
Abstract
Macrophage heterogeneity in human atherosclerotic plaques has been recognized; however, markers for unequivocal identification of some subtypes are lacking. We found that the platelet chemokine CXCL4 induces a unique macrophage phenotype, which we proposed to call ‘M4’. Here, we sought to identify suitable markers that identify M4 macrophages in vitro and in vivo. Using a stringent algorithm, we identified a set of potential markers from transcriptomic data derived from polarized macrophages. We specifically focused on matrix metalloproteinase (MMP)7 and S100A8, the co-expression of which has not been described in any macrophage type thus far. We found dose- and time-dependent MMP7 and S100A8 expression in M4 macrophages at the gene and protein levels. CXCL4-induced up-regulation of both MMP7 and S100A8 was curbed in the presence of heparin, which binds to CXCL4 and glycosaminoglycans, most likely representing the macrophage receptor for CXCL4. Immunofluorescence of post-mortem atherosclerotic coronary arteries identified CD68+MMP7+, CD68+MMP7−, CD68+S100A8+ and CD68+S100A8− macrophages. A small proportion of MMP7+S100A8+ macrophages most likely represent M4 macrophages. In summary, we have identified co-expression of MMP7 and S100A8 to be a marker combination exclusively found in M4 macrophages. This finding may allow further dissection of the role of M4 macrophages in atherosclerosis and other pathologic conditions.
Introduction
Monocyte-derived macrophages are essential mediators of innate immunity. 1 Similar to macrophages that can be found constitutively in tissues like skin (Langerhans cells), lung (alveolar macrophages) or liver (Kupffer cells), monocyte-derived macrophage can contribute to acute or chronic inflammatory processes. 2 Atherosclerosis is an inflammatory disease in which macrophages are critical for initiation and progression of the disease.3,4 Atherosclerotic plaque macrophages differentiate from blood monocytes that have transmigrated into the subendothelial space. In addition, local proliferation of macrophages may also promote the disease development. 5 Monocyte macrophage differentiation is a complex process accompanied by substantial changes in gene and protein expression, and cellular function.6,7 Thus, monocyte-derived macrophages can sustain the disease process by ingesting oxidized lipids or by secreting pro-inflammatory mediators leading to recruitment of further immune cells. 8 Also, macrophages can secrete matrix metalloproteinases (MMPs), which degrade the extracellular matrix, resulting in thinning or rupture of the fibrous cap of the atherosclerotic lesion leading to myocardial infarction or stroke. 8
Over the last few years, it has become evident that monocyte-derived macrophages are not a homogeneous population of cells, but may assume different polarization types (M1 and M2). 9 Accordingly, M1 macrophages are induced by IFN-γ or LPS, whereas M2 macrophages can be generated through activation by IL-4, IL-10 or IL-13.10,11 While gene expression of both M1 and M2 macrophage markers have been shown in vivo, 12 it is extremely likely that additional phenotypes exist in atherosclerotic lesions.13,14 Thus, granulocyte macrophage colony stimulating factor (GM-CSF)-induced macrophages have been described to be CD68+CD14−, while macrophage colony stimulating factor (M-CSF)-induced macrophages express both markers. 13 Also, a novel hemoglobin-induced macrophage type has been described characterized by high expression levels of CD163.14–16
We have previously studied monocyte-derived macrophages induced by the platelet chemokine CXCL4. 17 CXCL4 plays an important role in atherosclerosis. It can be found in human atherosclerotic lesions; its presence in carotid plaques has been shown to correlate with histological and clinical variables, such as lesion grade or ischemic symptoms. 18 In conjunction with CCL5, CXCL4 promotes monocyte adhesion to endothelial cells. 19 Furthermore, CXCL4 induces macrophage differentiation from human peripheral blood monocytes. 20 Finally, knocking out the Pf4 gene encoding CXCL4 in Apoe− / − mice leads to reduced atherosclerotic lesion size. 21 Taken together, these data suggest a pro-atherogenic role for CXCL4.
We have demonstrated that CXCL4-induced macrophages display a unique transcriptome that significantly differs from that of M-CSF-induced macrophages, or M1 or M2 polarized macrophages. 17 Functionally, we were able to demonstrate that CXCL4-induced macrophages lose the hemoglobin–haptoglobin scavenger receptor CD163 and are unable to up-regulate atheroprotective heme oxygenase-1 in response to haptoglobin–hemoglobin complexes. 22 While we found an inverse correlation between PF4 and CD163 in human carotid plaques, suggesting that our findings are important in vivo, a suitable combination of markers to identify CXCL4-induced macrophages in vivo is still lacking.
Macrophage heterogeneity may not only comprise phenotypic, but also functional aspects. Thus, it is very possible that certain macrophage types act in a pro-atherogenic manner (e.g. by secreting pro-inflammatory cytokines or MMPs), while others may play an atheroprotective role (e.g. by limiting inflammation through secretion of anti-inflammatory cytokines). Accordingly, macrophage heterogeneity may represent an interesting therapeutic target in atherosclerosis, for example driving macrophages towards an atheroprotective phenotype may help to limit disease progression. However, in order to develop such strategies it is necessary to understand exactly the quantity, location or cellular interaction of the specific macrophage types within human atherosclerotic lesions. This is at least partly known for M1 or M2 macrophages; 12 however, there are no such data regarding M4 macrophages.
Therefore, we sought to identify a marker combination that may facilitate identification and study of CXCL4-induced macrophages in human atherosclerotic coronary arteries. Based on transcriptomic data from M1, M2 and M4 macrophages, we used a stringent algorithm to identify potential candidate markers and subsequently validated expression of these markers both in vitro and in human atherosclerotic lesions.
Materials and methods
Monocyte-derived macrophages
Monocyte-derived macrophages were generated in serum-free media with M-CSF as described previously.17,22,23 M1 and M2 polarization of macrophages was performed as described. 6 Recombinant human M-CSF, CXCL4, IFN-γ and IL-4 were purchased from Peprotech (Wiesbaden, Germany). LPS was purchased from Sigma (Taufkirchen, Germany). For dose–response experiments, monocyte-derived macrophages were induced with 100 ng/ml M-CSF for 3 d and increasing concentrations of CXCL4 for another 3 d (0.125–1.000 µM), as described previously. 24
Quantitative PCR
Quantitative PCR was performed as described. 22 Primer sequences were obtained from Primer Bank: 25 GAPDH forward GGCTCATGACCACAGTCCAT; GAPDH reverse GCCTGCTTCACCACCTTCT; MMP7 forward CATGAGTGAGCTACAGTGGGA; MMP7 reverse CTATGACGCGGGAGTTTAACAT; S100A8 forward TTTCCATGCCGTCTACAG; S100A8 reverse ACGCCCATCTTTATCACC. Gene expression was calculated using GAPDH as the housekeeping gene.
Flow cytometry
Intracellular flow cytometry was performed as described previously. 22 MMP7 was stained using rabbit anti-human MMP7, IgG, polyclonal (Abcam, Cambridge, UK).
ELISA
ELISA kits to measure secretory forms of MMP7 and S100A8 were purchased from Raybiotech (Norcross, GA, USA). Soluble protein was measured as indicated by the manufacturer’s instructions.
Immunofluorescence
Cells were washed with PBS, fixed with 4% paraformaledehyde/PBS for 20 min and permeabilized with 0.25% triton. Three percent BSA/PBS was used for blocking; cells were then treated with 1/50 mouse monoclonal IgG1 anti-human S100A8 (clone: 8-5C2; BMA Biomedicals, Augst, Switzerland) for 1 h. After washing with PBS, a Texas-Red-labeled secondary Ab (1/250; Dianova, Hamburg, Germany) was added for 1 h and cells were covered with Roti-Mount FluorCare DAPI (Roth, Karlsruhe, Germany). For controls, the primary Ab was omitted.
Coronary arteries were embedded in paraffin and 5 μm sections were prepared. After heat-induced antigen retrieval using antigen unmasking solution (Vector Laboratories), sections were incubated with antibodies against CD 68 (clone KP-1, Santa Cruz Biotechnology). Antibody was detected using the ABC method (Vector Laboratories). Antibodies were visualized using DAB (DAKO corporation). After counterstaining with hematoxylin (Sigma) sections were analyzed using a Nikon 80i microscope and Image Pro Plus 3.0 software (Media Cybernectics). For immunofluorescence staining of human samples, the following primary Abs were used: mouse monoclonal IgG1 anti-human S100A8 (clone: 8-5C2, 1/50; BMA Biomedicals), polyclonal rabbit anti-human MMP7 (1/20; Abcam), mouse monoclonal IgG3 anti-human CD68 (clone: PGM1, 1/50; Biogenex, San Ramon, CA, USA). Isotype- and concentration-matched control Abs (Dianova) served as negative controls. Fresh frozen human coronary arteries that were provided by the tissue bank for inflammatory diseases Heidelberg (GEZEH, www.gezeh.de) were cut into 5 -µm sections. Slides were fixed with acetone (Roth) for 10 min at –20℃. The primary Ab was added for 1 h at 20℃ (room temperature) in TBS/0.2% BSA (both from Sigma), which also served as washing buffer. Sheep anti-mouse (1/250) and biotinylated donkey anti-rabbit Abs (1/200; both Dianova) were used as secondary Abs.
Double immunofluorescence staining was performed using a combination of S100A8 and CD68 or MMP7 and CD68, respectively. Donkey anti-sheep DyLight488 (1/50; Dianova) and streptavidin-conjugated Cy3 (1/1000; Dianova) were used as secondary reagents (30 min at room temperature). Slides were mounted with a 4′,6-diamidin-2-phenylindol (DAPI)-containing mounting medium (Roti-Mount FluorCare DAPI; Roth) and viewed with a Laserscan microscope (Leica Microsystems, Mannheim, Germany) applying suitable filter combinations.
Statistical analysis
Gene expression changes for M1, M2 or M4 macrophage differentiation were analyzed by comparing previously published transcriptome data sets (GDS2430 6 and GSE20484 17 ) with their corresponding M-CSF-induced ‘M0’ macrophage transcriptomes using the local pooled error (LPE) test. 26 For genes overexpressed in M4 macrophages with a false discovery rate < 0.01, an overexpression score was calculated as follows: The fold-change M4 versus M0 was multiplied by the average expression level in M4 macrophages divided by 1.000. Similar overexpression scores were calculated for M1 and M2 macrophages. For all genes with an M4 overexpression score of ≥10, a normalized heat map was calculated using R (http://www.r-project.org). Quantitative PCR and flow cytometry data were analyzed by a paired t-test; groups were compared by non-parametric Kruskal–Wallis one-way ANOVA with post hoc Dunn’s test. P-values of < 0.05 were considered statistically significant.
Results
Identification of a potential set of M4 marker genes
Based on previously published transcriptomic data derived from M1, M2 and CXCL4-induced M4 macrophages,6,17 we sought to identify a set of potential specific M4 marker genes (Figure 1A). Briefly, we identified all genes significantly overexpressed in M1, M2 or M4 macrophages compared with their M-CSF-induced non-polarized counterparts (M0). Overexpression was defined by a false discovery rate of < 0.01, as determined by LPE testing.
26
Only genes specifically overexpressed in M4 macrophages were considered suitable (n = 60). Subsequently, an overexpression score was calculated for these genes normalizing for their expression level by multiplying the fold-change overexpression of M4 versus M-CSF with the intensity level in M4 macrophages given by Affymetrix. In order to ensure optimal discriminatory power, only M4 genes with an overexpression score >10 were included. The remaining 27 genes were plotted in a heat map that allowed for free clustering of genes in the x and y direction (x axis = condition, y axis = gene), resulting in a final cluster of nine genes (Figure 1B).
Identification of suitable candidate markers for M4 macrophages. (A) Work flow for the identification of suitable M4 macrophage markers. (B) Heat map indicating the expression of 27 potential M4 marker genes [as defined in (A, b.)] in M1-, M2- and M4-polarized macrophages. Gene expression of each condition is normalized to unpolarized (M0) macrophages derived from the same donor.6,17 FDR: false discovery rate.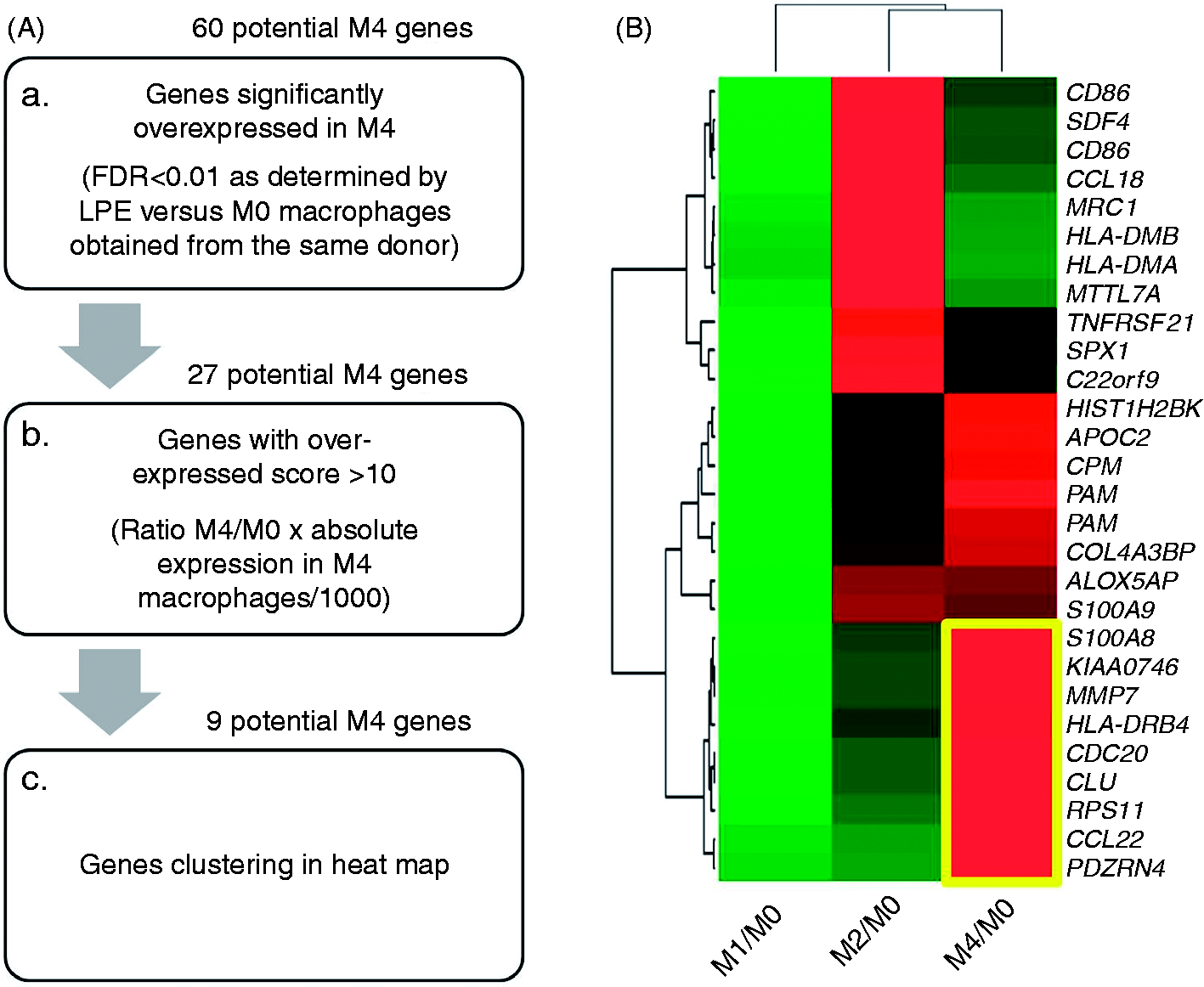
Accordingly, out of the 375 genes differentially expressed in CXCL4- and M-CSF-induced macrophages, this very stringent approach allowed identification a small number of genes that were potential specific markers of M4 macrophages. This set included four genes coding for secreted proteins [CCL22, CLU (clusterin), MMP7, S100A8], two genes coding for membrane-bound molecules [HLA-DR4, SEL1L3 (sel-1 suppressor of lin-12-like 3)] and three genes coding for intracellular proteins [CDC20 (cell division cycle homologue 20), PDZRN4 (PDZ domain containing ring finger 4), RPS11 (ribosomal protein S11)].
CXCL4-induced macrophages overexpress MMP7 and S100A8
To further narrow down the number of potential candidates, two strategies were applied. First, we looked for markers associated with atherosclerotic disease; second, we looked for markers for which suitable reagents for protein detection are commercially available. Based on a literature search, we decided to further follow up MMP7 coding for MMP7 and the calcium binding protein S100A8 coding for migration inhibitory factor-related protein (MRP)-8. For both genes, overexpression in M4 macrophages was highly significant (Figure 2A, B). Quantitative RT-PCR confirmed a dose-dependent increase for both MMP7 and S100A8 mRNA expression after treatment with CXCL4, reaching significance at 1 µM, a physiologically relevant concentration
27
that has been demonstrated to prevent monocyte apoptosis efficiently and promote macrophage differentiation (Figure 2C, D).
20
MMP7 and S100A8 mRNA expression in CXCL4-induced macrophages. (A, B) Gene expression of S100A8 or MMP7 in monocyte-derived macrophages induced over 6 d with M-CSF (100 ng/ml, M0) or CXCL4 (1 µM, M4) as determined by Affymetrix gene array.
17
***P < 0.001 by local pooled error testing. (C, D) MMP7 or S100A8 gene expression in monocyte-derived macrophages after 6 d in culture with M-CSF (100 ng/ml) or 3 d with M-CSF (100 ng/ml), and additional 3 d with indicated concentrations of CXCL4 as determined by quantitative RT-PCR; n = 3–5, **P < 0.01, *P < 0.05 versus M-CSF. (E, F) MMP7 or S100A8 gene expression in monocyte-derived macrophages after 6 d in culture with M-CSF (100 ng/ml), M-CSF plus heparin (2 U/ml), CXCL4 (1 µM) or CXCL plus heparin (2 U/ml), as determined by quantitative RT-PCR; n = 4–9, **P < 0.01, *P < 0.05 versus CXCL4 plus heparin.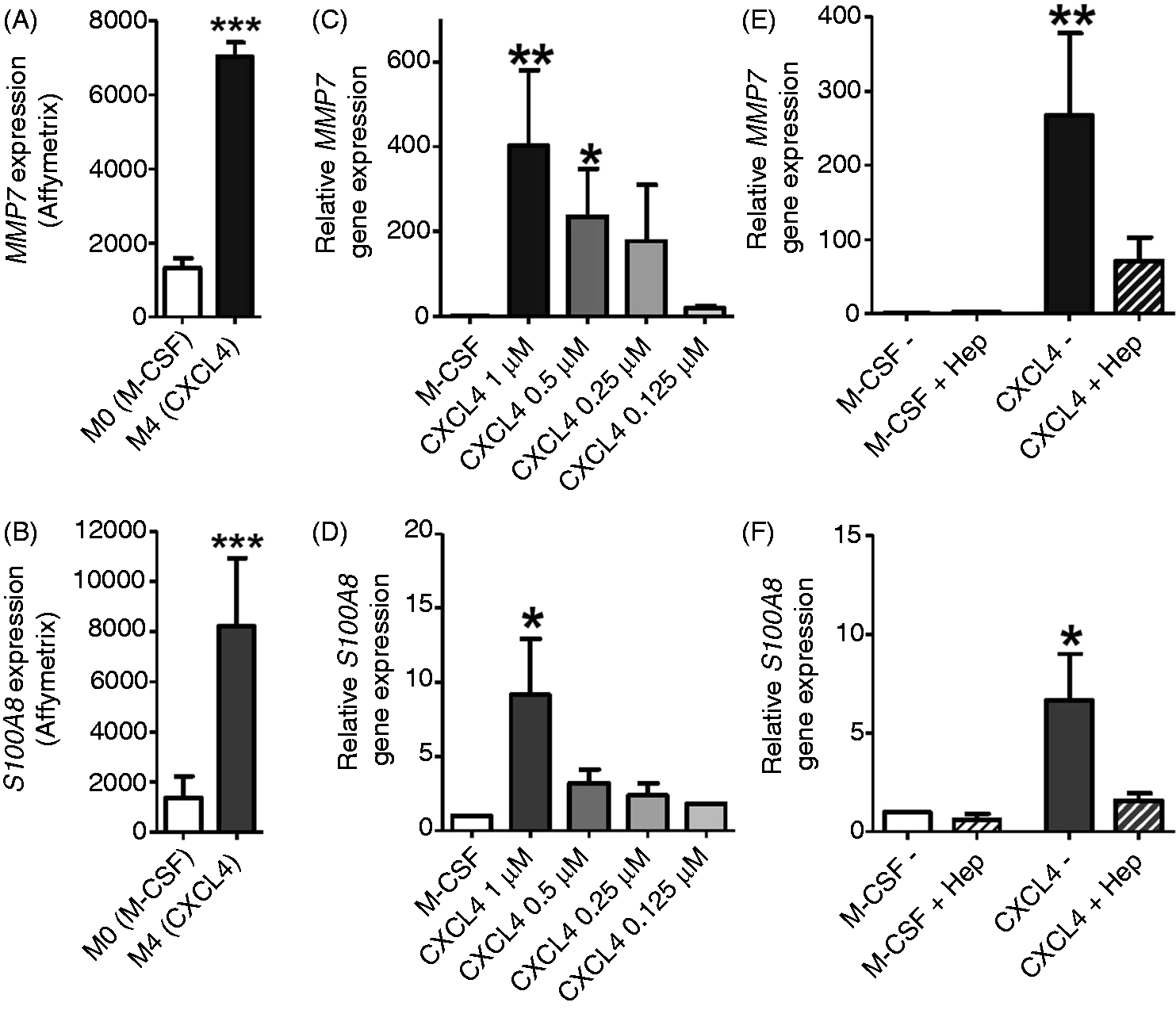
CXCL4 effects on macrophages are most likely mediated by binding to surface glycosaminoglycans. 22 Heparin is known to bind to both CXCL4 and glycosaminoglcans. 28 To test whether CXCL4-dependent induction of MMP7 and S100A8 was specific, we added heparin to monocyte cultures treated with M-CSF for 6 d or those switched to 1 µM CXCL4 on d 3. While addition of heparin had no effect on MMP7 and S100A8 gene expression in M-CSF cultures, addition to CXCL4 cultures resulted in significantly lower MMP7 and S100A8 gene expression levels suggesting that overexpression of both MMP7 and S100A8 was specifically mediated by CXCL4 (Figure 2E, F).
MMP7 and S100A8 expression is specific for CXCL4-induced macrophages
Specificity of both MMP7 and S100A8 expression in M4 macrophages could be demonstrated by radar charts plotting gene expression determined by Affymetrix gene arrays (Figure 3A, B) or quantitative RT-PCR (Figure 3C, D). Some of the markers overexpressed in M4 macrophages have also been associated with M1 or M2 polarization (e.g. TNF with M1 or CCL18 with M2).
17
As a specificity control, we therefore tested whether established marker combinations for M1 or M2 macrophages would accidentally identify M4 macrophages. Radar charts of gene expression data clearly demonstrate sensitivity and specificity for M1 markers (CCR7, TNF, CXCL11, CCL15),
6
while M2 markers [MRC1 (CD206), CD163, CD209 (DI-SIGN), CD36]
6
showed less sensitivity and specificity; in particular, MRC1 expression was also high in M4 macrophages (Figure 3E, F).
MMP7 and S100A8 are specific markers for M4 macrophages. (A, B) Radar plots indicating the ratio of S100A8 or MMP7 gene expression in M1-, M2- and M4-polarized macrophages versus unpolarized macrophages derived from the same donor, as determined by Affymetrix gene array.6,17 (C, D) Radar plots indicating the ratio of S100A8 or MMP7 gene expression in M1-, M2- and M4-polarized macrophages versus unpolarized macrophages derived from the same donor as determined by quantitative RT-PCR. (E, F) Radar plots indicating the gene expression of typical marker genes for M1 or M2 macrophage polarization in M1-, M2- and M4-polarized macrophages. In all cases, gene expression was determined by real time RT-PCR and normalized unpolarized macrophages derived from the same donor.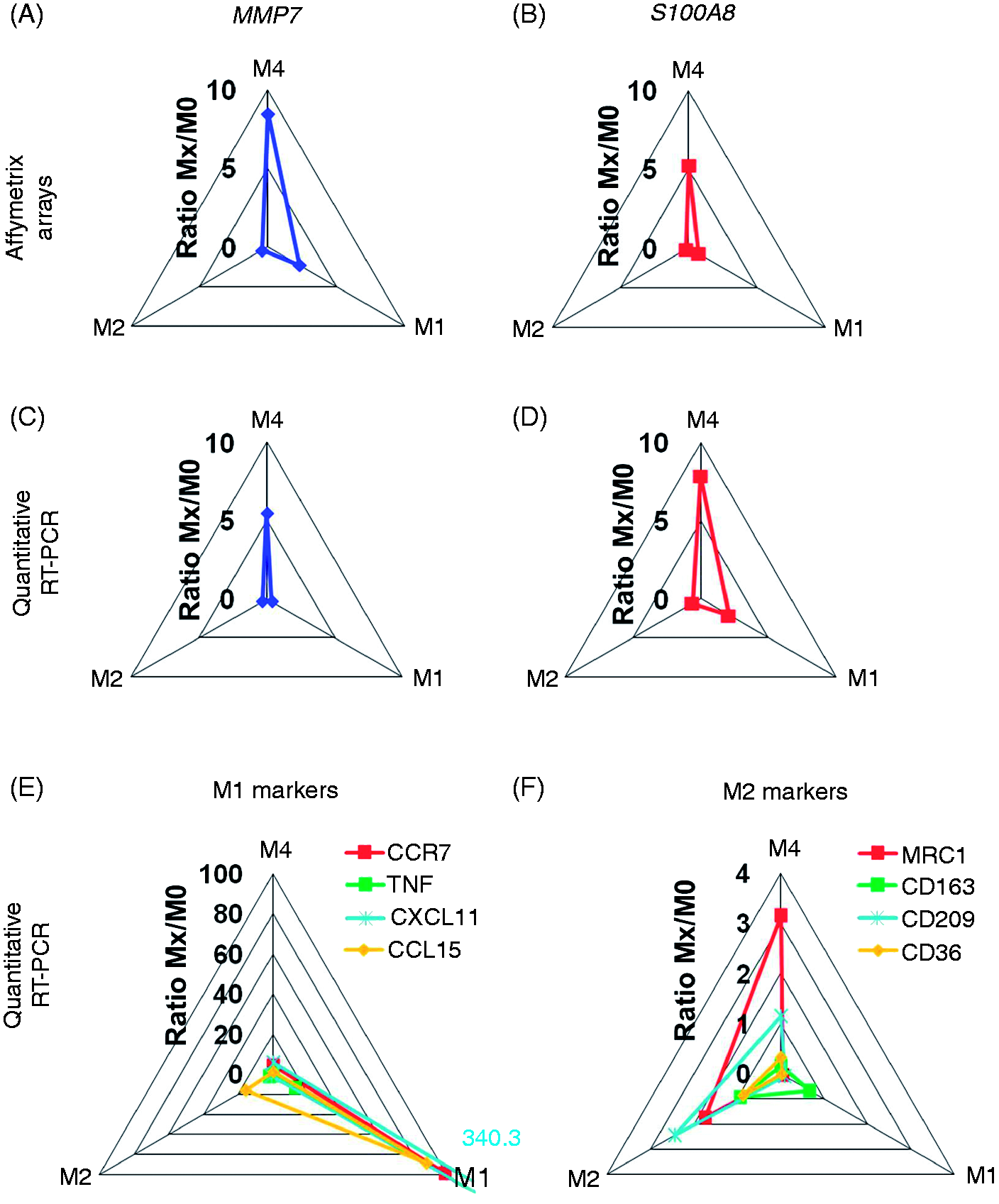
M4 macrophages co-express MMP7 and S100A8 protein in vitro and in vivo
In a next step, we sought to confirm our findings at the protein level. Using intracellular flow cytometry, we found that the levels of MMP7 increased about 1.5-fold in CXCL4-induced macrophages (Figure 4A). Soluble MMP7 measured in cell culture supernatants was not affected (Figure 4B). Immunofluorescence staining revealed significant overexpression of S100A8 protein in CXCL4-induced macrophages compared with M-CSF-induced control cells (Figure 4C). Other than MMP7, soluble S100A8 protein was also increased in supernatants derived from CXCL4-induced macrophages (Figure 4D). These findings demonstrate that MMP7 and S100A8 gene regulation also translates into increased protein levels in M4 macrophages and—at least for S100A8—in increased levels of secreted protein.
In vitro validation of MMP7, S100A8 as M4 macrophage markers. (A) Representative histograms of intracellular measurement of MMP7 protein in monocyte-derived macrophages differentiated with M-CSF or CXCL4. Bar graph indicating relative fluorescence in M-CSF- or CXCL4-induced macrophages, as determined by flow cytometry; n = 6, *P < 0.05 versus M-CSF. (B) Soluble MMP7 in supernatants of monocyte-derived macrophages differentiated with M-CSF (100 ng/ml) or CXCL4 (1 µM) as determined by ELISA; n = 4, P = not significant. (C) Representative images of S100A8 immunostaining in M-CSF- or CXCL4-induced macrophages. (D) Soluble S100A8 in supernatants of monocyte-derived macrophages differentiated with M-CSF (100 ng/ml) or CXCL4 (1 µM) as determined by ELISA; n = 4, *P < 0.05.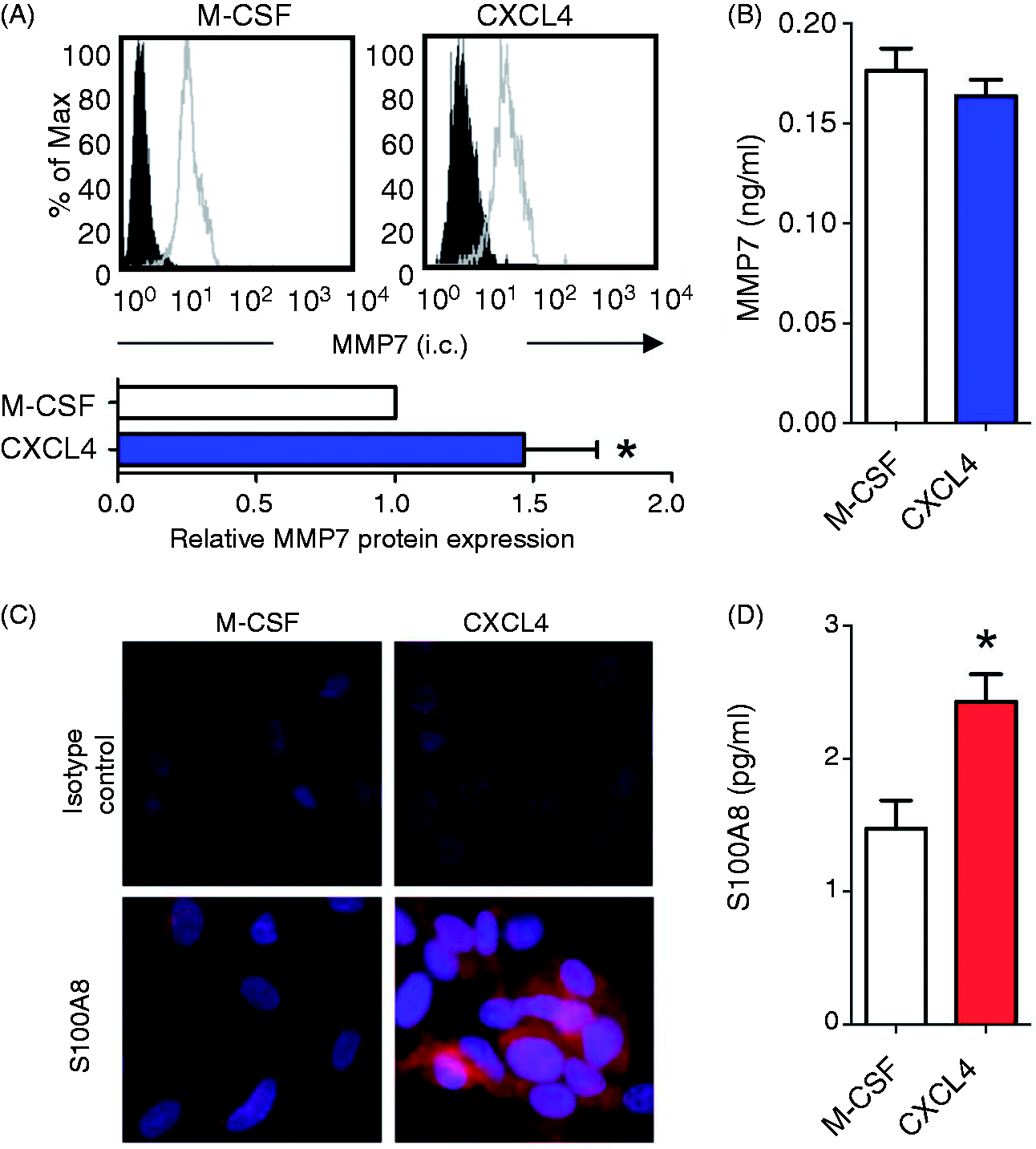
To assess whether there is differential macrophage expression of MMP7 and S100A8 in vivo, human atherosclerotic coronary arteries from eight individual donors were stained post mortem with Abs against the pan-macrophage marker CD68, and either MMP7 or S100A8 in (Figure 5A). In fact, we found both CD68+MMP7+ and CD68+MMP7− macrophages. Similarly, both CD68+S100A8+ and CD68+S100A8− macrophages were present, suggesting substantial heterogeneity for both markers. In order to confirm that MMP7 and S100A8 are co-expressed in a distinct macrophage subset, we performed co-staining, which revealed a large proportion of MMP7−S100A8+ cells (93 ± 9%) and a smaller proportion of MMP7+S100A8+ cells (18 ± 14% of all CD68+ macrophages) within atherosclerotic plaques (Figure 5B), the latter most likely representing M4 macrophages.
In vivo evidence for the presence of M4 macrophages in atherosclerotic lesions. Human coronary arteries from patients with coronary artery disease were stained post mortem with (A) hematoxylin and eosin staining or (B) with Abs against CD68 (FITC) and MMP7 (left) or S100A8 (right, both Texas red, magnification 40×). (C) Human coronary arteries from patients with coronary artery disease were stained post mortem with Abs against MMP7 (FITC) and S100A8 (Texas red, magnification 40×). White boxes are magnified on the right giving examples of (a.) MMP7−S100A8+ or (b.) MMP7+S100A8− macrophages. *Vessel lumen.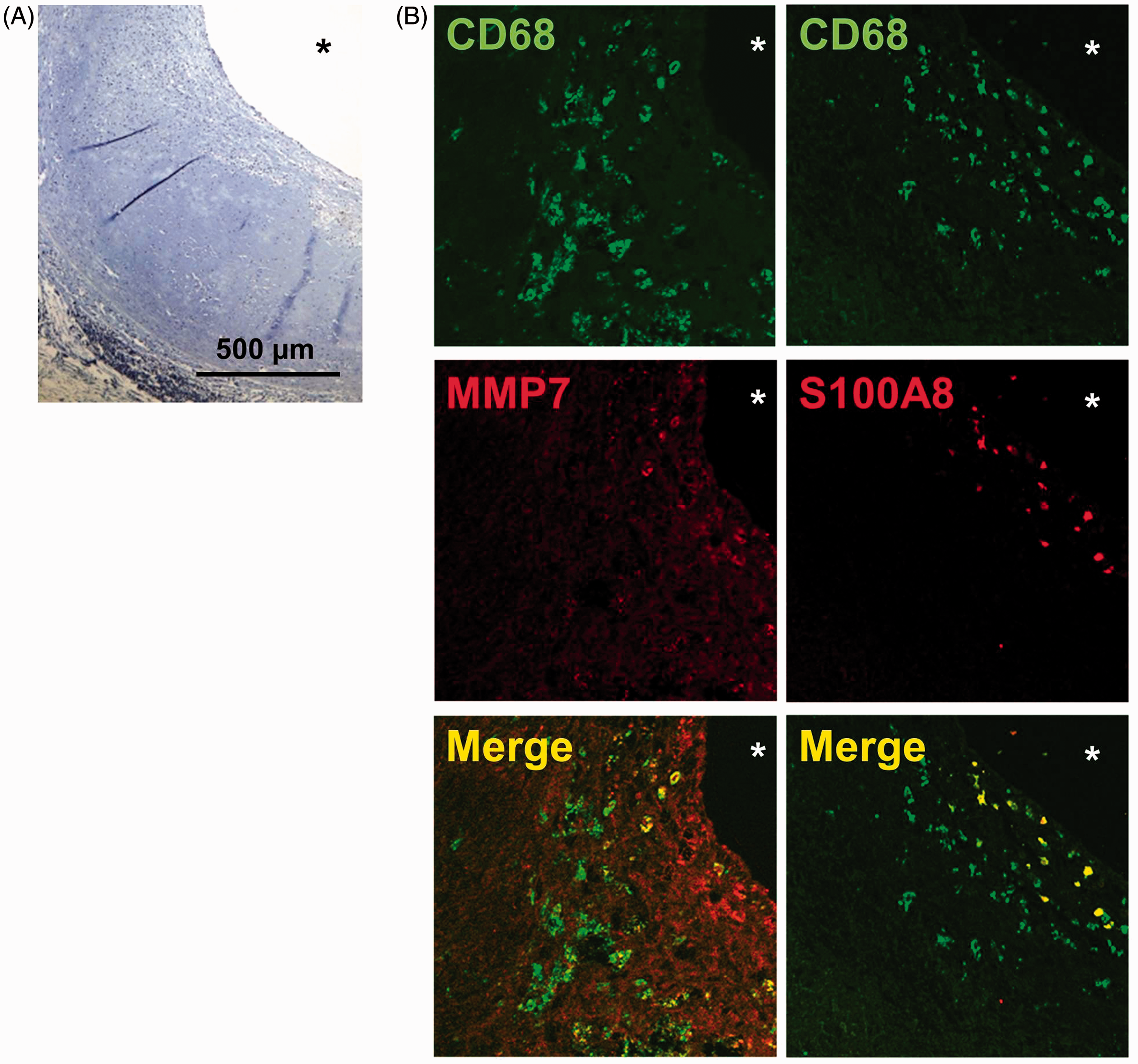
Discussion
Macrophage heterogeneity in atherosclerosis has become a topic of increasing relevance.29–31 The essential role of monocyte-derived macrophages in the disease process was recognized early on, along with the fact that macrophages represent an interesting therapeutic target. 32 Recent evidence for substantial macrophage heterogeneity has made this concept more complicated, but also more promising: assuming that certain macrophage types may act in an atheroprotective manner and that others may promote the disease could offer new therapeutic perspectives.
While in vitro M1- or M2-polarized macrophages can be readily identified by using one or two markers (such as CD206 for M2 macrophages 33 ), identification of macrophage subtypes in vivo is more difficult. This is mainly owing to the fact that in vivo macrophage polarization is oftentimes less distinctive and may lead to intermediate polarization types. Thus, for histological identification of macrophage types, other approaches are necessary, which may include staining for polarization-specific transcription factors or staining for more than one or two markers. 34
We have recently described CXCL4-induced macrophages as a specific macrophage type distinct from previously characterized macrophage polarization types and therefore suggested calling these macrophages ‘M4’. 17 M4 macrophage display a transcriptome distinct from classically (M1) or alternatively polarized (M2) macrophages. 17 Furthermore, these CXCL4-induced macrophages are characterized by a distinct cytokine profile. 17 M4 macrophages lack the hemoglobin receptor CD163 and are unable to up-regulate atheroprotective heme oxygenase-1 in response to exposure to hemoglobin or hemoglobin–haptoglobin complexes. 17
Interestingly, CXCL4-dependent loss of CD163 was irreversible suggesting that—other than M1 or M2 polarization—M4 macrophages represent a final stage of cell differentiation. 22 Accordingly, it seems reasonable to study the role of M4 macrophages within human atherosclerotic lesions in more detail.
While lack of CD163 could be shown to be a characteristic feature of these cells, detailed information and suitable markers to clearly identify M4 macrophages in vitro or in vivo have been lacking. However, knowing these markers is indispensable in order to exploit M4 macrophages as a therapeutic target in human atherosclerosis. As using merely one or two markers may not be sufficient to identify polarized macrophages in vitro or in vivo, 34 we sought to identify a combination of three markers that sensitively and specifically identify M4 macrophages in vitro and in vivo. Using a statistical approach based on transcriptomic data on M1, M2 and M4 macrophages,6,17 we here identify co-expression of MMP7 and S100A8 as a specific feature of CXCL4-induced macrophages in vitro and in vivo. To our knowledge, neither in vitro nor in vivo MMP7+S100A8+ macrophages have been described in the past. Thus, the combination of these two markers is specific for the identification of M4 macrophages.
Our study did not specifically focus on the functional aspects of CXCL4-dependent induction of MMP7 and S100A8, even though both markers have been associated with atherosclerosis. Thus, MMPs, such as MMP7, are enzymes that are able to degrade extracellular matrix. 35 This is of special importance in atherosclerotic plaques, where degradation of structural proteins leads to thinning of the fibrous plaque cap and subsequently to plaque rupture and blockage of the artery. 35 The exact role of MMP7 in this context is still under investigation. Using Mmp7−/− mice, as well as selective MMP7 inhibitors, Williams et al. 36 could demonstrate that MMP7 induces smooth muscle cell apoptosis, resulting in destabilization of atherosclerotic plaques. Similarly, a correlation between smooth muscle cell apoptosis and MMP7 expression could be demonstrated in human atherosclerotic lesions, supporting a role for MMP7 in human disease. One could speculate that increased MMP7 expression by M4 macrophages leads to plaque destabilization and may thereby promote disease progression.
S100A8 is a calcium binding protein and forms heterodimers with S100A9 in human plasma. 37 S100A8/S100A9 plasma levels have been associated with various inflammatory diseases, including diabetes, 38 chronic inflammatory bowel disease or rheumatoid arthritis. 39 There is evidence suggesting a role for S100A8/S100A9 as biomarker for cardiovascular events. 40 Thus, plasma levels have been associated with both myocardial infarction and cardiovascular death. Also, the risk for recurrent adverse cardiovascular events was associated with increased S100A8/S100A9 plasma levels. Myeloid cells represent a major source of S100A8/S100A9. 37 Interestingly, S100A8 has been reported to control the cell cycle. 41 Considering that lesional macrophage accumulation within atherosclerotic plaques depends on macrophage proliferation, 5 increased expression of S100A8 may affect lesion progression through decreased macrophage proliferation.
Within human atherosclerotic plaques, we were able to demonstrate the presence MMP7+S100A8+ macrophages. While both markers individually are not specific for macrophages, but expressed by various other cell types, the combination of CD68, MMP7 and S100A is macrophage-specific. Based on our in vitro data, MMP7+S100A8+ macrophages most likely represent M4 macrophages induced by CXCL4, even though one cannot fully exclude that there may be other mediators that induce a similar macrophage phenotype. However, to the best of our knowledge, co-expression of MMP7 and S100A8 has not been demonstrated in any macrophage polarization type thus far.
While we find CD68+MMP7+S100A8+ macrophages in human atherosclerotic plaques ex vivo, focusing on human samples constitutes a limitation regarding the study of these cells in vivo. However, one has to be aware that mouse models may be not ideal to study macrophage heterogeneity relevant to human disease. Thus, LPS induces increase uptake of modified low-density lipoprotein (LDL) in murine macrophages 42 but, at the same time, it inhibits LDL uptake in human monocyte-derived macrophages. 43 Similarly, a recent study comparing the inflammatory response in humans and mice has revealed substantial differences. 44 We therefore believe that focusing on human macrophages is reasonable in order to assure the potential clinical relevance of the findings.
Based on our findings, we propose a combination of CD68, MMP7 and S100A8 to identify M4 macrophages in vivo. We here show that this combination is specific and does not show great overlap with other macrophage polarization types. Also, we demonstrate that established marker combinations to identify M1 or M2 macrophages do not accidentally identify M4 macrophages. Accordingly, our data may represent the foundation for further studies investigating the prevalence and function of M4 macrophages in human atherosclerosis, potentially leading to novel therapeutic approaches.
Footnotes
Funding
This work was funded by the Innovation Fund FRONTIER (University of Heidelberg) to CAG, by the DZHK (German Centre for Cardiovascular Research) and by the BMBF (German Ministry of Education and Research) to HAK, and the DFG (German Research Foundation) to FL (SFB938/TP Z2).
Acknowledgements
We thank Nadine Wambsganss and Jutta Scheuerer for excellent technical assistance.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.