of macrophage polarization, and possesses potential anti-inflammatory and protective activity on early-stage sepsis induced by LPS. The M1/M2 balance has been showed to take part in multiple clinical settings such as inflammatory bowel disease, cardiovascular disease and autoimmune disease,32–34 suggesting that rTsP53 has therapeutic potential for sepsis and other inflammatory diseases through the modulation of macrophages. It has to be noted that although the pro-inflammatory response of sepsis is generally the major cause of organ failure, the later anti-inflammatory stages, also described as chronic immune paralysis, cause substantial sepsis-related death. 4 This stage may reflect the self-regulatory mechanism in response to severe systemic inflammation. The effective anti-inflammatory property and early intervention of rTsP53 may attenuate the systemic inflammation at an early stage and may therefore prevent later immune paralysis, and hence represents an ideal therapeutic candidate.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by scientific research projects (Bureau of science and Information Technology, Guangzhou, s/n 201300000160; Provincial Scientific Project, Guangdong, s/n 2012B031800291) and the open-end fund (National Key Laboratory of Trauma, Burn and Compound Wounds, The 3rd Military Medical University, s/n SKLKF201201). The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.