
Abstract
Innate immune activation with expression of pro-inflammatory molecules such as TNF-α is a hallmark of the chronic inflammation associated with periodontal disease (PD). Porphyromonas gingivalis, a bacterium associated with PD, engages TLRs and activates MyD88-dependent and TIR-domain-containing adapter-inducing IFN-β (TRIF)-dependent signaling pathways. IFN regulatory factor (IRF) 3 is activated in a TRIF-dependent manner and participates in production of cytokines such as TNF-α; however, little is known regarding IRF3 and the host response to PD pathogens. We speculated that IRF3 participates in the host inflammatory response to P. gingivalis. Our results show that bone marrow macrophages (MØ) from WT mice respond to P. gingivalis with activation and nuclear translocation of IRF3. Compared with WT, MØ from IRF3−/−, TRIF−/−, and TLR4−/− mice responded with reduced levels of TNF-α on P. gingivalis challenge. In addition, full expression of IL-6 and RANTES by MØ to P. gingivalis was dependent on IRF3. Lastly, employing MØ from IRF3−/− and IRF7−/− mice we observed a significant role for IRF3 and a modest role for IRF7 in the P. gingivalis-elicited TNF-α response. These studies identify a role for IRF3 in the inflammatory response by MØ to the periodontal pathogen P. gingivalis.
Keywords
Introduction
Periodontal disease (PD) is a common chronic oral inflammatory disease. 1 The host response during PD is complex and much of the hard and soft tissue destruction observed is attributed to immune recognition and subsequent inflammatory responses to periodontal pathogens.2–5 An array of cytokines and chemokines, including TNF-α, IL-1, IL-8, RANTES and others, have been detected at higher levels in samples from individuals with PD than from healthy samples.4,6,7 Experimental studies identify various cell types, including monocytes, and epithelial and endothelial cells, as sources of cytokines, chemokines and other inflammatory molecules when exposed to oral pathogens, including Porphyromonas gingivalis.8–10
TLRs are a family of host innate immune receptors that detect the presence of conserved microbial antigens, such as LPS, peptidoglycan and others. 11 These receptors are expressed by a variety of cells, including bone marrow macrophages (MØ). TLR engagement activates signal transduction pathways governed by adaptor molecules MyD88 and TIR-domain-containing adapter-inducing IFN-β (TRIF), culminating in transcription of a variety of genes encoding molecules such as cytokines and chemokines. 12 All TLRs with the exception of TLR3 signal via MyD88. 11 TLR3 utilizes TRIF exclusively, while TLR4 uses both MyD88 and TRIF pathways. 11 TLR signaling pathways are implicated in host responses to P. gingivalis, including cytokine and chemokine production,13,14 osteoclastogenesis 15 and oral bone loss.16,17 Moreover, MyD88-dependent and TRIF-dependent pathways participate in the host response to this organism.18,19 Little is known about the signaling molecules downstream of TRIF that contribute to host immune response to periodontal pathogens.
IFN regulator factor (IRF) 3 is activated in a TLR/TRIF-dependent manner, 20 following activation of TANK-binding kinase (TBK)-1. 21 IRF3 binding sites have been identified in the promoter region of NF-κB-dependent genes, such as TNF-α, 22 thus identifying a potential mechanism in which IRF3 can influence expression of this and other pro-inflammatory cytokines. IRF3 is one member of a family of nine IRFs identified in humans. 20 IRF3 and IRF7 are among the best-studied IRF family members and participate in immune signaling. 23 In the context of influenza A virus infection, IRF3 is necessary for the expression of IFN-β and a variety of inflammatory mediators, including MCP-1 and, to a lesser extent, TNF-α. 24 Bacterial LPS challenge requires intact IRF3 to induce endotoxic shock response in mice. 25 Use of purified enteric LPS identified TLR-mediated activation of IRF3/IRF7 in a TLR4-mediated, TRIF- and TRIF-related adaptor molecule (TRAM)-dependent manner. 26 This latter finding supports an important role for IRF3 in host response to bacterial pathogens. In the context of host response to periodontal pathogens, there is limited information regarding the involvement of IRF. Dendritic cells cultured with recombinant P. gingivalis hemagglutinin B protein increase transcription of the IRF3 gene, 27 while MØ response to P. gingivalis LPS involves IRF7. 19 The aim of the present study is to define the contribution of IRF3 in the host cytokine and chemokine responses to P. gingivalis.
Materials and methods
Mice and MØ collection
Male 6–8-wk-old C57BL-6 wild type (WT) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). IRF3−/−28 and TRIF−/−29 mice were obtained from Dr Egil Lien (University of Massachusetts Medical Center, Worchester, MA, USA), TLR4−/−30 were obtained from Dr Caroline Genco (Boston University Medical Center, Boston, MA, USA), and IRF7−/−31 mice were from Dr Ian Rifkin (Boston University Medical Center, Boston, MA, USA). MyD88−/− mice 32 were from Dr Shizuo Akira (Osaka University, Osaka, Japan). All studies were performed in accordance with Boston University Institutional Animal Care and Use Committee approvals, and animals received chow diet and water ad libitum. Bone marrow cells were flushed from femurs of humanely sacrificed mice. The red blood cells were removed by 1 × RBC lysis buffer (eBioscience, San Diego, CA, USA). Remaining bone marrow cells were differentiated into MØ over 7 d in RPMI-1640 + 10% FBS supplemented with 20% conditioned medium from L929 cells (source of M-CSF, a kind gift of Dr Robin Ingalls, Boston University Medical Center, Boston, MA, USA), and penicillin/streptomycin in a CO2 incubator, under standard cell culture conditions. Following washing and replacement with antibiotic-free and M-CSF-free cell culture medium, adherent MØ were collected and added to wells of tissue culture plates at 5 × 105 cells/ml 2 h prior to challenge.
P. gingivalis cultivation
P. gingivalis strain 381 was primarily used in these studies; however, where indicated, P. gingivalis strain 33277 was also employed to study host response to different P. gingivalis strains. In all cases, bacteria were grown in an anaerobic environment on brain–heart infusion (BHI) yeast extract–blood agar plates for 3–5 d, followed by 24 h cultivation in BHI yeast extract broth, as previously described. 33 Broth-grown bacteria were harvested by centrifugation, washed, adjusted to an OD of 1 at 660 nm (approximately 1 × 109 bacteria/ml) with antibiotic-free RPMI-1640 + 10% FBS, and were added to MØ cultures at the indicated multiplicities of infection (MOI). Gram staining was used to assess purity of all broth cultures.
MØ challenge assays
For IRF3 activation studies MØ from WT mice were placed into wells of tissue culture dishes (5 × 106 total cells), and were incubated with antibiotic-free cell culture medium alone, or medium containing P. gingivalis MOI 100. PolyI:C (1 µg/ml; InvivoGen, San Diego, CA, USA) 1 h treatment served as positive control. 34 After 1, 2 and 4 h of incubation, cells were washed and used for isolation of cytoplasmic and nuclear fractions to determine IRF3 activation. For studies investigating cytokine and chemokine production, MØ from WT and mutant mice were placed into wells of tissue culture dishes, and were incubated with fresh antibiotic-free cell culture medium alone, or medium containing P. gingivalis at MOIs ranging from 1 to 100 as indicated. In some experiments ultrapure preparations of Pam3CSK4 (100 ng/ml; TLR2/1 agonist; InvivoGen, San Diego, CA, USA) or Escherichia coli LPS (10 ng/ml; TLR4 agonist; InvivoGen) were used as specific ligands. Culture supernatant fluids were harvested as indicated, and stored frozen at −80℃ until ELISA or multiplex assays were performed.
MØ viability assay
For the MØ viability assay, MØ from WT and IRF3−/− mice were placed into wells of tissue culture dishes (six-well) and were incubated with fresh antibiotic-free cell culture medium alone, or medium containing P. gingivalis at a MOI of 100 for 24 h. Culture supernatant fluids were aspirated and MØ were washed with antibiotic-free cell culture medium. One milliliter of fresh medium with out antibiotics was added to each well and MØ were gently collected using cell scraper. Viability of recovered MØ was determined by Trypan blue dye exclusion method using hemocytometer.
Detection of IRF3 activation
Cytoplasmic and nuclear fractions were prepared from MØ using a nuclear extraction kit according to the manufacturer’s instructions (Active Motif, Carlsbad, CA, USA). Briefly, medium was aspirated from MØ and the adherent cells were washed with ice-cold PBS containing phosphatase inhibitors. Cells were removed from culture dishes by scraping, collected, pelleted by centrifugation at 55 g and were suspended in 1 × hypotonic buffer. After 15 min incubation, cells were solubilized by the addition of detergent provided as part of the kit. The cytoplasmic fraction was collected following centrifugation at 14,000 g at 4℃, and the cell pellet was suspended in complete lysis buffer and gently mixed for 30 min on ice. The nuclear fraction was obtained from the supernatant by centrifugation at 14,000 g at 4℃. Protein concentrations of cytoplasmic and nuclear fractions were calculated using ProStain-Protein Quantification Kit (Active Motif). Levels of IRF3 in cytoplasmic and nuclear fractions were determined on 7 µg of total protein, and presented as arbitrary units at 450 nm using mouse TransAM IRF3 kit, according to the manufacturer’s instructions (Active Motif).
Cytokine and chemokine detection
Frozen cell culture supernatant fluids were thawed and the levels of TNF-α, IL-6 and RANTES were determined from unchallenged and challenged MØ using commercially available ELISA kits (R & D Systems, Minneapolis, MN, USA), or Luminex-based multiplex assays (Invitrogen, Grand Island, NY, USA) as per manufacturer’s instructions.
Statistical analysis
All studies were performed using MØ from individual animals, and all experiments were performed on two separate occasions. Data were imported into Prism statistical analysis software (Graphpad Inc., La Jolla, CA, USA) and are presented as means ± SEM. Comparisons between groups were determined using unpaired t-test, or one-way and two-way ANOVA with Tukey’s or Bonferroni multiple comparisons respectively. P < 0.05 was considered significant.
Results
P. gingivalis induces IRF3 activation and nuclear localization in MØ
Here, we investigated IRF3 activation and nuclear translocation in WT mouse MØ to P. gingivalis challenge. Medium treatment established baseline IRF3 levels in nuclear and cytoplasmic fractions. A P. gingivalis MOI of 100 induced IRF3 levels in nuclear protein fraction compared with unchallenged control (Figure 1A). Moreover, activation of IRF3 was observed at 1, 2 and 4 h of P. gingivalis challenge (Figure 1A). As anticipated, 1 h PolyI:C treatment (positive control) induced IRF3 activation and nuclear localization
34
(Figure 1A). Cytoplasmic fractions from medium, P. gingivalis and PolyI:C-treated MØ generally contained similar, low-level IRF3 detection (Figure 1B).
Activation of IRF3 in MØ cultured with P. gingivalis. MØ from C57BL-6 WT mice were cultured for 1, 2 and 4 h in medium alone or with P. gingivalis strain 381 (Pg) at a MOI of 100. One-hour PolyI:C (1 µg/ml) treatment was used as a positive control for IRF3 activation. MØ cultured in medium alone (open bars), and MØ cultured with P. gingivalis strain 381 (Pg) or PolyI:C (filled bars). MØ IRF3 activation was measured in nuclear fractions (A) or cytoplasmic fractions (B) by spectrophotometric assay per manufacturer’s instructions. Data are presented as mean ± SEM of two independent experiments (n = 1 mouse per experiment).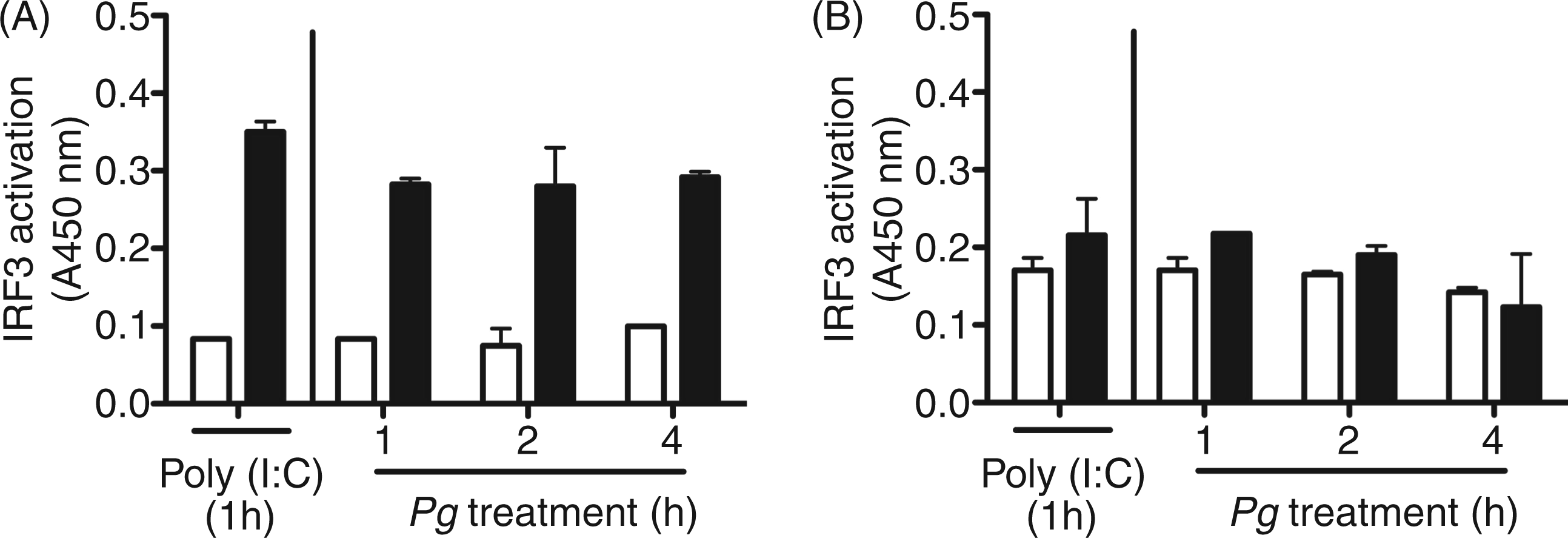
Stimulation of MØ TNF-α response by P. gingivalis is, in part, mediated by IRF3
As P. gingivalis induced IRF3 activation, we speculated that IRF3 may participate in cytokine and chemokine production. To test this we compared TNF-α produced by MØ from WT and IRF3−/− mice cultured with P. gingivalis. IRF3−/− MØ cultured with P. gingivalis produced significantly less TNF-α than observed with WT (P < 0.05; Figure 2A). Based on this finding with P. gingivalis, we speculated that an upstream pathway involving TLR4 and TRIF could be involved in host response to this organism. Employing MØ from WT, TRIF−/− and TLR4−/− mice, we observed that TNF‐α levels elicited by P. gingivalis were significantly reduced in MØ from TRIF−/− and TLR4−/− mice compared with WT respectively (Figure 2B and 2C). In addition we challenged MØ from WT and MyD88−/− mice, and observed a significant reduction in P. gingivalis-elicited TNF-α by MØ deficient in MyD88 compared with WT status (P < 0.05; Figure 2D).
IRF3, TRIF, TLR4 and MyD88 contribute to the production of TNF-α by murine MØ cultured with P. gingivalis. MØ from C57BL-6 WT, IRF3−/− (A), TRIF−/− (B), TLR4−/− (C) and MyD88−/− (D) mice were cultured for 24 h in medium alone (open bars) or with P. gingivalis strain 381 at a MOI of 100 (filled bars). ELISA was used to measure TNF-α levels in harvested culture supernatant fluids. Data are presented as mean ± SEM from two separate experiments; n = 3–6 individual mice per group in total. *P < 0.05 as determined by unpaired t-test.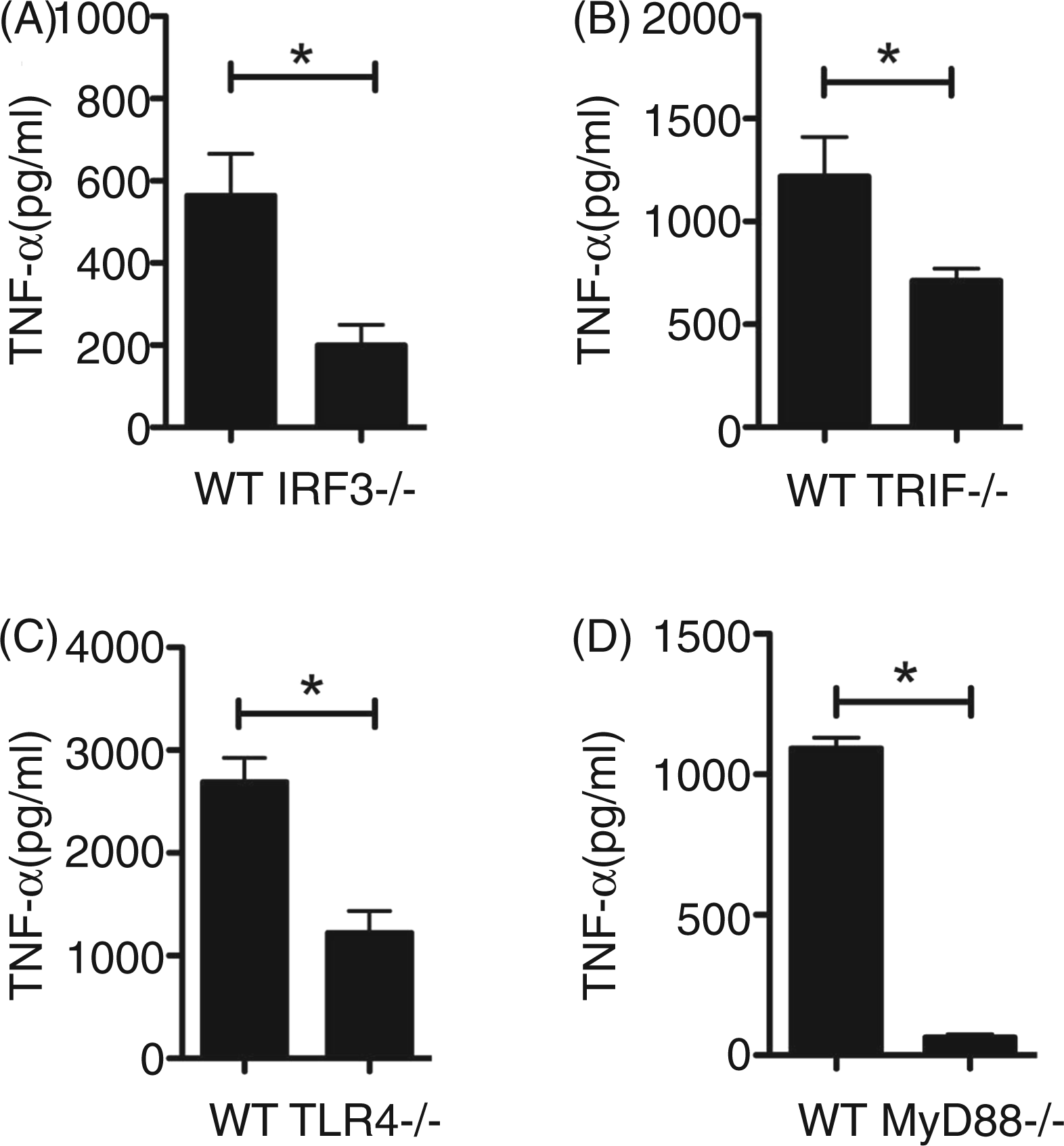
Role of IRF3 in TNF-α by MØ is conserved between P. gingivalis strains
To ensure that the observed reduction in TNF-α levels from IRF3−/− mouse MØ was not specific only to P. gingivalis strain 381, we challenged MØ from WT and IRF3−/− with either P. gingivalis 381 or P. gingivalis 33277 at a MOI of 100, and measured the levels of TNF-α. WT MØ responded less robustly to P. gingivalis strain 33277 compared with P. gingivalis 381. Although magnitude differences in elicited TNF-α were observed between strains, similar patterns showing significantly reduced levels of TNF-α as a consequence of IRF3−/− were observed (Figure 3).
Role of P. gingivalis strains on the production of TNF-α in MØ from IRF3−/− mice. MØ from C57BL-6 WT (open bars), and IRF3−/− (filled bars) mice were cultured for 24 h with P. gingivalis (Pg) strain 381 and P. gingivalis strain 33277 at a MOI of 100. ELISA was used to measure TNF-α levels in harvested culture supernatant fluids. Data are presented as mean ± SEM from two separate experiments; n = 4 individual mice per group in total. *P < 0.05 by two-way ANOVA with Bonferroni post-test.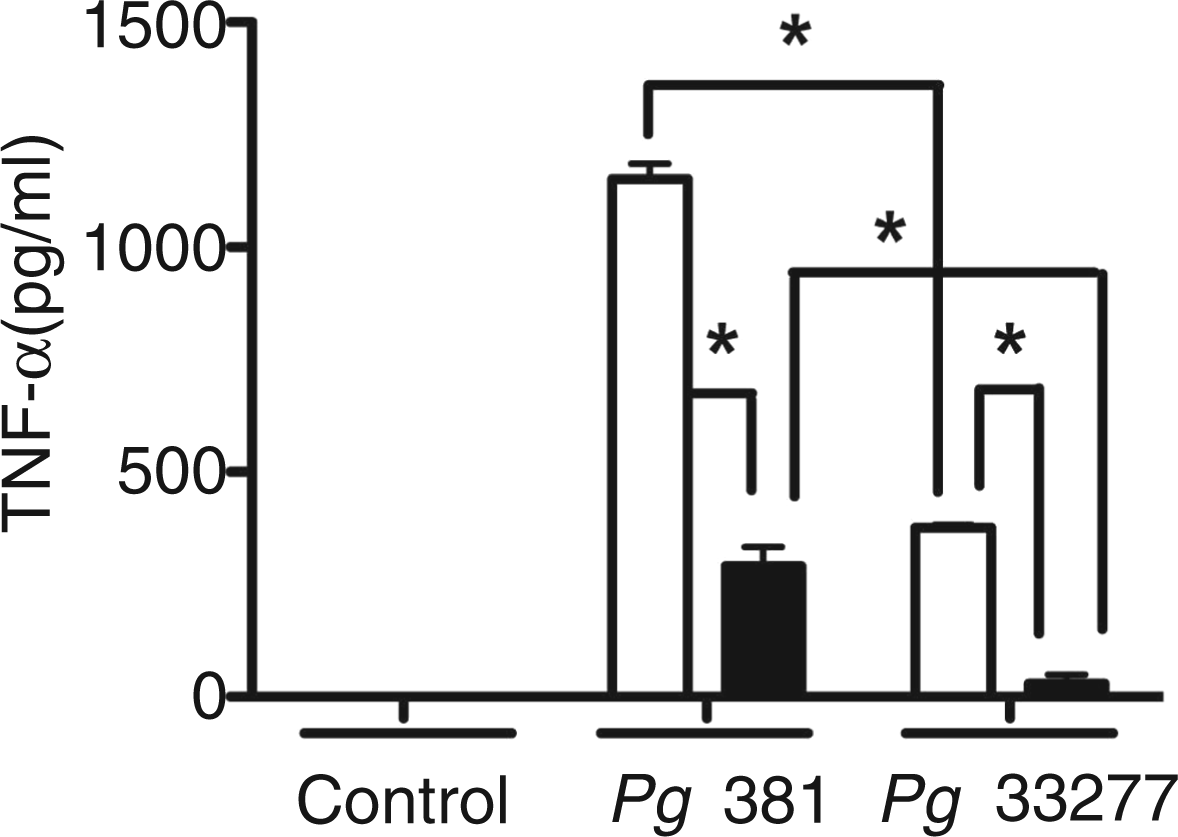
IRF3 ablation limits TNF-α, IL-6 and RANTES production from MØ cultured with P. gingivalis
As IRF3 was involved in TNF-α production by MØ response to P. gingivalis, we next investigated the influence of IRF3 on the production of other inflammatory molecules from MØ to P. gingivalis. We challenged MØ from WT and IRF3−/− mice with various doses of P. gingivalis for 24 h, and measured TNF-α, IL-6 and the IRF3-dependent chemokine RANTES.
35
As anticipated, ablation of IRF3 revealed its role in P. gingivalis-elicited TNF-α at MOIs of 100 and 10 (Figure 4A). Little TNF-α was detected with a MOI of 1 in WT, and differences between genotypes at this MOI were not observed. At a MOI of 100, a significant decrease in IL-6 was observed from IRF3−/− MØ compared with WT (Figure 4B). We also observed a reduction in IL-6 from IRF3−/− mouse MØ cultured with P. gingivalis at a MOI of 10; however, this did not reach a level of significance (P > 0.05; Figure 4B). Focusing on RANTES, a consistent and significant reduction was observed for IRF3−/− at all P. gingivalis MOIs studied (P < 0.05; Figure 4C). To ensure that MØ from IRF3−/− mice were not intrinsically defective in developing inflammatory responses to bacteria, we challenged WT and IRF3−/− mouse MØ with ultrapure preparations of Pam3CSK4 (100 ng/ml) or E. coli LPS (10 ng/ml), and measured TNF-α production by ELISA. IRF3 ablation resulted in a significant reduction in E. coli LPS elicited TNF-α, while levels of TNF-α to Pam3CSK4 were similar between WT and IRF3−/− MØ (Figure 4D).
Cytokine and chemokine response of MØ deficient in IRF3 to P. gingivalis challenge. MØ from C57BL-6 WT (open bars) and IRF3−/− (filled bars) mice were cultured with medium alone (Cont), medium containing P. gingivalis 381 at MOIs of 100, 10, and 1 for 24 h. ELISA was used to measure TNF-α (A), IL-6 (B) and RANTES (C) in culture supernatant fluids. For ligand treatments, MØ from C57BL-6 WT (open bars) and IRF3−/− (filled bars) mice were cultured with medium alone (Cont), medium containing ultra pure E. coli 0111:B4 LPS (Ec-LPS, 10 ng/ml) or medium containing ultra pure Pam3CSK4 (100 ng/ml) for 24 h. TNF-α levels in culture supernatant fluids was measured by ELISA (D). Bar represents mean ± SEM from two separate experiments; n = 3–5 individual mice per group in total. *P < 0.05 by two-way ANOVA with Bonferroni post-test. ND: none detected; NS: not significant.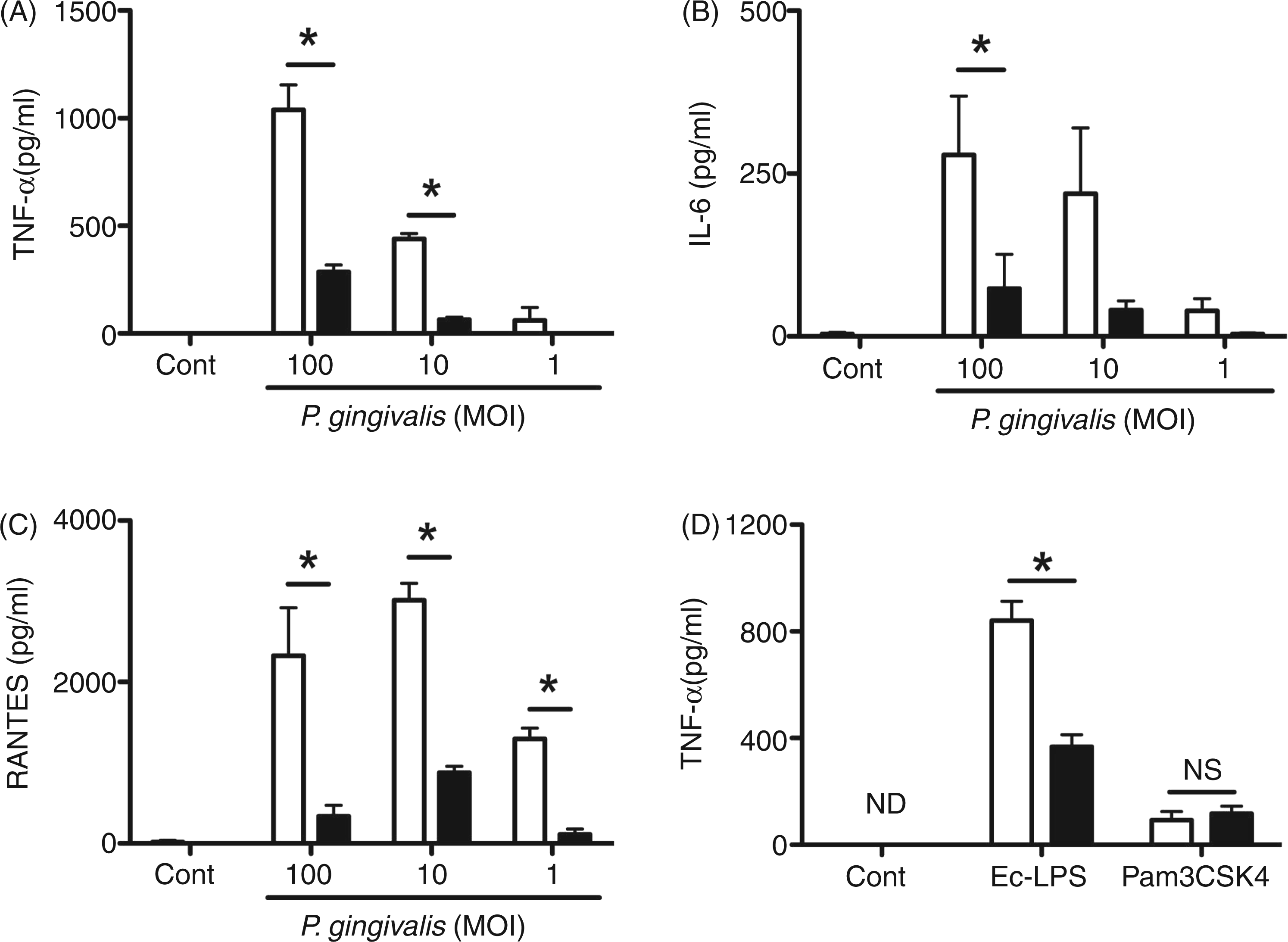
Viability of MØ from WT and IRF3−/− mice in response to P. gingivalis.

Viability of WT (n = 3) and IRF3−/− mouse MØ (n = 3) recovered after 24 h P. gingivalis challenge was determined by Trypan blue dye exclusion method using hemocytometer. Comparisons between groups were performed using one-way ANOVA with Bonferroni post-test. No significant differences were observed between any groups.
IRF3 influences MØ TNF-α response to P. gingivalis more strongly than IRF7
Lastly, we were interested to know if MØ cytokine production in response to P. gingivalis was specific for IRF3 or was shared by other IRFs. At 6 h challenge, we observed significant reduction in P. gingivalis-elicited TNF-α from IRF3−/− MØ, while MØ from IRF7−/− mice produced levels of TNF-α similar to WT (Figure 5A). By 24 h, TNF-α levels from IRF3−/− MØ remained significantly reduced; however, a partial and mild role for IRF7−/− was observed (Figure 5B).
Comparison of IRF3 and IRF7 involvement in TNF-α production by MØ cultured with P. gingivalis. MØ from C57BL-6 WT (open bars), IRF3−/− (black filled bars), and IRF7−/− (gray filled bars) mice were cultured for 6 h (A) or 24 h (B) with P. gingivalis strain 381 at a MOI of 100. Cell culture supernatant fluids were harvested and ELISA was used to measure TNF-α levels. Data presented as a percentage of WT levels from two separate experiments; n = 6 individual mice per group in total. *P < 0.05 by one-way ANOVA with Tukey’s post test. NS: not significant.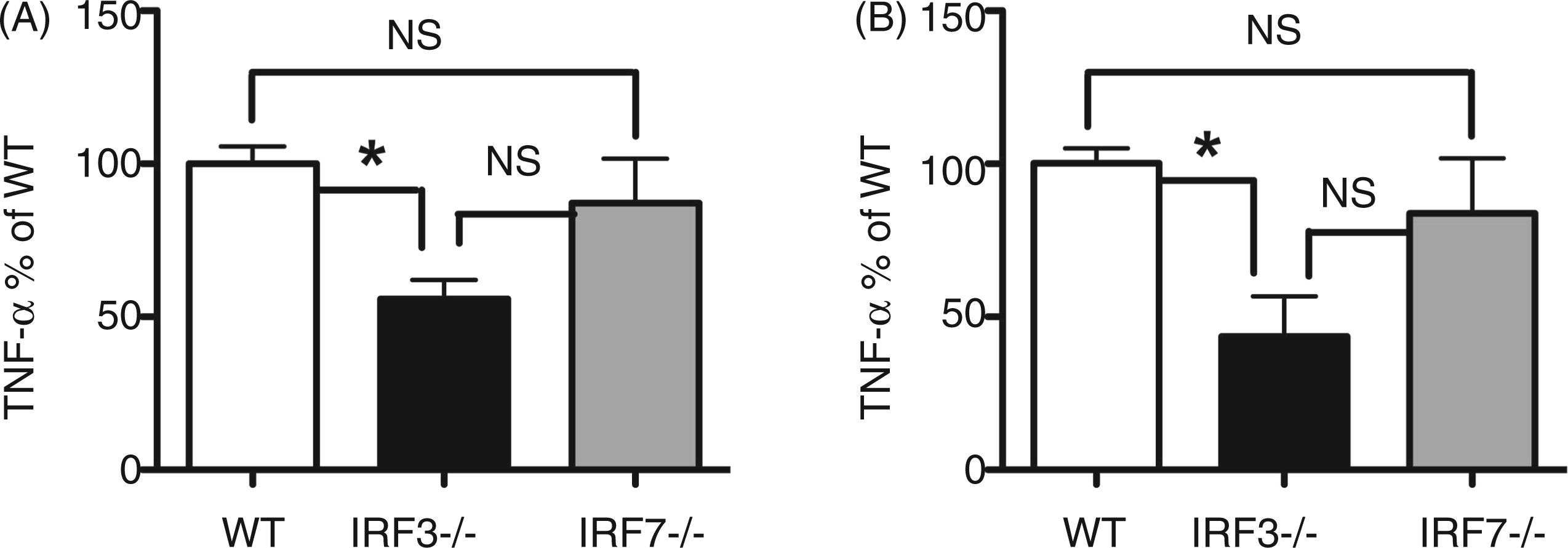
Discussion
Herein, using a murine bone marrow MØ system we report that IRF3 is activated in response to P. gingivalis challenge, and IRF3 functionally participates in the development of cytokine and chemokine response to P. gingivalis. We observed that the TNF-α response of MØ to P. gingivalis is partially dependent on TLR4, TRIF and IRF3. Our data from MyD88−/− mouse MØ is in agreement with others who implicate MyD88 in TNF-α production by the host to P. gingivalis challenge.19,36 The ability of MØ to produce IL-6 and RANTES in response to P. gingivalis challenge also involved IRF3. Employing two distinct strains of P. gingivalis, we observed that the expression of TNF-α to either P. gingivalis strain 381 or P. gingivalis strain 33277 by MØ required intact IRF3. Although differences in maximally achieved TNF-α levels were observed between P. gingivalis strains, a similar contribution of IRF3 to TNF-α expression was observed. Lastly, the use of MØ from IRF3- and IRF7-deficient mice point to an important role for IRF3 in TNF-α response to P. gingivalis.
Our biochemical approach identified activation and nuclear translocation of IRF3 in WT mouse MØ in response to P. gingivalis challenge. These data are the first to show activation of IRF3 in primary MØ from mice in response to a pathogen and are in agreement with a previous report using RAW 264.7 cells, where activation of IRF3 was observed. 37 IRF3 binding sites have been identified in the promoter of TNF-α. 22 Our data support that IRF3 is functionally involved in the development of a full TNF-α response by MØ to P. gingivalis; however, we did not confirm IRF3 binding to the TNF-α promoter. Similar to that observed with MØ from IRF3 deficient mice, MØ TNF-α response to P. gingivalis was also partially dependent on TLR4 and TRIF. TRIF signaling is critical for MØ IRF3 phosphodimerization. 38 Thus, our findings of IRF3 activation and nuclear localization are in agreement with the model that MØ from TRIF−/− and TLR4−/− mice mimic the response seen with IRF3−/− mouse MØ to P. gingivalis challenge, and it is in agreement with previous studies that TRIF, IRF3 and TLR4 molecules signal through a common pathway.39,40 Prior studies using purified ligands including LPS identify a similar trend to what we observed in the context of TNF-α response by MØ when using MyD88−/−, TRIF−/− and TRIFlps2 mice. Kawai et al. 41 reported a strong reduction in TNF-α response by MyD88−/− MØ to E. coli LPS. Yamamoto et al. 29 reported a significant reduction in TNF-α produced from TRIF−/− MØ cultured with LPS. This later finding was substantiated by Hoebe et al. 42 where MØ from TRIFlps2 mice also produced little TNF-α to E. coli LPS exposure. These findings add complexity to the interpretation of host response in the presence/absence of MyD88 signaling as the three systems report very strong reductions in TNF-α to LPS, with the latter two systems limiting TRIF signaling capacity in the presence of an intact MyD88 pathway. In the context of MyD88 and TRIF, our findings are in broad agreement with those of Burns et al., 18 who concluded, using an in vivo chamber model, that TLR2-dependent host pro-inflammatory cytokine response to P. gingivalis challenge was not dependent on the MyD88 pathway but that MyD88 was necessary for clearance of P. gingivalis. A recent study by Liang et al. 43 identified TLR2/CR5a interaction and influence of this receptor complex on MØ response to P. gingivalis, as well as P. gingivalis-induced oral bone loss. In the present study our results indicate that host response to P. gingivalis activates IRF3 and the ablation of IRF3 contributes to reduction in secreted levels of TNF-α in response to P. gingivalis.
Implication of IRF3 in the development of full immune response to bacterial challenge has been reported for several bacteria. In a lung epithelial cell model, IRF3 knockdown revealed its role in controlling Legionella pneumophila replication. 44 IRF3-deficient mice are more resistant to Listeria monocytogenes infection.45 MØ cultures and lung infection models support a role for IRF3 in full expression of TNF-α to Pseudomonas aeruginosa. 39 The effect of IRF3 deficiency on host response to P. aeruginosa was not limited to TNF-α, as defective responses in MØ expressing RANTES and IL-10 were also observed. 39 Our data, that IRF3 is involved in full host cytokine and chemokine response to P. gingivalis, parallel findings in studies employing P. aeruginosa. 39 Our data support that P. gingivalis stimulates RANTES production from MØ cultured with P. gingivalis and that this response is partially IRF3-dependent. In the context of IRF3 involvement in TNF-α expression, our results also parallel findings of a recent study employing the influenza A virus, which reported partial involvement of IRF3 in pathogen-elicited TNF-α from MØ. 24
Recent data identify IRF activation in response to P. gingivalis challenge. Gaddis et al. 27 reported activation of IRF3 by dendritic cells cultured with recombinant P. gingivalis HagB. Our studies using whole P. gingivalis challenge are in broad agreement with the results of this study, which suggest involvement of IRF3 in the host response to P. gingivalis. Our data, however, differ from a recent study reporting involvement of IRF7 along with TLR2 and TLR7 in host response by MØ to P. gingivalis and its LPS, 19 and no apparent role for IRF3. 19 In our hands we observed activation and nuclear localization of IRF3 in MØ as a consequence of P. gingivalis challenge. Moreover, using MØ from IRF3−/− mice, we observed that IRF3 contributed to full TNF-α, IL-6 and RANTES response of these cells to P. gingivalis challenge. Lastly, IRF3 played a consistent role in TNF-α response at 6 and 24 h, while only a partial role for IRF7 was detected. The differences observed between our studies and those of Zhou and Amar, 19 may reflect differences in systems used. Both studies utilized murine MØ and employed P. gingivalis strain 381; however, we utilized MØ from IRF3 and IRF7 knockout mice, while Zhou and Amar 19 utilized several strains of knockout mice, including TLR2, TLR7 and MyD88 mice, but did not employ IRF3−/− and IRF7−/− mice. Regardless, when taken together, these studies do support a role for IRFs in development of the host response to P. gingivalis. More detailed studies are needed to understand the significance of these differences, in the context of other periodontal disease-associated pathogens and to elucidate the bacterial antigens driving IFN pathway activation.
Employing an in vitro P. gingivalis mouse MØ challenge model, our data provide new information involving IRF3 in the development of full host inflammatory responses to this organism. Future studies are needed to determine the precise roles of IFN signaling pathways in the host response to P. gingivalis and the contribution these pathways have in the pathogenesis of PD.
Footnotes
Funding
These studies were supported by PHS through NIH/NIDCR grants DE018318 and DE021497 (FCG), and NIH/NIAID grant P01AI078894 (subcontract to FCG), and through a BU Department of Medicine Pilot grant (FCG).
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
Acknowledgments
The authors would like to acknowledge Dr Tadatsugu Taniguchi (University of Tokyo, Tokyo, Japan) for making IRF3−/− and IRF7−/− mice available for these studies.