
Abstract
Microglia respond to focal cerebral ischemia by increasing their production of the neuromodulatory cytokine tumor necrosis factor, which exists both as membrane-anchored tumor necrosis factor and as cleaved soluble tumor necrosis factor forms. We previously demonstrated that tumor necrosis factor knockout mice display increased lesion volume after focal cerebral ischemia, suggesting that tumor necrosis factor is neuroprotective in experimental stroke. Here, we extend our studies to show that mice with intact membrane-anchored tumor necrosis factor, but no soluble tumor necrosis factor, display reduced infarct volumes at one and five days after stroke. This was associated with improved functional outcome after experimental stroke. No changes were found in the mRNA levels of tumor necrosis factor and tumor necrosis factor-related genes (TNFR1, TNFR2, TACE), pro-inflammatory cytokines (IL-1β, IL-6) or chemokines (CXCL1, CXCL10, CCL2); however, protein expression of TNF, IL-1β, IL-6 and CXCL1 was reduced in membrane-anchored tumor necrosis factorΔ/Δ compared to membrane-anchored tumor necrosis factorwt/wt mice one day after experimental stroke. This was paralleled by reduced MHCII expression and a reduction in macrophage infiltration in the ipsilateral cortex of membrane-anchored tumor necrosis factorΔ/Δ mice. Collectively, these findings indicate that membrane-anchored tumor necrosis factor mediates the protective effects of tumor necrosis factor signaling in experimental stroke, and therapeutic strategies specifically targeting soluble tumor necrosis factor could be beneficial in clinical stroke therapy.
Introduction
The pleiotropic cytokine tumor necrosis factor (TNF) is involved in the regulation of physiological and pathophysiological processes in the central nervous system (CNS).1,2 TNF occurs both as transmembrane TNF (mTNF), a 26 kDa membrane-anchored form, and soluble TNF (solTNF), a 17 kDa soluble form which is released into the extracellular space after cleavage of mTNF by the metalloproteinase TNFα converting enzyme (TACE/ADAM17). 3 The cellular functions of TNF are mediated by TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2), which differ in expression, ligand affinity, cytoplasmic tail structure and downstream signaling pathways. SolTNF binds to TNFR1 with higher affinity than TNFR2, and studies indicate that binding of solTNF to TNFR2 does not result in receptor activation.4,5 TNFR1 primarily mediates apoptosis and inflammation, 6 but has also been shown to have anti-apoptotic functions via activation of the NF-κB signaling pathway.7–9 Conversely, TNFR2, which is only activated by mTNF, promotes cell survival, resolution of inflammation, immunity and myelination.9–11
TNF is constitutively expressed in the brain where it plays an important physiological function as a modulator of neuronal activities, among others through regulation of neurotransmitter production. 12 Glial-derived TNF is required for synaptic scaling and preservation of synaptic strength.13,14 Furthermore, TNF and its receptors are also essential for normal cognitive function under naive conditions. 15 It has been shown that TNF deficiency improves spatial learning and memory 16 and decreases anxiety levels in mice, and knockout of TNF receptors results in increased exploration and anxiolytic-like effects in mice.17,18
Studies in humans and animal models have yielded significant insight into the function of TNF in neurological diseases, indicating distinct and often opposite roles for solTNF and mTNF.1,9,19–22 TNF has been implicated in many aspects of stroke pathology,23,24 and it is increased in the blood and cerebrospinal fluid of stroke patients.25–29 In some studies, this increase has been found to correlate with stroke severity,27,30,31 in line with recent findings of an association between TNF-α 238G/A, 308G/A, or rs1800628 polymorphisms, resulting in increased serum TNF levels, and an increased risk of stroke.32–34 In mice, TNF mRNA is upregulated 4–6 h after the ischemic insult.35,36 Acutely after stroke, TNF is mainly produced by activated microglia36–38 and at later times additionally sustained by infiltrating macrophages, which invade the peri-infarct and infarct areas.36–38 TNF receptors are also upregulated after ischemia. 39 Specifically, TNFR1 has been shown to mediate ischemic tolerance 40 and neuroprotection,37,41 presumably through the TNFR1-NF-κB-FLIPL pathway. 42
In the present study, the function of solTNF and mTNF in focal cerebral ischemia was investigated using genetically modified mTNFΔ/Δ mice that express only the membrane-anchored form of TNF. The mTNFΔ/Δ mice were generated by replacing the endogenous TNF allele with the Δ1-9, K11E TNF allele. This combined deletion/mutation guarantees loss of TACE-mediated cleavage preventing shedding of solTNF,19,43 but maintains normal cell-surface expression and function of mTNF. 19 We show that transgenic mTNFΔ/Δ mice have no abnormal behavioral or histopathological phenotypes when compared to mTNFwt/wt wild-type (WT) littermates. Importantly, we show that absence of solTNF in these mice translates into a neuroprotective effect after focal cerebral ischemia, as mTNFΔ/Δ mice have significantly reduced cortical infarcts. This effect is accompanied by improved functional outcomes and reduction of the inflammatory response in the brain of mTNFΔ/Δ mice.
Materials and methods
Mice
Homozygous mTNFΔ/Δ19 breeders, on a C57BL/6 background, were obtained from the Department of Biochemistry, University of Lusanne, kindly provided by Dr. Tacchini-Cottier and established as heterozygous mTNFΔ/wt breeding colonies at the animal facility of the Biomedical Laboratory, University of Southern Denmark (BL-SDU). All experiments were performed blinded on age-matched (8–12 weeks old) homozygous mTNFΔ/Δ male mice and mTNFwt/wt littermates. TNF knockout (TNF−/−) mice 44 were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Age-matched C57BL/6 WT mice were purchased from Taconic A/S (Ry, Denmark). Animals were housed in ventilated cages with 1–3 cage-mates at a 12 h light/dark cycle, under controlled temperature and humidity, and with free access to food and water. Mice were cared for in accordance with the protocols and guidelines approved by the Danish Animal Health Care Committee; experiments are reported in accordance with the ARRIVE guidelines, and all efforts were made to minimize pain and distress (J. No. 2011/561-1950 and 2013-15-2934-00924).
Body composition measurements
Body composition was examined immediately before surgery on fully sedated mTNFΔ/Δ (n = 18) and mTNFwt/wt (n = 15) mice using non-invasive Dual-Energy X-ray Absorptiometry (DXA) (PIXImus 2, Version 1.44, GE Lunar, Madison, WI, USA). This method provides accurate assessments of total tissue mass (g), bone mineral density (BMD, g/cm2), bone mineral content (BMC, g), bone area (cm2), fat mass (g), % fat tissue and lean tissue mass (g). 45
Behavioral assessments
Open field test
The open field test was performed using a non-transparent plastic square arena measuring 45 (W) × 45 (D) × 40 (H) cm. The arena was divided into three zones (wall, intermediate and center), and spontaneous movements in each zone were tracked using the SMART 2.5 Video Tracking System (Panlab Harvard Apparatus, Barcelona, Spain) connected to a high-resolution color video camera (SSC-DC378P, Biosite, Stockholm, Sweden). Mouse behavior was recorded over a 10 min period and total distance travelled (m), as well as time spent in each zone were automatically recorded. Rearing, as a measure of stereotypical mouse behavior, was manually recorded and expressed as total number of events 45 (n = 6/group).
Rotarod
The accelerating rotarod test was performed in two parts: a pre-training and a four-trial test using the LE8200 system (Panlab Harvard Apparatus). The pre-training was performed the day prior to the rotarod test and conducted at constant speed of 4 r/min. Inclusion criteria required that, during pre-training, mice kept their balance on the rotating rod for a minimum of 30 s. Mice that did not meet these criteria were excluded. The rotarod test was conducted with accelerating speed from 4 to 40 r/min over a 5 min period, with each mouse performing in total four trials. After each trial, mice were transferred back to their cage and allowed to rest for 20 min to prevent exhaustion or stress. The time spent on the rod before fall was automatically recorded and the total time spent on the rod was calculated 46 (n = 8–10/group (mTNFΔ/Δ and mTNFwt/wt) and n = 6/group (TNF−/− and WT)).
Grip strength test
Grip strength was measured prior to (baseline) and three and five days after surgery using the grip strength meter (BIO-GT-3, BIOSEB, Paris, France) connected to a stainless steel grid. The mouse was pulled along the horizontal plane until the grip was released. The force applied by the mouse was automatically detected and recorded. Individual (right and left) and total (both) front paw strengths were measured before and after permanent middle cerebral artery occlusion (pMCAO) and results plotted as Δ grip strength in order to calculate asymmetry (changes in grip strength at days 3 and 5 compared to baseline). Each mouse was tested in five sequential trials, and the highest force measured was selected as the final score for each mouse.37,47 No asymmetry was observed under baseline conditions (data not shown) (n = 6–15 mice/group).
Rung walk test
Rung walk analysis was performed as described by Novrup et al. 22 two days after induction of focal cerebral ischemia. Mice were allowed to traverse the rungs and were filmed using a handheld GoPro HD camera with 48 fps. Data were evaluated frame by frame with a VLC Mediaplayer (2.1.2, Rincewind, Paris, France). Left and right scores were calculated as follows: six, complete miss; five, touching the rung, but sliding off and losing balance; four, touch, miss but no loss of balance; three, replacement, mouse placed paw on rung but quickly removed it; two, re-correction, aimed for rung but changed direction; one, anterior or posterior placement; 0, perfect step. 48 The total number of mistakes on each paw was plotted for analysis of asymmetry. Prior to surgery, mice were pre-trained in the rung walk test, and no asymmetry was observed under baseline conditions (data not shown) (n = 7–13 mice/group).
Y-maze test
Spontaneous alternation behavior and hence working memory was tested using the Y-maze test in naive mTNFΔ/Δ and mTNFwt/wt as previously described. 45 Each mouse was placed in the arm designated (A) of the Y-maze field. Except for the first two, the number of entries into each arm (A, B, C) was recorded manually over an 8 min period and spontaneous alternation calculated based on these numbers (n = 6 mice/group (mTNFΔ/Δ and mTNFwt/wt) and n = 15–18/group (TNF−/− and WT)).
Permanent focal cerebral ischemia
The distal part of the left middle cerebral artery (MCA) was permanently occluded under surgical anaesthesia consisting of a mixture of Hypnorm® (fentanyl citrate 0.315 mg/ml and fluanisone 10 mg/ml, VetaPharma Ltd, Leeds, UK), Stesolid® (Diazepanum 5 mg/ml, Actavis, Gentofte, Denmark) and distilled H2O in a 1:1:2 ratio, as previously described.35,37 The surgery was conducted on a 37 ℃ heating pad to maintain a stable body temperature. An incision was made from the lateral part of the orbit to the external auditory meatus. The underlying superior pole of the parotid gland and the upper part of the temporal muscle were pushed in the distal direction after partial resection. A craniotomy was performed directly above the distal part of the MCA using a 0.8 mm high-speed microdrill. The bone was removed and the dura carefully opened. The distal part of the MCA was coagulated using bipolar forceps coupled to an electrosurgical unit (ICC 50, Erbe, Tübingen, Germany) ensuring a restricted cortical infarct. Post-operative treatment consisted of supplying the mice with physiological saline (0.9% NaCl) and Temgesic® (Buprenorphinum 0.3 mg/ml, RB Pharmaceuticals, North Chesterfield, VA, USA) three times during the first 24 h. The mice were allowed to survive for one or five days (n = 7–18 mice/group). Mortality was < 2% and only related to anaesthesia.
Sham mice were subjected to the same procedure, except the bipolar forceps were applied in the superficial brain parenchyma next to the MCA, without coagulating the MCA (n = 5/group).
Tissue processing and infarct volume analysis
Mice with 3 h (n = 5/group), one or five days post-surgical survival, as well as naive control (n = 5/group) mice, were anesthetized with pentobarbital (200 mg/ml)/lidocain hydrochloride (20 mg/ml) (Glostrup Apotek, Glostrup, Denmark) prior to euthanasia. The brains were removed, fresh-frozen in CO2 snow and coronally cryo-sectioned into 30 μm thick sections in six parallel series, three series were placed on microscope slides and three series in eppendorf tubes. Every sixth series was stained with Toludine Blue (TB) for visualization of the infarct area. 49 The infarct was estimated on TB-stained serial sections from mice with one and five days survival utilizing the computer-assisted stereological test system CAST© 2000 (Olympus, Ballerup, Denmark) and applying Cavalieri's principle for volume estimation. 50 The rostrocaudal distribution of the infarct was analyzed as previously described 51 using the anterior commissure as an anatomical landmark. In addition, to correct for edema, the volume of the contralateral and the non-ischemic ipsilateral cortex and the volume of injury spanning from 1080 μm anterior to 1080 μm posterior to the anterior commissure was compared in mTNFΔ/Δ and mTNFwt/wt mice at one and five days after pMCAO using an indirect method of infarct volume estimation.37,52
Immunohistochemistry
Naive mice (n = 6 mice/group) were anesthetized with pentobarbital (200 mg/ml)/lidocain hydrochloride (20 mg/ml) and transcardially perfused with 4% paraformaldehyde (PFA) through the left ventricle. Brains were quickly removed and left in 4% PFA overnight, changed to 1% PFA and finally 0.1% PFA before they were cut horizontally on a vibratome (VT100S, Leica Microsystems A/S, Ballerup, Denmark) into six 60 μm-thick parallel series of free floating sections and cryoprotected in de Olmo's solution. 53 One series from each animal was blocked for endogenous peroxidase activity using methanol and peroxide in tris-buffered saline (TBS), then incubated with 10% fetal bovine serum in 0.05 M TBS + 0.1 % Triton-X100 (TBS-T) for 30 min. Sections were incubated with anti-Iba1 primary antibody (rabbit, 1:600, Wako, Neuss, Germany) in TBS-T for two days at 4 ℃, followed by biotinylated anti-rabbit secondary antibody (1:200, GE Healthcare, Buckinghamshire, UK) in TBS-T for 1 h at RT. Sections were rinsed with TBS-T, incubated with horseradish peroxidase (HRP)-conjugated streptavidin (1:200, GE Healthcare) and developed using diaminobenzidine and hydrogen peroxide diluted in TBS. Finally, sections were transferred to gelatin-coated glass slides, counter-stained with TB, dehydrated and coverslipped using Depex.
Neocortical volume estimation and quantification of Iba1+ cells
The volume of the neocortex was estimated on one series of horizontally cut vibratome sections from naive control mice using Cavalieri's principle for volume estimation. 37 The neocortex was delineated using a 2 × magnification and included the following regions: anterior part of the agranular insular cortex, orbital cortex, cingulate cortex, frontal cortex, pre-limbic cortex, granular insular cortex, retrosplenial agranular cortex, anterior parts of retrosplenial granular cortex, ectorhinal cortex, parietal cortex, temporal cortex and all areas of the occipital cortex. 54 Immunolabeled Iba1+ cells were counted at a 60 × magnification in the right hemisphere of 10–14 sections from each mouse by the use of CAST1 (Visiopharm, Hørsholm, Denmark). The number of Iba1+ positive cells was recorded by systematically counting cells using a counting frame of 1012.6 μm2 and a X-Y step size of 350 μm resulting in an area (a(step)) of 122,500 μm2 to ensure that a representative sample was counted. 54 The number of microglial cells per area unit was calculated as previously described 54 and expressed as Iba1+ cells/mm2. The person performing the cell counting was blinded to the genotype.
Total RNA isolation and real-time RT-PCR
Total RNA was extracted from one series of brain tissue sections with TRIZOL® according to manufacturer's instructions (Invitrogen, Grand Island, NY, USA). To ensure complete elimination of genomic DNA, RNA was further purified with RNeasy MinElute Cleanup Kit (Qiagen, Valencia, CA, USA) in combination with DNA digestion using RNase-free DNase (Qiagen). Reverse transcription was performed with Omniscript (Qiagen), according to manufacturer's protocols. cDNA equal to 50 ng of initial total RNA was used as a template in each PCR reaction. Real-time PCR was performed in the Rotor-Gene 3000 Real-Time Cycler (Corbett Life Science, Sydney, Australia) with QuantiTect SYBR Green PCR MasterMix (Qiagen). Relative expression was calculated by comparison with a standard curve, after normalization to β-actin gene expression. Primers for gene amplification are listed in Supporting Table 1.
Protein extraction and Western Blotting
One series of brain tissue sections, parallel to those used for real-time RT-PCR, was homogenized in 300 μl of RIPA buffer supplemented with Complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA) and phosphatase inhibitor cocktail 1 (Sigma, St. Louis, MO, USA). The total protein concentration was quantified utilizing the Lowry assay. Proteins were resolved by SDS-PAGE on 8–11% gels transferred to nitrocellulose or polyvinylidene fluoride membranes and blocked in 5% non-fat milk in TBS-T. Membranes were probed overnight with anti-TNFR1 (mouse, 1:500, Santa Cruz Biotechnology, Dallas, TX, USA), anti-MHCII (mouse, 1:1000, Dako, Carpinteria, CA, USA), and anti-β-tubulin (mouse, 1:5000, Sigma), followed by HRP-conjugated secondary antibodies (GE Healthcare/Amersham, Brøndby, Denmark). Proteins were visualized with Super Signal West Pico chemiluminescent substrate (Thermo Scientific, Waltham, MA, USA) and bands quantified with Quantity One software (Biorad, Hercules, CA; USA). Data were normalized to β-tubulin.
Multiplex analysis
Cytokine and chemokine protein concentrations were determined by multiplex technology in brain samples (25 μl/sample), parallel to those used for Western Blotting and real-time RT-PCR, from mTNFΔ/Δ and mTNFwt/wt mice using a MSD Mouse Pro-inflammatory V-Plex Plus Kit (Mesoscale Discovery, Rockville, MD, USA) and a SECTOR Imager 6000 (Mesoscale Discovery) Plate Reader according to the manufacturer's instructions. Samples were diluted two-fold in Diluent 41 prior to measurement. Data were analyzed using MSD Discovery Workbench software. Groups consisted of naive mTNFΔ/Δ and mTNFwt/wt mice, and mice with 3 h, one day or five days survival following focal cerebral ischemia (n = 5–6/group).
Flow cytometry
One day after pMCAO, mice (n = 5–6 mice/group) were anaesthetized with an overdose of pentobarbital (200 mg/ml)/lidocaine hydrochloride (20 mg/ml) and transcardially perfused with phosphate-buffered saline (PBS). Ipsilateral cortices were quickly removed and placed in RPMI medium containing 10% foetal calf serum. Single-cell suspensions were obtained as previously described,37,38 and non-specific binding was blocked at 4 ℃ with anti-CD16/32 FcR. Cells were then incubated with the following antibodies: rat anti-mouse CD45-PerCP-Cy5.5 (BD Biosciences, clone 30-F11), rat anti-mouse CD11b-PE (BD Biosciences, clone M1/70), rat anti-mouse Ly6G/Ly6C-PE-Cy7 (Gr1) (Biolegend, Copenhagen, Denmark, clone RB6-8C5) and hamster anti-mouse CD3-APC (BD Biosciences, clone 145-2C11). Isotype controls used were hamster IgG1κ (BD Biosciences, clone A19-3) and rat IgG2b (BD Biosciences, clone A95-1 or Biolegend, clone RTK4530). Prior to fixation, suspensions were stained for live/dead cells for 30 min at 4 ℃ using the Fixable Viability Dye eFluor 506 (eBioscience, Aarhus, Denmark) diluted in PBS. Positive staining for microglia [CD45dimCD11b+], macrophages [CD45highCD11b+ Gr1−], granulocytes [CD45highCD11b+Gr1+] and T cells [CD45highCD3+] was determined based on fluorescence levels of the respective isotype controls and fluorescence minus one (FMO) controls, as previously described. 47 Samples (n = 5–6/group) were run on a FACSVerse flow cytometer (BD Biosciences) equipped with FACSuite software for data analysis. 47
Statistics
Real-time PCR and multiplex cytokine measurements were analyzed with parametric one-way ANOVA followed by Tukey's or Bonferroni's test for multiple comparisons. For single comparisons, Student's t-test was applied. Correlation analysis was performed using Pearson's correlation analysis. P values ≤ 0.05 were considered statistically significant. Data are presented as mean ± SEM.
Results
No phenotypical abnormalities in naive mTNFΔ/Δ mice
Since TNF has been implicated in the regulation of spatial memory and locomotor activity in physiological conditions,15,16 we investigated whether absence of solTNF caused any phenotypical abnormalities in naive mTNFΔ/Δ mice. Radiographic assessment of total body mass, bone mineral density, bone mineral content, bone area, % fat tissue, and % lean mass using the DXA scanner showed no differences between mTNFΔ/Δ and mTNFwt/wt mice (Supporting Table 2). In the Y-maze test for spontaneous alternation, which measures the willingness of rodents to explore new environments and involves areas of the brain such as hippocampus, septum, basal forebrain and prefrontal cortex, we found the total number of Y-maze entries (Figure 1(a)) and the spontaneous alternation percentage (Figure 1(b)) to be similar between mTNFΔ/Δ and mTNFwt/wt mice. In the open field test for spontaneous locomotion and anxiety-related behavior, we found no differences in all parameters evaluated including number of rearings (Figure 1(c)), total distance traveled (Figure 1(d)), and center/perimeter ratio (Figure 1(e)). Finally, the rotarod test showed comparable motor coordination (Figure 1(f)), and the grip strength test comparable proprioception and neuromuscular function (Figure 1(g)) between mTNFΔ/Δ and mTNFwt/wt mice.
Genetic ablation of solTNF does not change microglial cell density and morphology or behavioral phenotype under naive conditions. (a, b) To assess exploratory behavior, the total number of arm entries in the Y-maze (a) and spontaneous alternation (b) were measured. (c–e) To assess spontaneous locomotor activity and anxiety-related behavior, total distance travelled (c), center/perimeter ratio (d) and number of rearings (vertical activity) (e) were measured in the open field. (f, g) Motor coordination was assessed with the rotarod test, (f) and strength was measured by grip strength analysis (g). No differences were observed between groups (Student's t test, n = 5–15 mice/group). (h) Representative immunohistochemical photomicrographs of Iba1+ cells in the cingulate gyrus of the neocortex (upper panel) and magnifications of Iba1+ microglia in the neocortex (lower panel). (i) Quantification of the number of Iba1+ microglial cells/area in the neocortex of naive mTNFwt/wt and mTNFΔ/Δ mice (Student's t test, n = 6 mice/group). (j) Quantification of the neocortical volume of naive mTNFwt/wt and mTNFΔ/Δ mice (Student's t test, n = 6 mice/group). Scale bars: 10 μm. Results are expressed as mean ± SEM.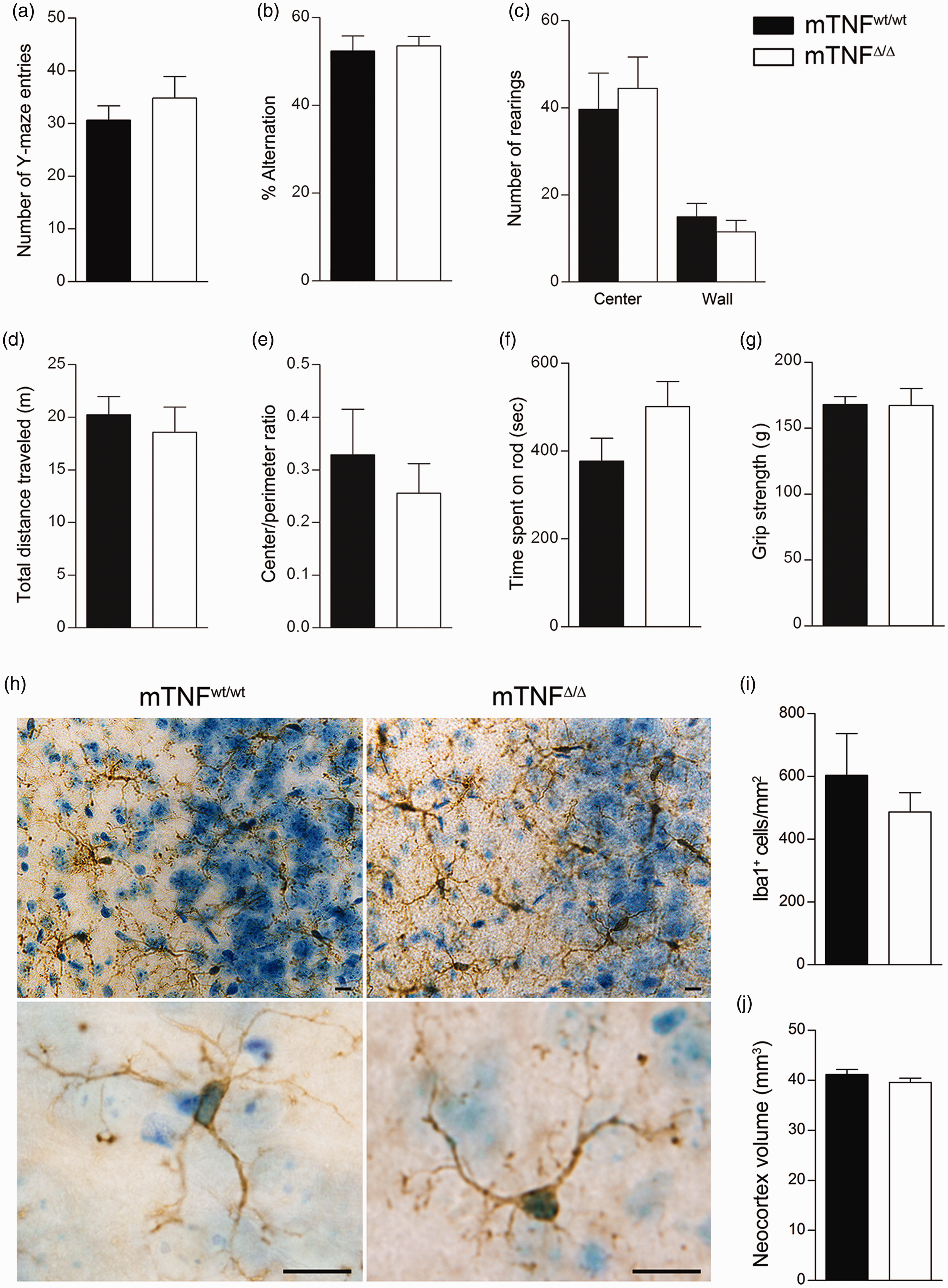
On the other hand, complete ablation of TNF in TNF−/− mice did result in behavioral abnormalities. In the Y-maze, TNF−/− mice showed a significant increase in the number of entries and total distance travelled (Supplementary Figure 1(a) and (b)), and a decrease in spontaneous alternation percentage (Supplementary Figure 1(c)) compared to WT mice. In the open field test, the number of rearings were comparable between TNF−/− and WT mice (Supplementary Figure 1(d)), whereas total distance travelled (Supplementary Figure 1(e)) and center/perimeter ratio (Supplementary Figure 1(f)) were significantly increased in TNF−/− mice. No differences were found in the rotarod and the grip strength tests, indicating that TNF−/− mice have normal proprioception and neuromuscular function (Supplementary Figure 1(h)). Collectively, these data show that while complete ablation of TNF leads to impairment of basal cognitive and motor function, absence of solTNF alone is not sufficient to alter these behaviors.
Extensive evidence has shown that TNF is primarily produced by microglia following focal cerebral ischemia in mice;35–38,55 therefore, we investigated whether genetic ablation of solTNF affected the total number of microglia in the neocortex. We found no difference in the number of Iba1+ microglial cells between the two genotypes (Figure 1(h) and (i)). Similarly, no obvious morphological differences were observed in cell shape or branching (Figure 1(h)). Total neocortex volume was also measured, and no differences were found (Figure 1(j)). These data indicate that absence of solTNF does not result in microglial alterations in the cortex and suggests that mTNF signaling is necessary and sufficient to maintain normal microglia homeostasis under naive conditions.
Absence of soluble TNF reduces infarct volume and improves asymmetry and neuromuscular function after pMCAO
To examine the effect of solTNF ablation on ischemic injury, we compared infarct volumes between mTNFΔ/Δ and mTNFwt/wt mice at one and five days after pMCAO (Figure 2(a) and (c)). The rostrocaudal distribution of the infarct showed that especially the sensory cortex, and in case of large infarcts also the motor cortex, is affected in this model (Figure 2(b), (d) and (e)). We found the volume of the injury to be significantly smaller in mTNFΔ/Δ mice one day after pMCAO (Figure 2(c)). Also at five days after pMCAO, the injury volume was still significantly reduced in mTNFΔ/Δ mice compared to mTNFwt/wt littermates (Figure 2(c)). The similar rostrocaudal distribution of the infarcts both at day 1 and 5 in mTNFΔ/Δ and mTNFwt/wt mice showed that ablation of solTNF had no effect on the regional distribution of the MCA territory in mTNFΔ/Δ mice (Figure 2(d) and (e)). When correcting for edema formation in the ischemic brain, the infarct was found to constitute 33% ± 6% of the ipsilateral cortex in mTNFΔ/Δ mice, whereas the infarct was found to constitute 43% ± 4% of the ipsilateral cortex in mTNFwt/wt mice one day after pMCAO. At five days, the infarct was found to constitute 14% ± 6% of the ipsilateral cortex in mTNFΔ/Δ mice and 33% ± 7% in mTNFwt/wt mice.
Genetic ablation of solTNF reduces infarct volumes after pMCAO with no changes in locomotor activity and anxiety-related behaviors assessed in the open field test. (a) Toluidine blue staining of frontal brain sections from mTNFwt/wt and mTNFΔ/Δ one and five days after pMCAO. (b) Schematic drawing of frontal sections of the mouse brain showing the location of motor (M) and sensory (S) areas in the cortex affected by pMCAO. (c) Estimation of cortical infarct volume in mTNFΔ/Δ mice and mTNFwt/wt littermates at one and five days after surgery (*P < 0.05, one-tailed Student's t test, n = 7–18 mice/group). (d,e) Rostrocaudal distributions of the infarcts at one day (d) and five days (e) after pMCAO. (f–h) Open field test on mTNFΔ/Δ mice and TNFwt/wt littermates two days after pMCAO. Locomotion was measured as total distance travelled (c), and stress-related behavior as center/perimeter ratio (d), and number of rearings (e). Results are expressed as mean ± SEM (n = 8–9 mice/group). No significant differences were measured between groups, Student's t test.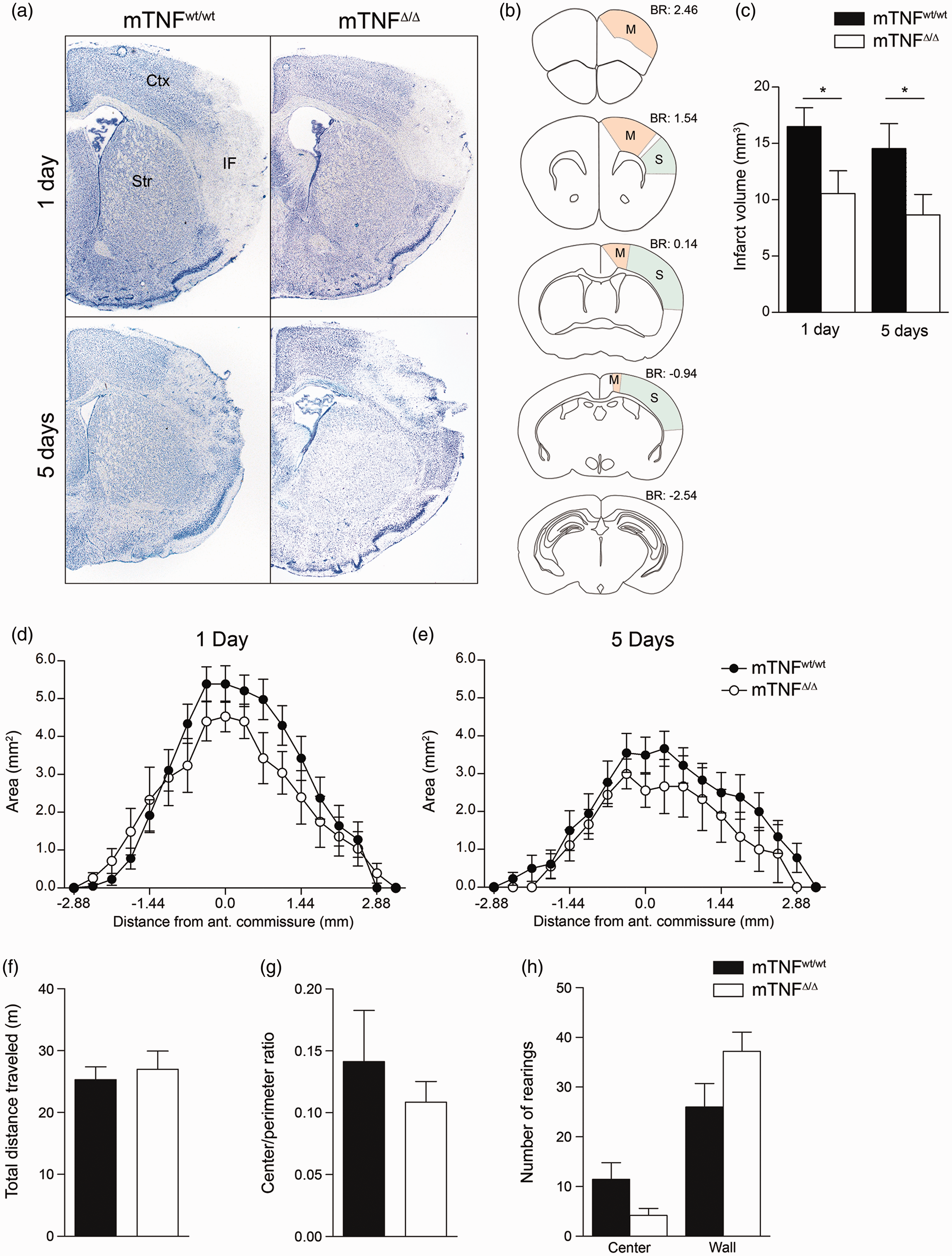
To assess whether the decreased infarcts after pMCAO in mTNFΔ/Δ mice translated into improved locomotor and/or sensory-motor function, we assessed mouse behavior two, three and five days after pMCAO. In the open field test two days after pMCAO, we found no difference between mTNFΔ/Δ and mTNFwt/wt mice in total distance traveled (Figure 2(f)), center/perimeter ratio (Figure 2(g)), and number of center and wall rearings (Figure 2(h)). Rung walk analysis on day 2 showed that mTNFwt/wt mice displayed a clear asymmetry with significantly more missteps of their right front limbs compared to the left front limbs (Figure 3(a)). This correlated with the large cortical infarct measured in mTNFwt/wt mice. In contrast, mTNFΔ/Δ mice did not show significant asymmetry of their front limbs (Figure 3(a)), which may be due to the smaller cortical infarcts. The reduced infarct volume in mTNFΔ/Δ did not result in improved motor coordination, as mTNFΔ/Δ and mTNFwt/wt mice performed similarly on the rotarod at both three and five days after pMCAO (Figure 3(b)). When analyzing proprioception and neuromuscular function using the grip strength test on the front paws, we observed significant asymmetry with loss of grip strength in the contralateral side (right, R) in mTNFwt/wt mice at both three and five days post pMCAO, compared to baseline values. On the contrary, mTNFΔ/Δ did not show loss of grip strength in the contralateral side as a consequence of the infarct and no significant asymmetry was measured between the front paws (Figure 3(c)).
Rung walk, rotarod and grip strength assessments of mTNFΔ/Δ and mTNFwt/wt mice after pMCAO. (a) Motor coordination and asymmetry by rung walk analysis was assessed two days after pMCAO and expressed as total number of missteps of the contralateral, right front limb compared to the unaffected left limb (paired Student's t test). (b) Motor coordination with the rotarod test was assessed three and five days after pMCAO as time spent on the rod (Student's t test). (c) Neuromuscular function was measured by grip strength analysis and expressed as Δ grip strength (change from baseline grip strength) (paired Student's t test). Results are expressed as mean ± SEM (n = 6–15 mice/group). *P < 0.05.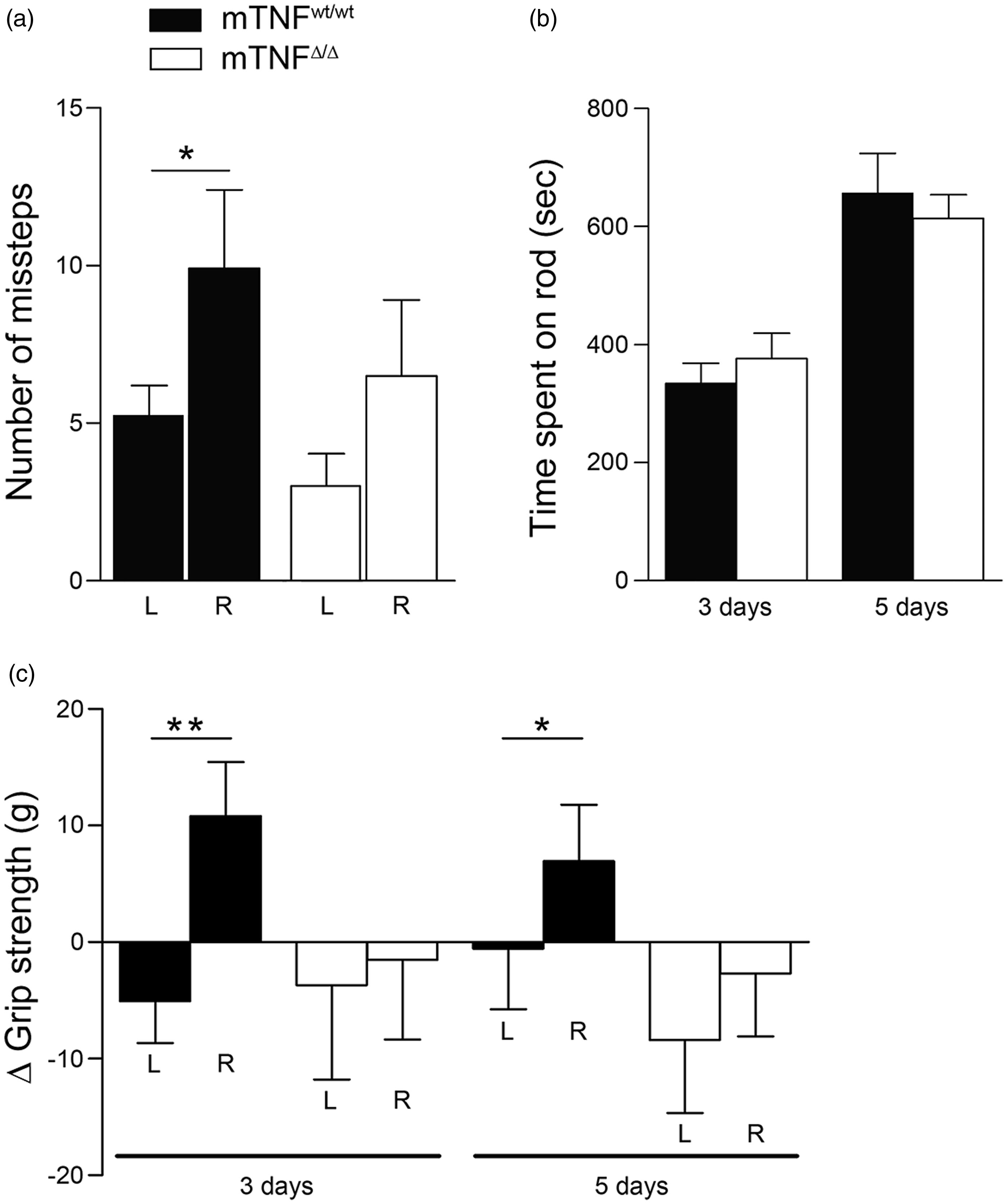
Absence of soluble TNF does not result in changes of pro-inflammatory gene expression after pMCAO
To investigate whether the functional improvement of mTNFΔ/Δ mice was associated with a modulation of the inflammatory response, we first evaluated the expression of pro-inflammatory cytokines and chemokines using real-time RT-PCR 1 day after pMCAO, the time point where expression of several cytokines relevant for infarct development is known to peak.
23
We found that IL-1β, IL-6, CXCL1, CXCL10 and CCL2 mRNA levels were significantly upregulated after stroke, but no significant difference was observed between mTNFΔ/Δ and mTNFwt/wt mice (Figure 4). TNF mRNA expression was also upregulated in sham-operated mice. In sham conditions, TNF mRNA upregulation was significantly higher in mTNFwt/wt controls as compared to mTNFΔ/Δ mice.
Gene expression profiling one day following pCMAO. Differential gene expression was evaluated one day after pMCAO in mTNFwt/wt and mTNFΔ/Δ mice, and compared to corresponding sham and naive mice brain tissue expression. For each gene, results are expressed as percent of WT ± SEM after normalization to β-tubulin gene expression (n = 5–6 mice/group).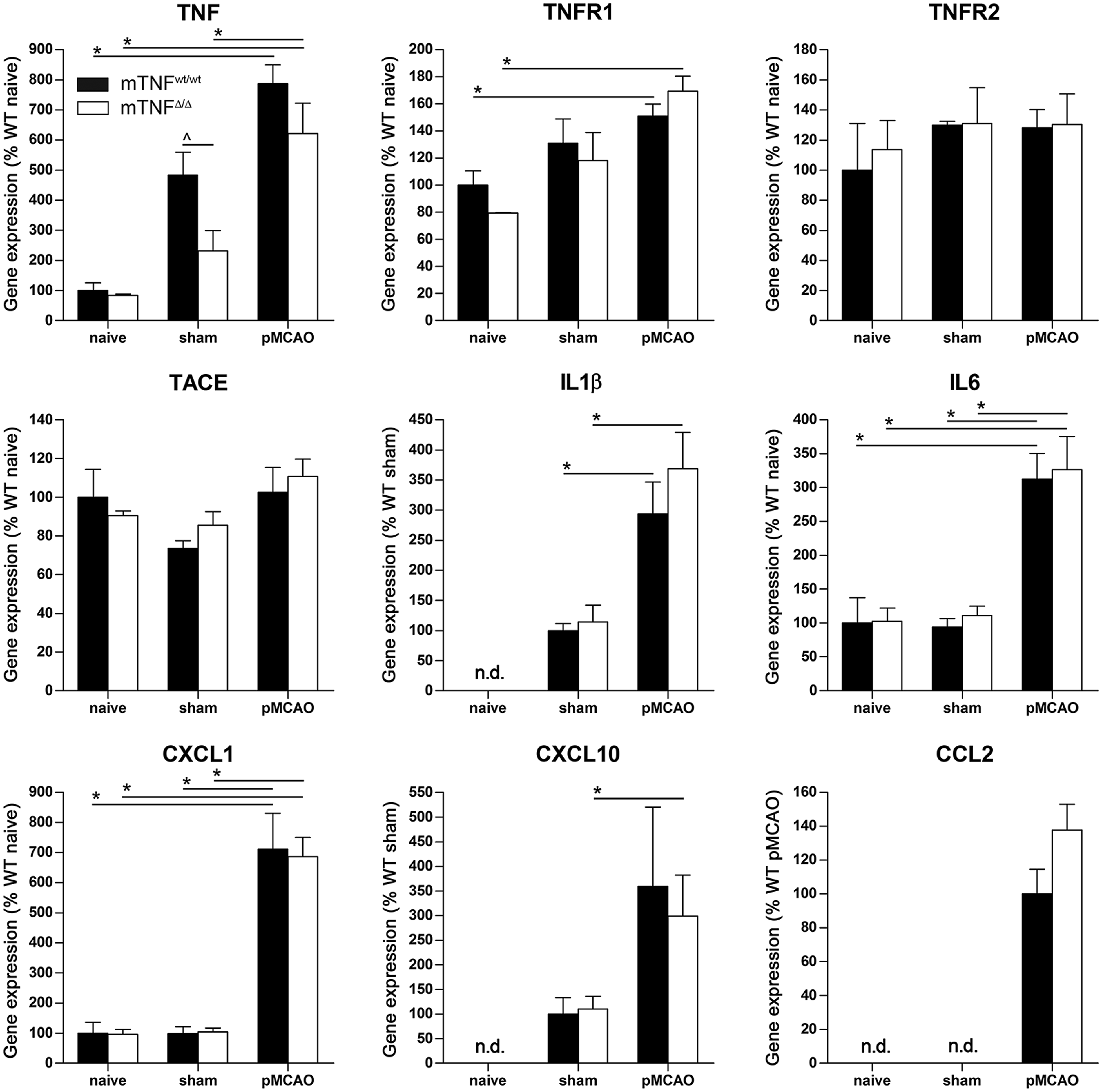
Absence of soluble TNF reduces protein levels of pro-inflammtory cytokines after pMCAO
To further investigate possible changes in the inflammatory response, we assessed the expression of pro- and anti-inflammatory cytokines at 3 h and one and five days after pMCAO using multiplex technology. We found the pro-inflammatory molecules TNF, IL-1β, IL-6 and CXCL1 to be significantly decreased in mTNFΔ/Δ mice compared to mTNFwt/wt controls one day after pMCAO (Figure 5(a)). IL-5 was upregulated in mTNFΔ/Δ mice one day after pMCAO, but no difference was found between mTNFΔ/Δ and mTNFwt/wt mice. In contrast, IL-2, IL-4 and IL-10, which have been associated with an anti-inflammatory function, did not change in the brain after pMCAO (Figure 5(a)). Correlation analysis (Supporting Table 3) showed a positive linear correlation between infarct volumes and IL-6 protein levels both at day 1 and 5. At day 1, IL-1β protein levels were also found to correlate with infarct volumes. In line with previous findings,
56
TNF protein levels correlated with infarct volumes five days after pMCAO, and so did CXCL1 and IL-2 protein (Supporting Table 3).
Cytokine protein expression profiling following pMCAO. (a) Cytokines and chemokines were quantified by multiplex technology in naive mice and 3 h, one day and five days after pMCAO in mTNFwt/wt and mTNFΔ/Δ mice. For each protein, results are expressed as mean ± SEM (n = 5–6 mice/group). ***P < 0.001 one-way ANOVA with Bonferroni multiple comparison test. (b) Western blot quantification of TNFR1 in the brain of naive, sham and pMCAO mice with one day survival. Results are expressed as mean ± SEM (n = 6 mice/group). *P < 0.05 and **P < 0.005, Student's t test.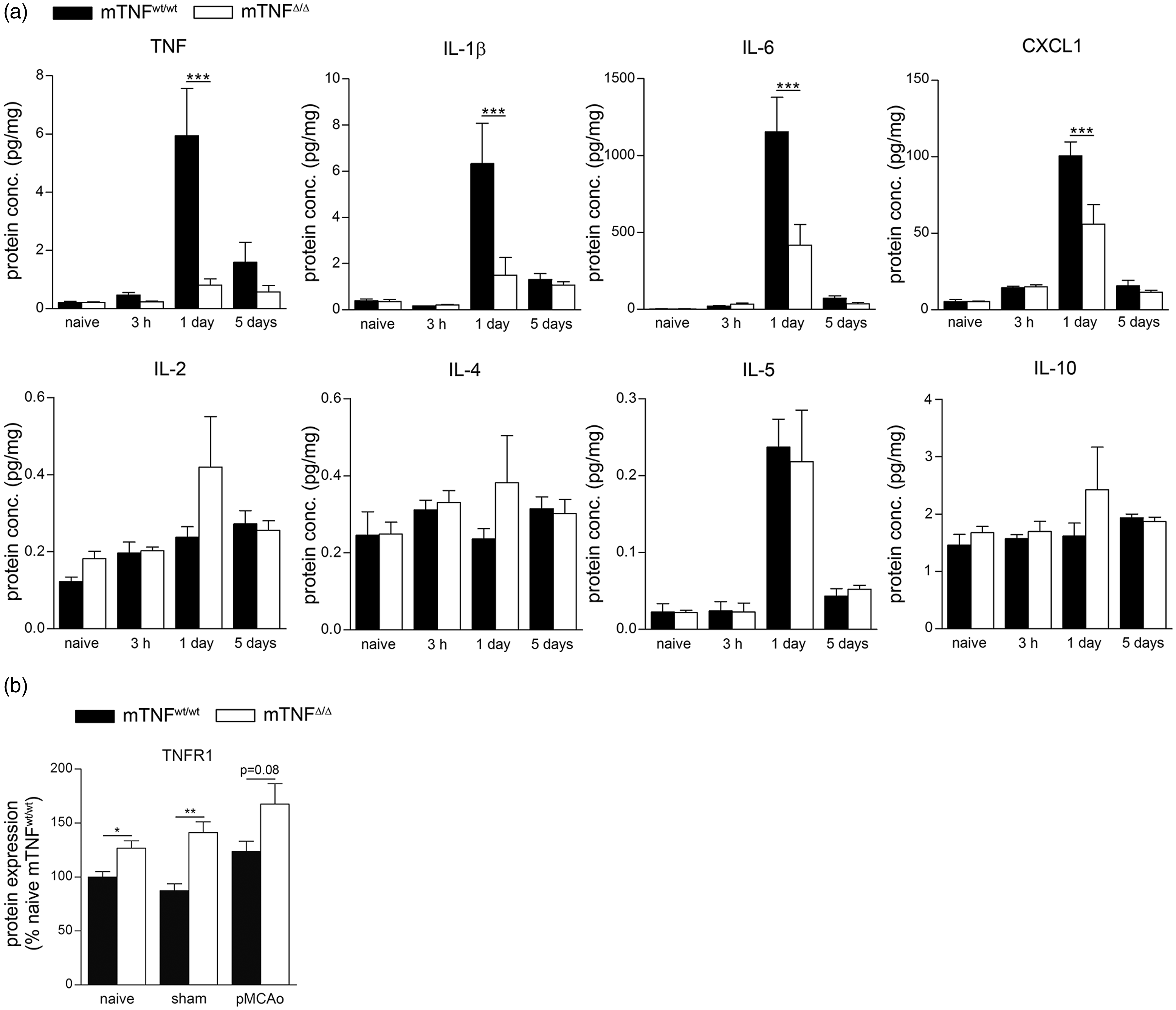
Since TNF has been shown to be neuroprotective through TNFR1,37,57 we evaluated TNFR1 expression by Western Blot. We found significant upregulation in mTNFΔ/Δ naive and sham mice compared to mTNFwt/wt mice. Despite tendencies to increased TNFR1 protein levels in mTNFΔ/Δ mice one day after pMCAO, no significant difference was observed between mTNFΔ/Δ and mTNFwt/wt mice (P = 0.08) (Figure 5(b)). These results suggest that genetic ablation of solTNF leads to suppression of the inflammatory response in the ischemic brain, contributing to an environment more favorable to functional recovery.
Absence of soluble TNF reduces macrophage infiltration into the ischemic cortex after pMCAO
Based on our previous findings of increased leukocyte infiltration in TNF−/− mice one day after experimental stroke
37
as well as altered microglial activation following anti-TNF treatment,
47
we investigated changes in microglia and infiltrating leukocytes in the ipsilateral cortex of mTNFΔ/Δ mice and mTNFwt/wt controls one day after pMCAO (Figure 6). We found that the number and percentage of infiltrating CD45highCD11b+Gr1− macrophages were significantly decreased in mTNFΔ/Δ mice, whereas the number and percentage of CD45highCD11b+Gr1+ granulocytes and CD45highCD3+ T cells were comparable between the two genotypes (Figure 6(a) to (c)). With respect to microglia, both number and percentage of CD45dimCD11b+ microglia were comparable between mTNFΔ/Δ mice and mTNFwt/wt controls (Figure 6(d) and (e)), consistent with findings of comparable numbers of Iba1+ cells (Figure 1(i)). Finally, we evaluated by Western Blot MHCII expression as a marker of microglial activation (Figure 6(f)), and found a significant reduction in mTNFΔ/Δ mice compared to mTNFwt/wt one day after pMCAO (Figure 6(f)). These findings suggest that genetic ablation of solTNF, while preserving mTNF, reduces the inflammatory response following stroke possibly by dampening the infiltration of macrophages into the ischemic brain.
Flow cytometry analysis of microglia and infiltrating leukocytes following pMCAO. (a, b) Number and percentage of infiltrating macrophages [CD45highCD11b+Gr1−], granulocytes [CD45highCD11b+Gr1+] and T cells [CD45highCD3+] in mTNFwt/wt and mTNFΔ/Δ mice one day after pMCAO. (C) Representative flow cytometry plots comparing macrophage and granulocyte infliltration in mTNFwt/wt and mTNFΔ/Δ mice. (d, e) Number and percentage of microglia [CD45dimCD11b+] in mTNFwt/wt and mTNFΔ/Δ mice one day after pMCAO. (f) Western blot quantification of MHCII in the brain of naive and pMCAO mice with one day survival. Results are expressed as mean ± SEM (n = 5–6 mice/group).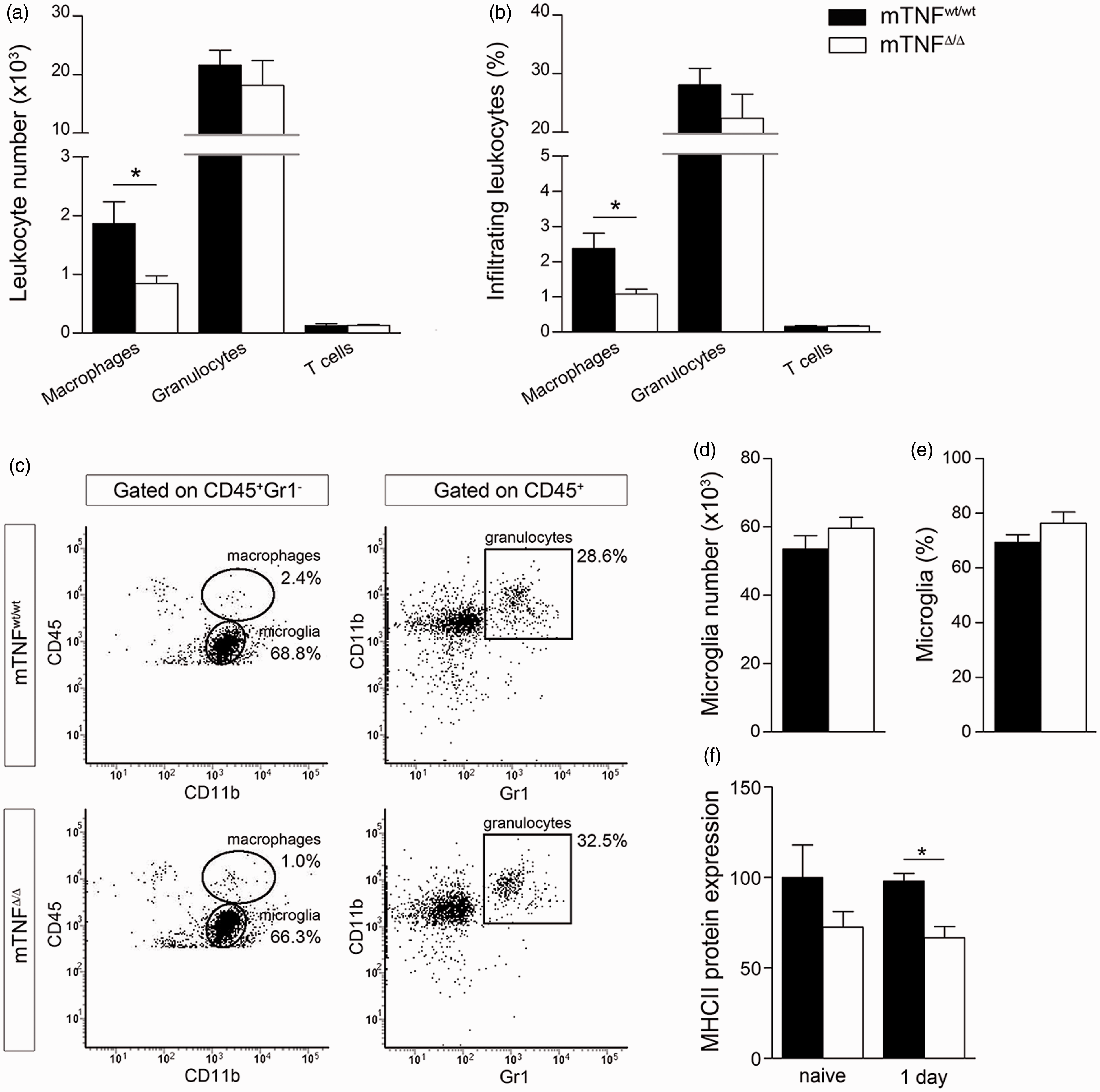
Discussion
In this study, we perused an in-depth investigation of the role of solTNF versus mTNF following focal cerebral ischemia. We used genetically modified mTNFΔ/Δ mice that express only the membrane-bound form of TNF and subjected these mice to experimental focal cerebral ischemia followed by assessment of functional outcome, estimation of lesion volumes and analyzed changes in leukocyte populations, genes and proteins relevant to stroke pathology.
Microglia have been demonstrated to regulate the number of neural precursor cells in the developing cerebral cortex, 58 and TNF has been shown to influence hippocampal development and function by regulating levels of neurotrophins available to neurons in the brain.59,60 Based on these reports and our own findings showing a reduction of microglial cell numbers in the brain of TNF−/− mice, 37 we set out to investigate whether genetic ablation of solTNF influenced microglial numbers in the mature mouse brain. We found that the total number of microglia per area in the neocortex was comparable between mTNFΔ/Δ and mTNFwt/wt mice suggesting that mTNF is sufficient to sustain microglial homeostasis in the developing and/or mature brain. The finding that absence of TNF leads to a reduction in microglial cell numbers in conventional TNF−/− mice may be ascribed to mTNF possibly acting as a survival factor via TNFR2 activation in the brain.61,62
TNF−/− mice have also been shown to display a skeletal phenotype with abnormal trabecular bone volume and increased bone mineral density, 63 and abnormal lipid homeostasis 64 therefore, we initially performed a radiographic assessment of body composition of the mTNFΔ/Δ mice using DXA scanning. We did not find differences in total body mass, % fat tissue, % lean mass, bone mineral density, bone mineral content and bone area, and can therefore conclude that under physiological conditions mTNF is sufficient to sustain normal lipid distribution and bone composition.
Numerous studies have investigated the role of TNF and its receptors in cognitive function,15,59,65,66 locomotor activity, 67 and anxiety-related behavior.66–69 In agreement with previous reports, we observed changes in cognitive function in TNF−/− mice with increased locomotor activity, decreased anxiety-related behavior and impaired short-term memory. Though alterations in these functions in TNF−/− mice were often ascribed to abnormal serotonine metabolism 69 or changes in nerve growth factor levels, 59 they are likely also associated with the important role of glial-derived TNF in homeostatic activity-dependent regulation of synaptic connectivity 13 and control of synaptic strength.14,70,71 Despite this evidence demonstrating a role for TNF in behavioral function, in the present study, we did not observe any changes in cognitive function in mTNFΔ/Δ mice compared to mTNFwt/wt littermates, suggesting that mTNF alone is necessary and sufficient for maintenance of cognitive function under physiological conditions.
Although the role of TNF in ischemic tissue injury and neurotoxicity has been widely reported (reviewed in literature 23 ), a clear understanding of the specific roles of solTNF versus mTNF in stroke pathology is still lacking. For the first time, our observations of decreased infarct volumes and improved functional outcome in mTNFΔ/Δ mice point at a neuroprotective role for mTNF in the brain after cerebral ischemia. These findings are consistent with studies showing that the neuroprotective effect is mediated through TNFR1 following stroke.37,72–74 TNFR1 is a high-affinity receptor allowing very low concentrations of TNF to have a significant impact on cellular responses. Thus, mTNF signaling through TNFR1, either constitutive or sustained mTNF production from microglia early after stroke, may have profound effects on the development of the ischemic infarct. In support of this, TNFR1 is known to be expressed constitutively on neurons in the brain, 75 making it possible for constitutively or early induced mTNF to signal directly to the ischemically vulnerable neurons. Nevertheless, it is also well documented that TNF may have neurotoxic effects, not only in focal cerebral ischemia76–80 but in other CNS traumatic pathologies as well, such as traumatic brain injury,81–85 and strategies to inhibit or reduce TNF have proved beneficial both in animal models and humans. Since solTNF is genetically ablated in our model, it is plausible that the neuroprotective effects observed in mTNFΔ/Δ mice could be ascribed, at least in part, to the beneficial effects of simply removing endogenous, microglial-derived solTNF, and not necessarily to neuroprotection through mTNF signaling. However, recent studies from various groups, including our own, have clearly shown that is not simply removal of solTNF that induces a neuroprotective effect, but the sustained presence of mTNF signaling. In experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis, blockade of solTNF with the selective inhibitor XPro1595, but not with the non-selective TNF blocker etanercept, resulted in improved locomotor function, neuroprotection and remyelination,21,57 indicating that removal of solTNF alone is not sufficient for a beneficial outcome, but the presence of mTNF signaling is required. In moderate contusive spinal cord injury, XPro1595, but not etanercept, also improved functional recovery and afforded neuroprotection but only when administered locally at the lesion site (epidural pump), and not when given systemically. 22 Similarly, recent studies from our group showed that a single, systemic injection of XPro1595 30 min after experimental stroke had no effect on the infarct size, 47 suggesting that for this compound to be effective a prolonged delivery may be necessary, like in spinal cord injury. For this reason, ongoing studies in our lab are addressing this very point using osmotic pumps to locally infuse XPro1595 at the infarct site.
Since mTNF is capable of activating TNFR1 and TNFR2, both receptors could be mediating the protective effects of mTNF observed in our model. While there is mounting evidence that in CNS cells TNFR2 is the receptor primarily associated with neuroprotection and remyelination,11,86–89 various studies also show such a role for TNFR1.37,42,57 Specifically in focal cerebral ischemia Taoufik and colleagues have shown that TNF is protective by activating the NF-κB-FLIPL signaling pathway downstream of TNFR1. In line with this report, our study is suggestive of a neuroprotective effect via TNFR1 signaling given the increased constitutive presence of this receptor in mTNFΔ/Δ mice compared to mTNFwt/wt.
In agreement with previous findings of increased cytokine and chemokine gene expression after stroke,37,39,90–94 we show significant upregulation of TNF, TNFR1, IL-1β, IL-6, CXCL1, CXCL10 and CCL2 mRNA levels in both mTNFΔ/Δ and mTNFwt/wt mice one day after pMCAO as compared to sham and naive mice. We did not find differences in gene expression between mTNFΔ/Δ and mTNFwt/wt mice in any of the genes investigated. This is in line with previous reports showing that chemokine gene expression is comparable in mTNFwt/wt and mTNFΔ/Δ mice following bacterial infections. 95 In contrast, using multiplex analysis, we found TNF, IL-1β, IL-6 and CXCL1 protein levels were unchanged in naïve mice or 3 h after pMCAO, but were significantly decreased in mTNFΔ/Δ mice one day after pMCAO. At this time after injury, we also found a significant reduction in MHCII expression and macrophage infiltration in mTNFΔ/Δ compared to mTNFwt/wt mice.
Since in this model the infarct is known to be fully developed within 6 h,35,49 the changes in cytokine levels that we observe at 24 h are unlikely to account for the reduction in infarct volume in mTNFΔ/Δ mice, rather they may be secondary to the reduced macrophage infiltration and activation. Collectively, this suggests that absence of solTNF results in efficient suppression of the inflammatory response in the ischemic brain, which might contribute to an environment more favorable to functional recovery.
In conclusion, the present study demonstrates that elimination of solTNF while maintaining mTNF is neuroprotective following focal cerebral ischemia. This finding may have implications in stroke therapy with the recent development of TNF inhibitors, such as XPro1595, that selectively target solTNF. Similarly to the successful therapeutic application of XPro1595 in experimental spinal cord injury, 22 local delivery of XPro1595 to the lesion site after stroke may prove beneficial in reducing lesion volume, inflammation and improving functional outcome.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institute of Health grant NS084303-01A1 (RB); The Miami Project To Cure Paralysis (RB); The Lundbeck Foundation (BHC (R67-A6383) and KLL (R54-A5539)); The Carlsberg Foundation (2007_01_0176), The Novo Nordisk Foundation (R153-A12550 & R168-A14120), and Fonden til Lægevidenskabens Fremme (KLL).
Acknowledgments
The authors acknowledge the technical assistance provided by Signe Marie Andersen, Dorte Lyholmer, Louise Lykkemark, and Mehran Taherian. The authors also thank Dr. Sedgwick for granting permission for the use of the mTNFΔ/Δ mice and Dr. Tacchini-Cottier (Department of Biochemistry, University of Lusanne, Switzerland) for the kind donation of the homozygous mTNFΔ/Δ breeding pairs.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
PMM performed experiments, interpreted results, performed statistical analysis and helped draft the manuscript. BHC conducted animal surgeries and performed experiments together with PMM and KLL, interpreted results and helped draft the manuscript. MD and LKK performed experiments and interpreted results. ST conducted the microglial cell counts and indirect infarct volumetric analysis. ND assisted with radiographic assessment of mice. BF gave useful input to the manuscript. TD and MS performed the multiplex analysis. RB performed experiments, interpreted results, performed statistical analysis and wrote the manuscript together with KLL. KLL conceived the study, performed animal surgeries, interpreted results, performed statistical analysis and wrote the manuscript together with RB.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.