
Abstract
Monocyte chemoattractant protein-1 (MCP-1) regulates monocyte accumulation in several macrophage-dependent experimental disease models. In the neonatal brain, activated microglia accumulate rapidly after hypoxic-ischemic injury. These cells produce potentially neurotoxic factors that may contribute to the progression of injury. To determine whether MCP-1 could be one of the molecular signals that influences the microglial response to hypoxic-ischemic injury in the neonatal brain, we examined the impact of acute hypoxic-ischemic injury on MCP-1 mRNA and protein expression. Seven-day-old rats underwent right carotid artery ligation, followed by 3 hours of 8% oxygen exposure, to elicit ipsilateral forebrain hypoxic-ischemic injury. To detect MCP-1 mRNA in situ hybridization assays were performed using 35S-labeled antisense riboprobes generated from rat MCP-1 cDNA. Animals were evaluated 0, 1, 2, 4, 8, 16, 24, 48, and 120 hours after hypoxic exposure (N ≥ 3/group). Immunocytochemistry (with a polyclonal rabbit antirat MCP-1 antibody) was used to determine the anatomic and temporal distribution of MCP-1, in samples obtained 10 minutes to 5 days after hypoxic exposure (N ≥ 3/group). Monocyte chemoattractant protein-1 mRNA was first detected in periventricular regions of the lesioned hemisphere 1 hour after hypoxia-ischemia; periependymal and intraparenchymal MCP-1 mRNA expression were detected at 4 hours; hybridization signal peaked at 8 to 24 hours; and no MCP-1 mRNA was detected at 48 and 120 hours. In lesioned forebrain, MCP-1 protein expression were consistently detected at 2.5 to 48 hours after hypoxia-ischemia. Many immunoreactive cells appeared to be neurons. These results suggest that in the developing brain, MCP-1 could represent a functionally important molecular signal for the microglial response to hypoxic-ischemic injury.
There is increasing recognition that activated CNS microglia contribute to the progression of neurodegeneration after diverse forms of brain injury (Giulian, 1987; McGeer et al., 1993). Microglia are a major cellular source of proinflammatory cytokines and other potentially neurotoxic-soluble products (Guilian et al., 1990; Thomas, 1992). It is unknown to what degree the functional properties of these activated cells vary in vivo, and whether the soluble products that they secrete are developmentally determined. In healthy 7-day-old rat brain, activated microglia (by morphological criteria) are readily detected, and both hypoxic-ischemic and excitotoxic injury elicit a rapid, relatively widespread microglial response (Acarin et al., 1996; Ivacko et al., 1996; McRae et al., 1995; Ohno et al., 1995). Blood-derived monocytes cannot be readily distinguished from activated microglia and the contribution of blood-derived monocytes to this cellular response has not been established. Distinctive developmental features of the microglial/monocyte response include the rapidity of onset and its relatively short duration, in comparison with similar adult brain injury models.
The molecular signals that initiate the microglial response are unknown. Monocyte chemoattractant protein-1 (MCP-1) is one of the best-characterized monocyte chemokines (Rollins et al., 1990; Carr et al., 1994; Oppenheim et al., 1991). It is a member of the “CC” chemokine gene family of cytokines, characterized by their chemotactic activity for mononuclear phagocytes. Monocyte chemoattractant protein-1 also induces a respiratory burst, and triggers monocyte cytokine secretion. In several diverse models of monocyte-dependent pathological disorders, MCP-1 expression precedes monocyte/macrophage accumulation (Flory et al., 1993), and functional inhibition of MCP-1 can limit the extent of tissue injury in monocyte/macrophage-dependent pathological processes (Flory et al., 1995).
The first evidence that CNS disease could lead to increased MCP-1 mRNA expression was reported in experimental allergic encephalitis (Hulkower et al., 1993; Ransohoff et al., 1993). Subsequently, MCP-1 was detected in brain tumor tissue (Takeshima et al., 1994), and transient increases in MCP mRNA expression were detected in temporary and permanent cerebral ischemia models in adult rats (Kim et al., 1995; Wang et al., 1995). Recently, Glabinski et al. (1996) showed that in adult mice a cortical stab wound stimulated astrocyte MCP-1 expression acutely. In this study, a similar stab wound elicited considerably less MCP-1 mRNA expression in the brains of neonatal animals, a finding that was interpreted as evidence of a developmentally regulated inflammatory response.
However, there is increasing recognition that inflammatory mediators may play an important role in the pathogenesis of hypoxic-ischemic injury in the immature rodent brain (Palmer, 1995). In the lesioned forebrain within the first 12 hours after injury, proinflammatory cytokine gene expression increases markedly (Szaflarski et al., 1995) and there is a robust microglial response (McRae et al., 1995; Ohno et al., 1995; Ivacko et al., 1996) in a neonatal rat stroke model elicited by unilateral carotid ligation followed by 2 to 3 hours of exposure to 8% oxygen (O2) in 7-day-old rats. In this model, drugs that are functional antagonists of specific components of the inflammatory cascade, including interleukin-1 receptor antagonist (Martin et al., 1994), and the PAF antagonist BN52021 (Liu et al., 1996) are neuroprotective.
We hypothesized that MCP-1 could be one of the molecular signals that regulated the acute microglial response to hypoxic-ischemic injury in neonatal rat brain. In situ hybridization assays with a rat-specific MCP-1 cDNA probe (Jones et al., 1992a) were used to evaluate the temporal and anatomic features of MCP-1 mRNA expression induced after hypoxic-ischemic injury and immunocytochemistry assays with a polyclonal rabbit antirat MCP-1 antibody (Jones et al., 1992b) were used to determine the distribution of the encoded protein.
METHODS
Animal lesioning
Sprague-Dawley rats were purchased from Charles Rivers Laboratories, and maintained under a 12-hour light/dark cycle with free access to food and water. Seven-day-old rats, anesthetized with methoxyfluorane, underwent right carotid ligation, using previously reported methods (Rice et al., 1981; Barks and Silverstein; 1992). After a 1-hour recovery period, they were placed in plastic chambers, partially submerged in a water-bath (36.5°C) and exposed to 8% oxygen/92% nitrogen for 3 hours. They were maintained in a warming incubator (set at 35.5°C) for a 30-minute recovery period, and then were returned to their dams. Surgical and animal use protocols were approved by the University of Michigan Committee on Use and Care of Animals.
For in situ hybridization assays, survival times of 0, 1, 2, 4, 8, 16, 24, 48, and 120 hours were evaluated (N ≥ 3/group). For the immunocytochemistry experiments, animals were killed at 10 minutes, 2.5, 4, 8, 12, 24, 48, and 120 hours after hypoxia-ischemia (N ≥ 3/group). For each time point, two unlesioned animals were also evaluated. Animals examined at each time interval were derived from at least two independent experiments. The time intervals selected for evaluation were based on information about the timing of the microglial response to injury (Ivacko et al., 1996) and on results of preliminary experiments that confirmed that these times encompassed the peak induction of MCP-1 expression.
In situ hybridization assays
Tissue preparation.
Animals were killed by decapitation and brains were removed intact and frozen on dry ice. Brains were stored at −70°C. 20-μm frozen coronal sections were mounted on poly-L-lysine (Sigma, St. Louis, MO, U.S.A.) coated slides and stored at −70°C. On the day of the assay, sections were thawed at room temperature and fixed for 1 hour in 10% phosphate-buffered saline (PBS) buffered formaldehyde (Mallinckrodt, Paris, KY, U.S.A.).
To confirm the adequacy of lesioning, cytochrome oxidase histochemistry assays were routinely performed in ≥10 sections/brain (as previously described, Nelson and Silverstein, 1994); there was ipsilateral suppression of cytochrome oxidase activity in all samples.
Probe preparation.
A linearized pBluescript 11 SK plasmid (Pharmacia, Piscataway, NJ, U.S.A.) that contained the complete rat MCP-1 cDNA insert (Jones et al., 1992a) was used to transcribe antisense and sense 35S-UTP-RNA probes (35S-UTP SA: 1200 Ci/mmol, NEN Dupont, Wilmington, DE, U.S.A.; MAXIscript kit, Ambion, Austin, TX, U.S.A.).
Assay procedures.
Slides were rinsed extensively in 1 × PBS, treated with Proteinase K (5 μg/mL, 37°C, 5 minutes), incubated sequentially in 0.1M TEA (3 minutes) and in 0.1 mol/L TEA/0.25% acetic anhydride (10 minutes), washed in 2 × SSC, dehydrated in ethanol gradients, and air dried. Sections were covered with 40 μL of hybridization solution containing 106 cpm of the 35S-UTP-labeled antisense or sense riboprobe (2/3 sections allocated to antisense group), cover-slipped, and incubated overnight (≥16 hours) in humidified chambers at 55°C. The hybridization solution consisted of 50% deionized formamide; 0.01 mol/L Tris, pH 8.0; 0.001 mol/L EDTA, pH 8.0; 0.35M NaCl; 10% dextran sulfate (molecular weight 400,000), and 0.1 mol/L dithiothreitol. Proteinase K and dithiothreitol were purchased from Boehringer Mannheim (Indianapolis, IN, U.S.A.); all other reagents were purchased from Sigma (St. Louis, MO, U.S.A.).
Coverslips were then removed, sections were washed in 2 × SSC/dithiothreitol and treated with 50% formamide/2 × SSC (55°C, 30 minutes). Unbound probe was digested with 50 μg/mL RNase A (37°C, 30 minutes), and removed by incubation in RNase A free buffer (60°C, 30 minutes). Slides were dehydrated in ethanol gradients, air dried, and exposed to X-ray film for 2 to 4 weeks (XAR, Kodak, Rochester NY, U.S.A. or REFLECTION, NEF, E.I. Dupont, Wilmington, DE, U.S.A.). Representative sections were prepared for emulsion autoradiography. The slides were dipped in emulsion (Kodak NTB-2, diluted 1:1 with deionized water), air dried, and exposed for 6 to 8 weeks at 4°C. They were developed (in Kodak D-19), and lightly counterstained with cresyl violet.
Immunocytochemistry
Tissue preparation.
Animals were deeply anesthetized with chloral hydrate (400 mg/kg) and perfused transcardially with 4% paraformaldehyde. Brains were postfixed (4% paraformaldehyde, ≥4 hours, 4°C), cryoprotected in graded sucrose solutions, and stored in 20% sucrose/0.02% sodium azide in PBS, pH 7.4. Immediately before tissue sectioning, brains were immersed in a 2:1 solution of 20% sucrose and OCT (Miles, Elkhart, IN, U.S.A.) for 45 minutes, and then were frozen, embedded in this solution, by immersion in isopentane, cooled to −40°C. Twenty μm frozen coronal sections were mounted on gelatin-coated slides, and stored for at least 4 hours at −20°C. Sections were thawed at room temperature, postfixed (4% paraformaldehyde, 30 minutes), and rinsed in 0.1 mol/L PBS, pH 7.4/0.1% Triton-X-100, for 15 minutes.
Antibody characterization.
The details of the preparation and characterization of the rabbit antirat MCP-1 antibody used in this assay were previously reported (Jones et al., 1992b). The antibody reacted specifically with recombinant rat MCP-1 species by Western immunoblot analysis, and neutralized both monocyte chemotaxis induced by recombinant MCP-1 and native monocyte-specific chemotactic activity secreted by tumor necrosis factor-α-stimulated rat endothelial cells. The molecular weight of the endothelium-derived monocyte-specific chemotactic activity corresponded with the expected size of native rat MCP-1 (Yoshimura et al., 1991).
Assay procedure.
Sections (≥24/brain) were preincubated with normal goat serum (150 μL/10 mL PBS/0.3% Triton-X, 20 minutes), and then incubated with anti-MCP-1 antibody (60 μL of solution containing 10 μg/mL PBS/0.3% Triton-X) overnight at 4°C. For negative controls, (≥4/brain), equivalent dilutions of rabbit IgG were substituted for the primary antibody. After three washes (PBS/0.1% Triton-X, 5 minutes), sections were incubated 30 minutes at room temperature with biotinylated goat antirabbit IgG (diluted according to manufacturer's instructions [Vectastain ABC Elite Kit, Vector Laboratories, Burlingham, CA, U.S.A.]). Sections were washed three times with PBS/0.1% Triton-X and immersed in 0.3% H2O2 in methanol for 10 minutes to block endogenous peroxidase activity. After three rinses, the sections were incubated with the avidin-biotin enzyme complex (Vectastain ABC Elite Kit, Vector Laboratories) for 30 minutes at room temperature, rinsed in PBS, and immersed in 3,3” diaminobenzidine in H2O2 (6 to 10 minutes). Sections were then rinsed in water, lightly counter-stained with cresyl violet, dehydrated through a graded series of alcohols, cleared with xylene, and coverslipped with Permount mounting media.
Lectin histochemistry.
The distribution of microglia and monocytes was evaluated with a lectin histochemistry assay, which enables detection of microglial activation and accumulation within the first 4 hours after hypoxia-ischemia (as previously described, Ivacko et al., 1996). A limitation of this assay is that it also labels endothelial cells which are distinguishable based on their morphology. Sections were immersed in 0.3% H2O2 in methanol for 10 minutes, washed in PBS/0.1% Triton-X, and then incubated with 60 μL of the Griffonia simplicifolia-B4-isolectin-horseradish peroxidase conjugate (Sigma), 10 μg/mL, overnight at 4°C. After three washes with PBS/0.1% Triton-X, sections were incubated with 3,3” diaminobenzidine-H2O2 for 5 to 8 minutes. Sections were counterstained with cresyl violet. Controls included omission of the lectin or incubation with 10 μg/mL lectin in 0.1 mol/L melibiose (a competitive inhibitor for lectin binding).
RESULTS
In this neonatal stroke model, tissue damage evolves predominantly ipsilateral to the carotid ligation. Typically, neuronal injury and substance loss are observed in the ipsilateral cortex, hippocampus, striatum, and thalamus. In the hippocampus, in contrast with adult cerebral ischemia models, both CA1 and CA3 pyramidal neurons are susceptible to injury, and dentate gyrus neurons are also damaged. Injury-induced microglial activation and accumulation proceed rapidly. Lectin histochemistry assays reveal subtle evidence of microglial activation within 10 minutes after the hypoxic-ischemic insult, and morphologic evidence of microglial activation is readily evident within 4 hours after the hypoxic-ischemic insult (Ivacko et al., 1996). Within 12 hours, many activated microglia accumulate in all lesioned forebrain structures. Results of a lectin histochemistry assay, illustrated in Fig. 1, show the distributions of resting and activated microglia in the left and right hippocampus 12 hours after lesioning; microglia are widely distributed bilaterally (panels A and B). Activated microglia, which are characterized morphologically by enlarged cell bodies and shortened, thickened cell processes, are readily discernible only in the lesioned hippocampus (panels D and F [arrows]). Of note, fully activated microglia cannot be morphologically distinguished from blood-derived monocytes. Activated microglia infiltrate the pyramidal cell layer only in the right hippocampus. This pattern is seen both in the CA1 sub-field (highlighted in panels D and F) and to a similar extent in CA3. In all perfusion-fixed brain tissue samples that were obtained between 4 hours and 5 days after hypoxia-ischemia, microglial activation was detected by lectin histochemistry in the lesioned forebrain.
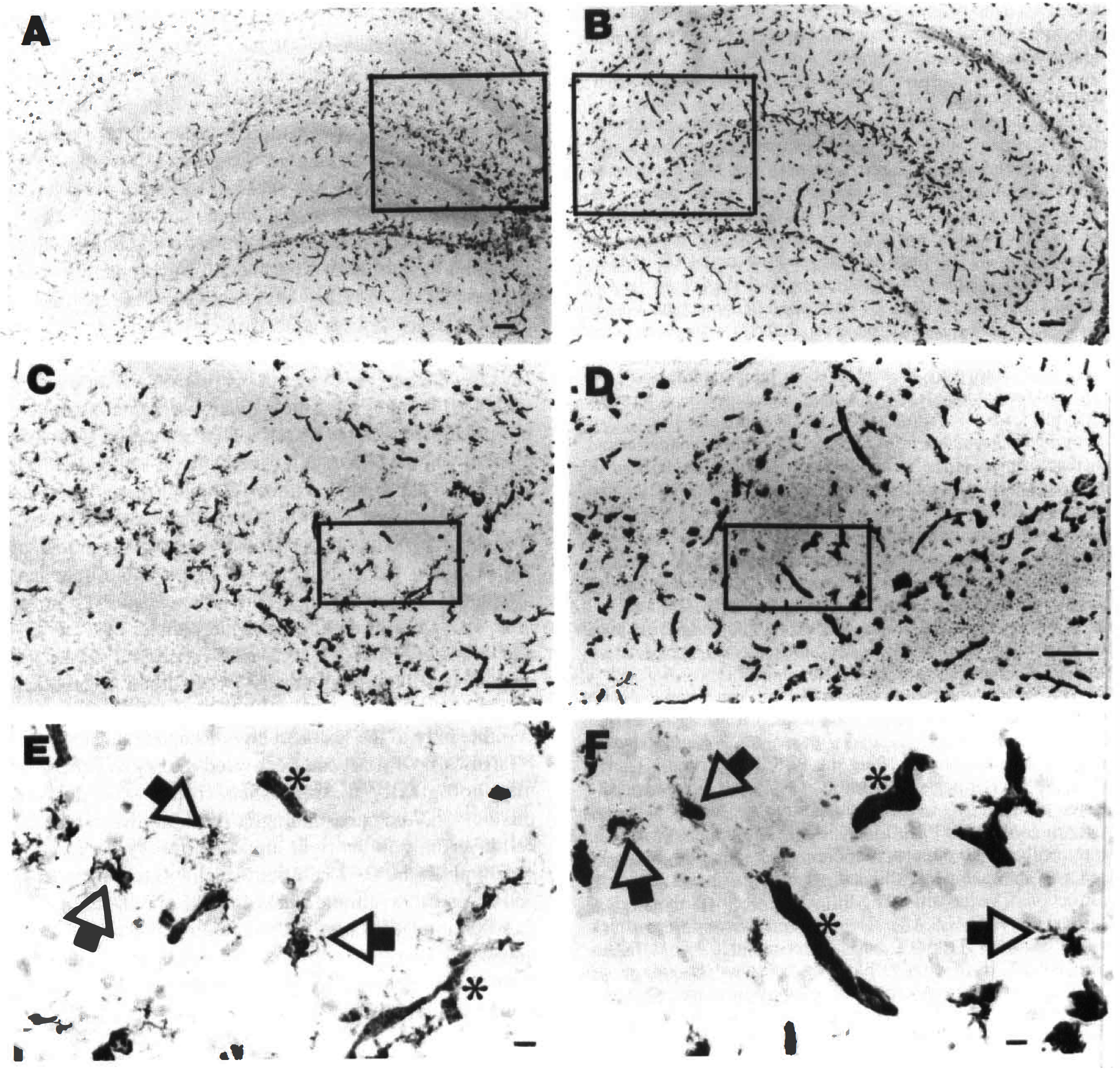
The typical distribution and morphology of microglia in the left (
In situ hybridization
X-ray autoradiography showed MCP-1 mRNA expression in the lesioned forebrain at 4 to 24 hours after hypoxia-ischemia. Immediately or 2 hours after hypoxia-ischemia, no hybridization was detected; at 4 hours, MCP-1 mRNA was consistently detected. Fig. 2 compares the distribution of MCP-1 mRNA at the level of the dorsal hippocampus, at 4, 8, and 24 hours after lesioning. At 4 hours, hybridization signal was consistently most intense in the choroid fissure, between the hippocampus and adjacent thalamus (Fig. 2A). By 8 hours (Fig. 2B), more extensive hybridization signal was detected within the parenchyma of the lesioned hemisphere, particularly in periventricular regions including the subiculum/CA1 subfield of hippocampus, habenula, and thalamus. At 24 hours (Fig. 2C), MCP-1 mRNA expression was routinely detected throughout the hippocampus, including the CA1 and CA3 subfields and the dentate gyrus. In addition, at 8 to 24 hours, MCP-1 mRNA expression was also detectable in the lesioned striatum (not shown). No hybridization signal was detected in any samples obtained at 48 hours or 120 hours after hypoxia-ischemia. No hybridization signal was detected in sense strand controls that were included in every assay (Fig. 2D).
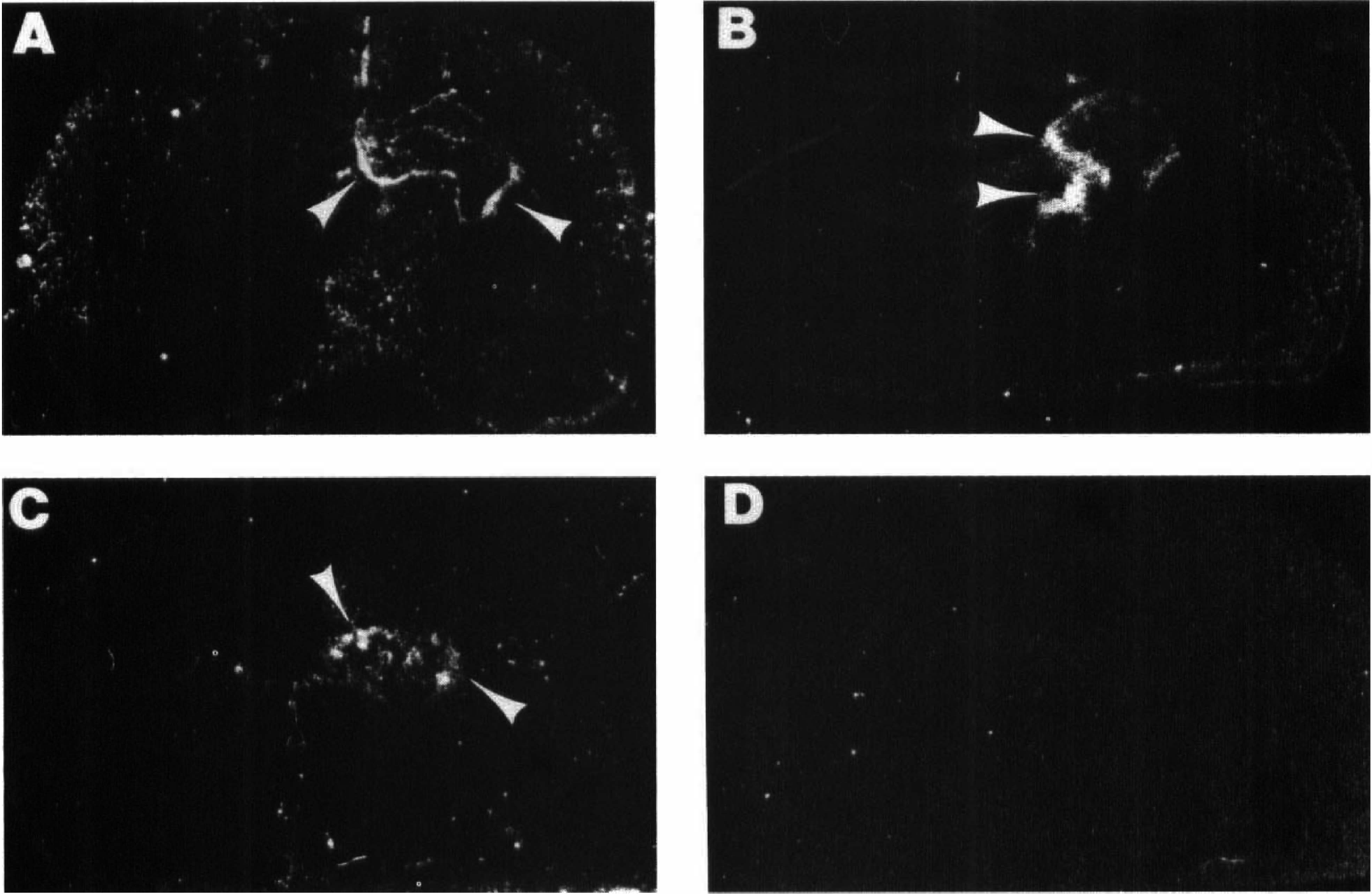
These dark-field autoradiograms show the distribution of monocyte chemoattractant protein-1 (MCP-1) mRNA in coronal brain sections from three animals, evaluated 4 hours
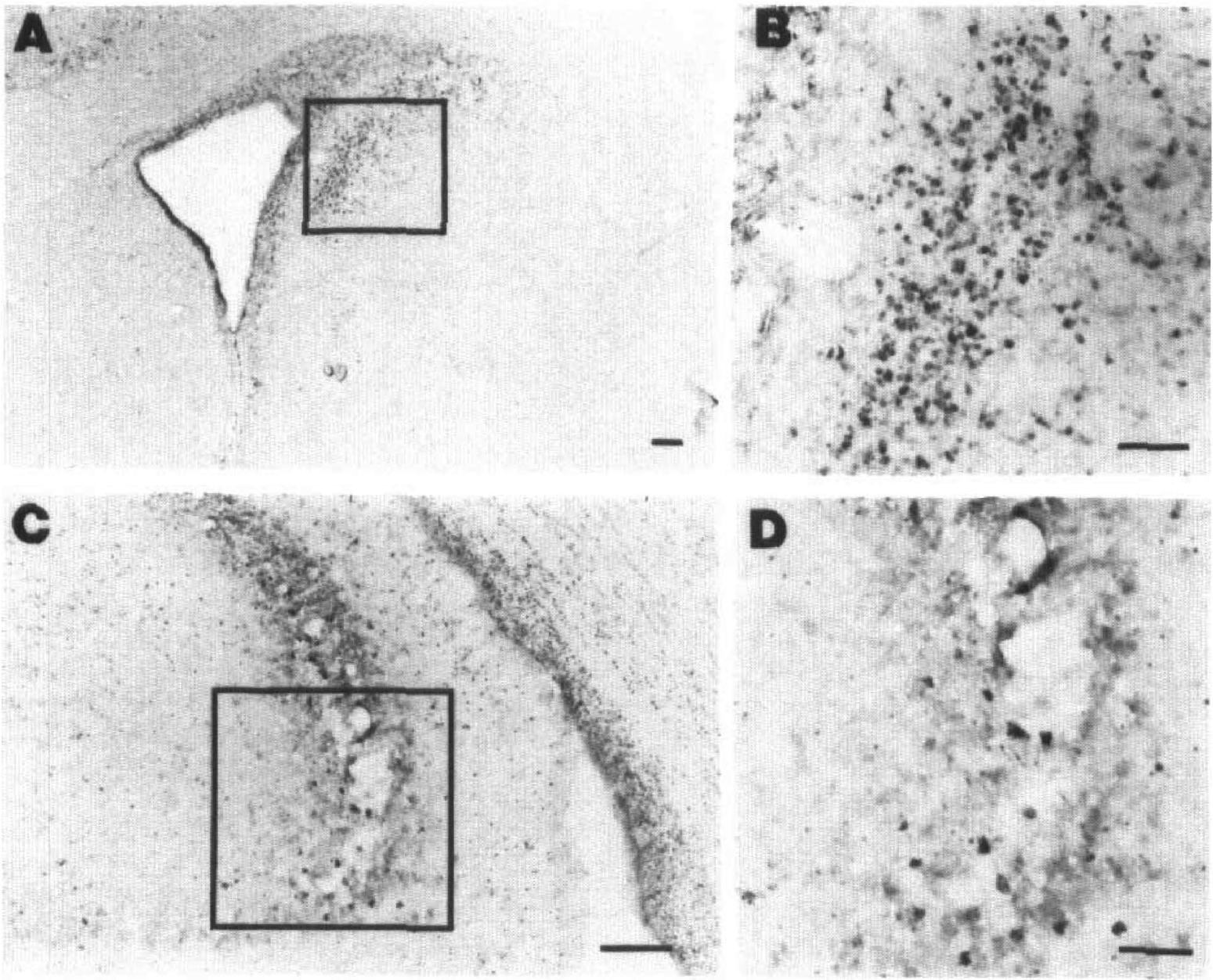
Emulsion autoradiography provided greater sensitivity, and MCP-1 mRNA expression was detected as early as 1 hour after lesioning (in 2 out of 3 samples evaluated). Hybridization signal was localized to ependymal and periependymal cells adjacent to the right choroid fissure (between the right hippocampus and thalamus). At 2 hours after hypoxia-ischemia, in all three samples evaluated, MCP-1 mRNA expression was evident in the periventricular ependymal cells of the lesioned hemisphere. In one of three, MCP-1 mRNA expression was also detected within brain parenchyma, in the lesioned thalamus, and cortex (data not shown). At 4 hours after hypoxia-ischemia (Fig. 3), intense hybridization signal was concentrated in periventricular ependymal cells. In six of seven animals evaluated, MCP-1 mRNA expression was also detected within the lesioned hippocampus. Based on their location, morphology, and cresyl violet staining pattern, many cells that expressed MCP-1 mRNA appeared to be neurons. For example, Fig. 3 illustrates scattered cells within the pyramidal cell layer of the CA1 that showed intense hybridization signal.
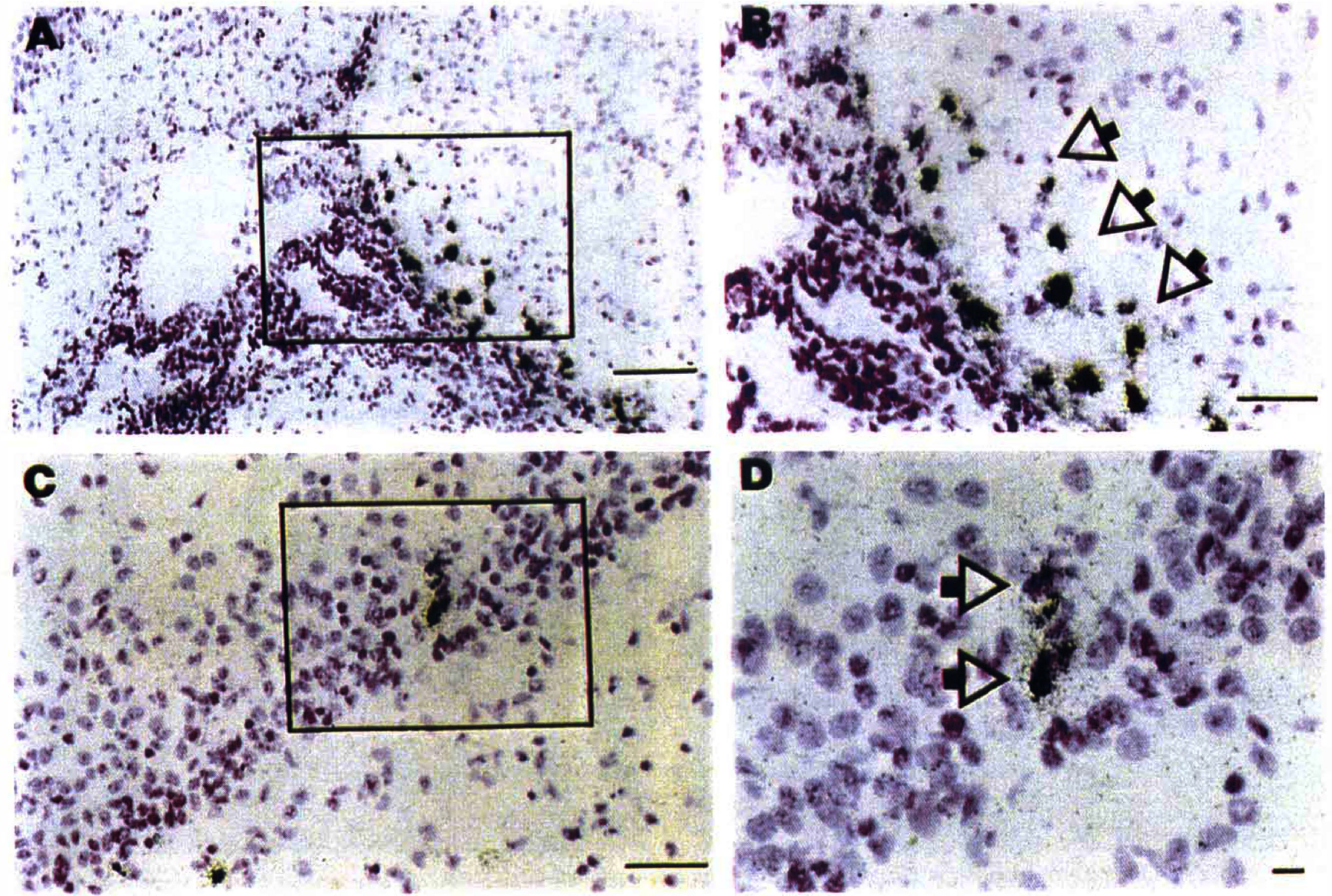
These emulsion autoradiograms document the distribution of MCP-1 mRNA expression at the level of the hippocampus in a 7-day-old rat that was evaluated 4 hours after right carotid ligation and 3 hours of 8% O2 exposure. An 35S-UTP-labeled rat MCP-1 antisense riboprobe was used for in situ hybridization assays of MCP-1 mRNA (see Methods). Hybridization signal is visualized by the distribution of silver grains. Sections were counter-stained with cresyl violet. The boxed areas in
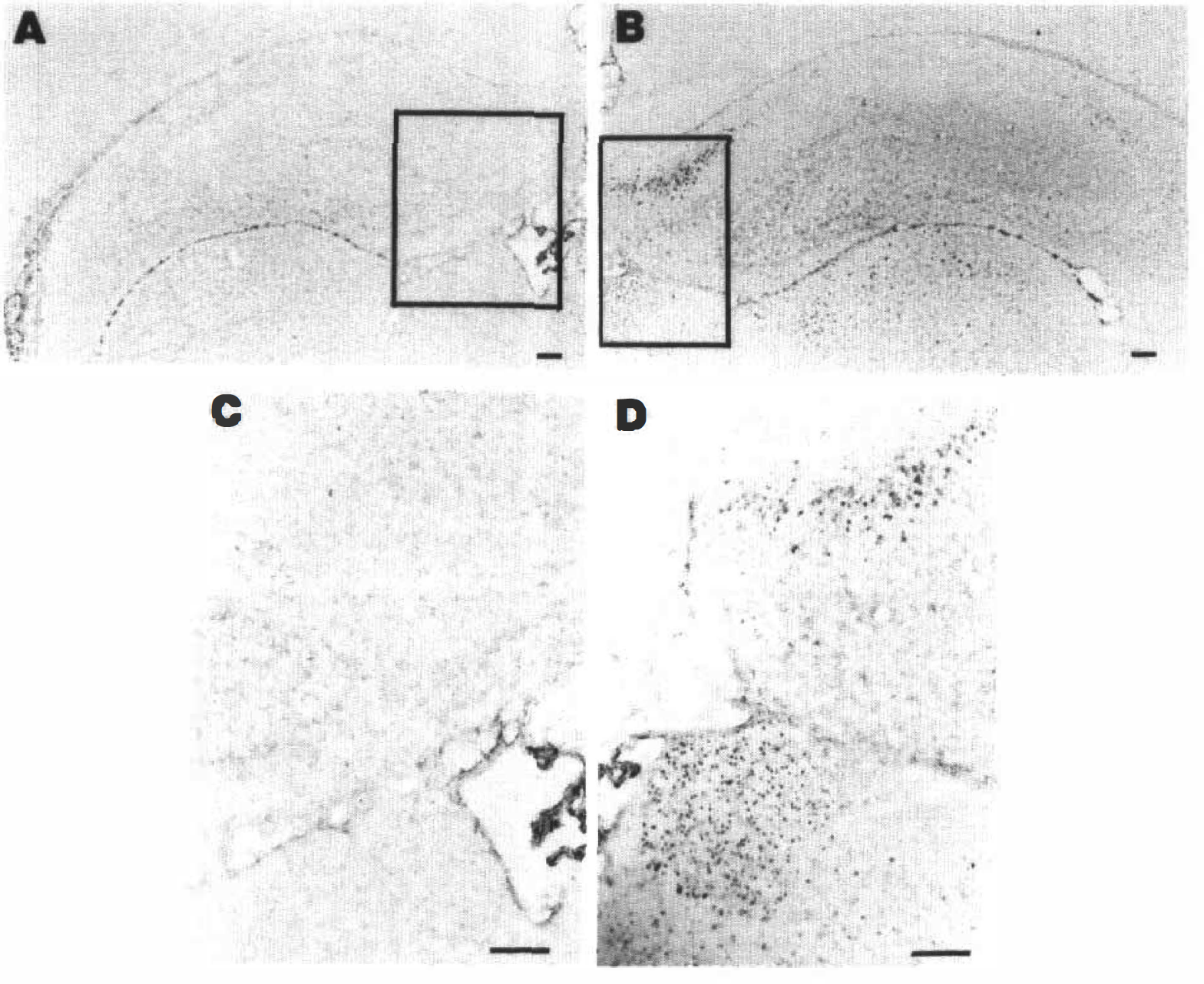
By 16 hours after lesioning, MCP-1 mRNA expression was detected throughout the lesioned cerebral hemisphere. Fig. 4 illustrates the distribution of MCP-1 mRNA in the pyramidal cell layer of the hippocampal CA3 subfield of hippocampus, the thalamus, and the cortex. In the lesioned cortex, typically hybridization was most intense in the middle layers. A columnar pattern of cortical neuronal injury has been described in this model (Rice et al., 1981); however, there was no columnar distribution of hybridization signal. The overall distribution of MCP-1 mRNA was similar at 24 hours. In contrast, no MCP-1 mRNA was detected in samples from animals evaluated at 48 or 120 hours after lesioning.
These emulsion autoradiograms show the distribution of MCP-1 mRNA expression in the right hippocampus (CA3)
Immunocytochemistry
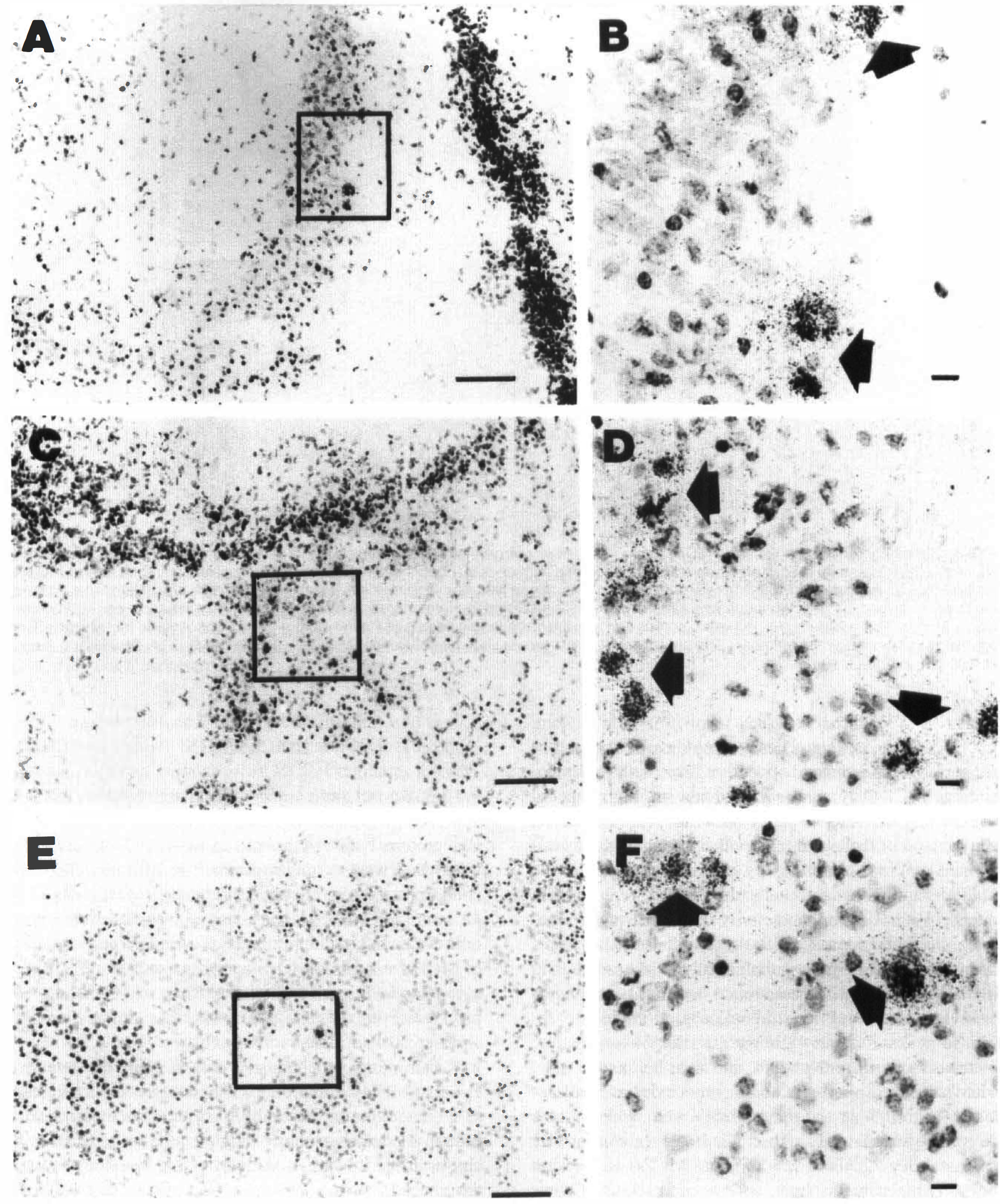
No MCP-1 expression was detected in animals evaluated at 10 minutes after hypoxia-ischemia. At 2.5 hours after hypoxic-ischemic lesioning, MCP-1 immunostaining was detected in the ependyma of the right hemisphere in three of three animals, and in one of three, scattered immunoreactive cells were also detected within the brain parenchyma (not shown). Both parenchymal and ipsilateral ependymal MCP-1 immunoreactive cells were consistently detected at 4 hours after lesioning (Fig. 5). In the lesioned right striatum, immunoreactive cells were concentrated adjacent to the ventricle; immunoreactive cells were also evident within the hippocampal pyramidal cell layer, particularly in the CA3 subfield.
These photomicrographs show MCP-1-immunoreactive cells in the right striatum
In all animals evaluated at 8, 12, or 24 hours after hypoxia-ischemia, MCP-1 immunoreactive cells were widely disseminated throughout the lesioned cerebral hemisphere. Based on their location and morphology, many of these cells seemed to be neurons. In the contralateral hemisphere, ependymal immunoreactive cells were sometimes seen, but no other cells expressed MCP-1. Fig. 6 shows the distribution MCP-1-immunoreactivity in the lesioned hippocampus and adjacent thalamus of an animal evaluated at 8 hours after hypoxia-ischemia. This section corresponds closely neuroanatomically with the section used for the in situ hybridization assay presented in Fig. 2B. There is a close correspondence between the distributions of MCP mRNA and protein in the two sections. Note again that the majority of immunoreactive cells in the hippocampus are within the pyramidal cell layer. Corresponding contralateral areas do not show MCP-1-immunoreactive cells. The distribution of MCP immunoreactivity in the lesioned hemisphere was similar at 12 and 24 hours (not shown). In the lesioned cortex, the neuroanatomic distribution of immunoreactive cells varied: there was no preferential distribution in specific cortical layers nor was there a columnar distribution of immunoreactive cells. In the lesioned hippocampus, immunoreactive cells were widely disseminated in CA1 and CA3 subfields and were also frequently noted within the dentate gyrus. No immunoreactive cells were seen in the corpus callosum or other white matter tracts.
This figure compares the distribution of MCP-1-immunoreactive cells in the left
In the first 48 hours after injury, brain regions with high concentrations of MCP-1-immunoreactive cells contained many activated microglia. Fig. 7 compares the distribution of MCP-1 immunoreactivity (panels A and B) and lectin-staining (panels C and D) in adjacent sections of the right cortex of an animal evaluated at 48 hours after lesioning. These complementary sections show a correspondence between regions with high densities of MCP-1 immunoreactive cells and high concentrations of activated microglia/monocytes. Based on comparison of these and many other sections, the lectin-stained microglia/monocytes were distinct from the MCP-1 immunoreactive cells. In general, regions with intense MCP-1 immunoreactivity contained many reactive microglia. However, the microglial/monocyte response was more homogeneous and widespread than MCP-1 expression in the lesioned cerebral hemisphere.
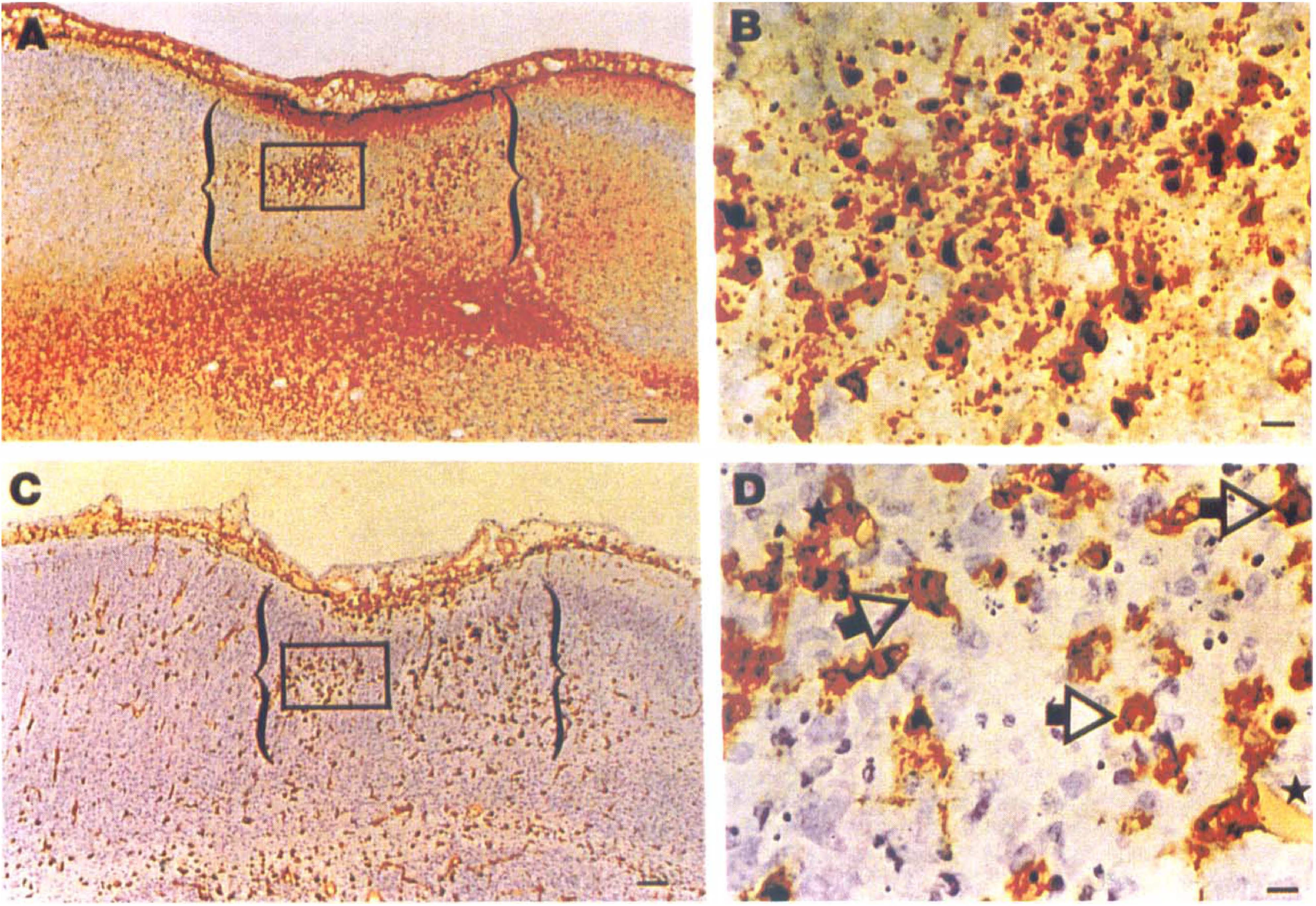
This figure compares the distribution of MCP-1-immunoreactivity
In lesioned forebrain, the microglial/monocyte response peaks at about 48 hours after hypoxia-ischemia, plateaus from 48 to 96 hours, and then begins to wane. Many activated microglia and/or monocytes are still detectable throughout the right cerebral hemisphere 5 to 6 days after lesioning (Ivacko et al., 1996). In contrast, by postnatal day 12, there was minimal MCP-1 expression in the lesioned hemisphere, and its expression was limited to endothelial cells. Fig. 8 illustrates MCP immunoreactivity in a small blood vessel in the right cortex of a lesioned 12-day-old animal. A similar pattern of immunostaining was also rarely seen in endothelial cells in a healthy, 12-day-old brain.
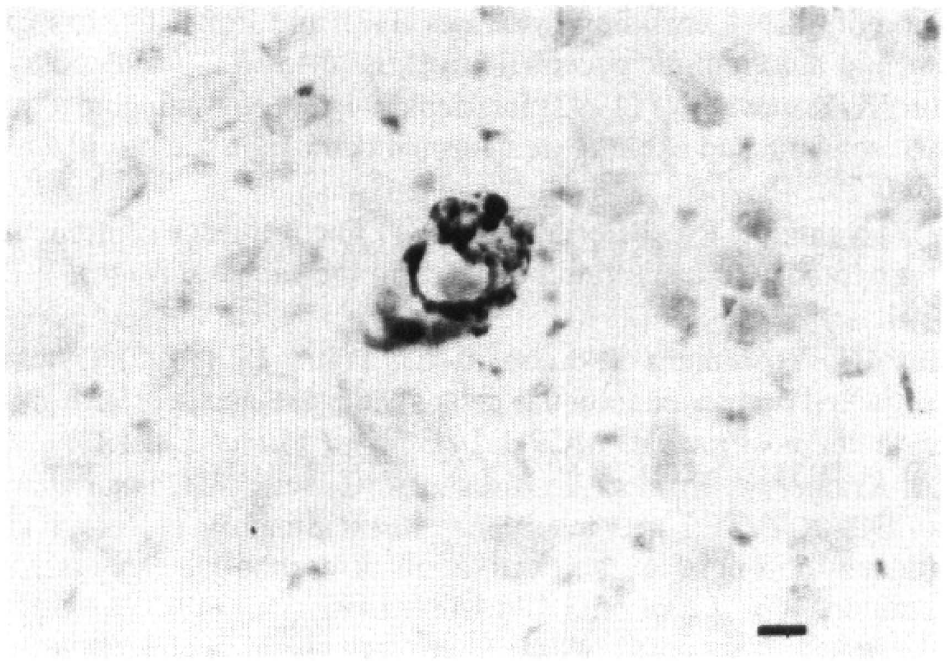
This photomicrograph shows an MCP-1-immunoreactive endothelial cell in the right cortex of an animal evaluated 5 days after hypoxic-ischemic lesioning. On postnatal day 12, the latest time interval after injury evaluated, no other cell types were immunoreactive. Magnification 40×. Scale bar 10 μm.
DISCUSSION
Hypoxic-ischemic injury stimulated MCP-1 mRNA and protein expression acutely. Monocyte chemoattractant protein-1 mRNA peaked in the first 24 hours after injury and was no longer detected at 48 hours. The encoded protein was consistently detected within the first 48 hours after injury, and expression subsequently declined. Overall, the temporal and neuroanatomic features of MCP-1 expression were congruent with the microglial/monocyte response elicited by hypoxic-ischemic injury. However, the cellular response was ultimately more widespread and more prolonged.
Our immunocytochemistry data support the hypothesis that MCP-1 could play a functionally important role as a chemokine in the brain. Monocyte chemoattractant protein-1 could potentially regulate microglial activation and/or influence egress of blood-derived monocytes into the brain. In a study of human malignant glioma tissue, the degree of macrophage infiltration was grossly correlated with the level of MCP-1 expression (Takeshima et al., 1994). With available methods blood-derived monocytes cannot be readily distinguished from fully activated microglia, and the specific temporal relationship between MCP-1 expression and monocyte infiltration into the injured brain cannot be precisely established. Currently, there is no direct evidence that MCP-1 regulates microglial activation in vivo, microglia do express MCP-1 receptors in vitro (Harrison et al., 1995).
In this neonatal stroke model, the earliest morphological features of microglial activation (evident as early as 10 minutes after hypoxia-ischemia [Ivacko et al., 1996]) precede the initial detection of MCP-1 expression. Whether the earlier recognition of microglial activation reflects limitations in the sensitivity of the assays used to detect MCP-1 or alternatively that other molecular signals initiate microglial activation is uncertain. Infiltration of activated microglia/monocytes into lesioned tissue increases rapidly in the first 24 hours after injury, a pattern which is congruent with the timing of MCP-1 expression.
Characteristic features of the distribution of MCP-1 in the lesioned cerebral hemisphere included its initial concentration in periventricular regions of the lesioned hemisphere, together with intense ependymal expression of MCP-1, particularly in the choroid fissure, and subsequent widespread expression within brain parenchyma. With respect to the early periventricular distribution, we had noted a similar ipsilateral periventricular microglial reaction previously (Ivacko et al., 1996). Of interest, in this perinatal stroke model, there is also early evidence of disruption of cytochrome oxidase activity, an indicator of neuronal metabolic integrity, in periventricular zones (Nelson and Silverstein, 1994). Although a periventricular effect might suggest exposure to a soluble factor disseminated in cerebrospinal fluid, the acute microglial response and loss of cytochrome oxidase activity are limited to the hypoxic-ischemic hemisphere, and MCP-1 expression is much more pronounced ipsilaterally. Thus, the factors that account for this unilateral periventricular pattern are uncertain. The distinctive and consistent ependymal expression of MCP-1 suggests that these cells may participate in the inflammatory response in the injured brain. Additional evidence that ependymal cells may play an active role in the immature brain's response to acute injury is provided by our preliminary results of in situ hybridization assays of a NMDA-lesioned 7-day-old brain, in which a similar pattern of ependymal MCP-1 mRNA expression was also seen acutely (Szaflarski and Silverstein, unpublished observation).
We relied on cellular morphology, pattern of Nissl-staining, and location to determine the identity of cells that expressed MCP-1 mRNA and protein. More definitive double-labeling methods have not been technically feasible in friable, lesioned neonatal tissue. Although we did not definitively establish the identity of these cells, their distribution, cresyl-violet staining properties, and morphology suggested that many were neurons. This interpretation is congruent with findings of a recent study of ischemia-induced expression of the proinflammatory cytokine tumor necrosis factor-α in adult brain, in which tumor necrosis factor-α was identified in neurons in the evolving infarct (Liu et al., 1994). A puzzling and unexplained feature of MCP-1 expression was that only a minority of (presumptive) neurons in the lesioned hemisphere were immunoreactive and these cells did not share identifiable neuroanatomic or neurochemical features.
Based on the morphology and distribution of immunoreactive cells, it was readily evident that astrocytes were not a significant source of MCP-1. In contrast, in a recent study in mouse brain, a cortical stab wound induced intense MCP-1 expression, localized predominantly to astrocytes (Glabinski et al., 1996). This study also addressed developmental factors in regulation of MCP-1 expression and reported that the MCP-1 response to acute brain injury was attenuated in the neonate, compared with the adult. This finding was interpreted as evidence of suppression of this component of inflammation in neonatal brain. Because no established ischemic brain injury models in the neonatal and adult rat are identical, it is not feasible to evaluate ontogenic differences in the magnitude of ischemia-induced stimulation of MCP-1. Nonetheless, our findings show robust induction of MCP-1 in the neonatal brain. Species differences, slight differences in maturational stage, and/or factors related to the mechanisms of eliciting brain injury could account for these outcome differences.
In comparison with patterns of MCP-1 expression induced by focal cerebral ischemia in adult brain, overall trends in the temporal pattern of hypoxia-ischemia–induced MCP-1 expression in the immature brain were similar. Wang et al. (1995) used tissue homogenate assays to measure MCP-1 mRNA content in the brain after permanent or temporary middle cerebral artery occlusion; after permanent occlusion, the onset of stimulation of MCP-1 mRNA expression was at 6 hours, the peak was at 24 hours, and there was minimal residual 5 days later; after temporary arterial occlusion, the onset was at 3 hours after reperfusion, and increased expression persisted for 2 days. These investigators did not evaluate cellular sources or measure protein content. Kim et al. (1995) found that focal cerebral ischemia induced MCP-1 mRNA expression from 6 to 48 hours after the insult, and that MCP-1 immunoreactivity was localized to the ischemic area, peaking at 48 hours in endothelial and macrophage-like cells. Whether the differences observed in the timing and cellular localization of the MCP-1 response predominantly reflect maturational-stage differences, or differences in the pathophysiology of cerebral ischemic injury among the models is uncertain. Whether neuronal MCP-1 expression is a response invariably restricted to the developing brain remains to be determined.
The molecular signals that induce MCP-1 expression in brain are unknown. Monocyte chemoattractant protein-1 was initially characterized as an early response gene (“JE”; Cochran et al., 1983). Although regulatory mechanisms have not been directly evaluated in the brain, in other systems expression of MCP-1 is not coordinately regulated with c-fos or c-myc expression (Hall and Stiles, 1987). Interleukin-1β is a potent inducer of MCP-1 in other tissues (Sica et al., 1990). In this stroke model, there are rapid transient increases Interleukin-1β gene expression, peaking at 4 hours after hypoxia-ischemia (Szaflarski et al., 1995). Whether Interleukin-1β is a major inducer of MCP-1 expression in the brain is an important unanswered question.
Our findings complement a rapidly growing body of evidence that proinflammatory mediators contribute to the progression of hypoxic-ischemic injury in the neonatal brain. Martin et al. (1994) reported that systemic treatment with Interleukin-1β receptor antagonist conferred substantial (>50%) neuroprotection from hypoxic-ischemic injury in this model. Of potential clinical relevance is the fact that delay of the onset of treatment up to 1 hours after hypoxia was still effective. Similarly, post-hypoxia-ischemia treatment with the platelet activating factor antagonist BN52021 conferred substantial neuroprotection in this model (Liu et al., 1996). Whether pharmacological antagonism of MCP-1 could represent an effective strategy for limiting the progression of acute brain injury remains to be determined.