
Abstract
The level of phosphodiesterase type 2 (PDE2) in the body is closely associated with human health. From the L6000 natural compound library, the natural coumarin compound coumestrol was identified as possessing PDE2 inhibitory activity. Using computer-aided drug design software, we modified and synthesized coumestrol derivatives and subsequently evaluated their PDE2 inhibitory activity. Among the synthesized compounds, B2-1 exhibited the strongest activity, with an IC₅₀ value of 1.09 ± 0.07 μM, approximately 7 times higher than that of coumestrol. Finally, ADMET prediction of coumestrol and B2-1 found that B2-1’s ability is equal to or better than coumestrol in terms of intestinal absorption, hepatotoxicity, protein binding, and so on.

Introduction
Phosphodiesterases (PDEs) form a large enzyme superfamily encoded by 21 distinct genes. Due to variations in transcription, translation, post-translational modifications, and alternative splicing, more than 80 different isoforms are ultimately produced from these genes. 1 PDEs are broadly classified into 11 families (PDE1–PDE11). They catalyze the hydrolysis of cyclic nucleotides—adenosine 3′,5′-cyclic monophosphate (cAMP) and guanosine 3′,5′-cyclic monophosphate (cGMP)—within cells, thereby terminating the diverse physiological processes mediated by cAMP and cGMP signaling pathways.2,3 Phosphodiesterase type 2 (PDE2) uniquely hydrolyzes both cAMP and cGMP, exhibiting approximately 30-fold selectivity for cGMP over cAMP. Furthermore, binding of cGMP to PDE2 allosterically enhances its catalytic hydrolysis of cAMP, thereby modulating the intracellular homeostasis of these secondary messengers and regulating associated signaling pathways. PDE2 is encoded by a single gene, PDE2A, which gives rise to three major isoforms: PDE2A1, PDE2A2, and PDE2A3. Within the central nervous system, PDE2 is predominantly expressed in regions critical for motor control, learning, and memory, including the cerebral cortex, hippocampus, striatum, amygdala, hypothalamus, and cerebellum.
Coumestrol, a prominent coumarin derivative, was first isolated from alfalfa in 1957 and is ubiquitously present in various legumes and foods such as soybeans, alfalfa sprouts, peas, Brussels sprouts, and spinach. Chemically, it possesses a rigid tetracyclic oxa-heterocyclic framework consisting of a coumarin moiety fused to a benzofuran ring. This fusion occurs via the six-membered lactone ring of the coumarin unit annulated with the furan ring, sharing a common carbon-carbon double bond. Coumestrol also features phenolic hydroxyl groups at both the 3 and 9 positions. Like other natural coumarins, coumestrol exhibits a broad spectrum of pharmacological activities, including estrogenic, 4 anti-osteoporotic, organ-protective, anticancer, 5 anti-inflammatory, 6 neuroprotective, 7 anti-diabetic and anti-obesity, antibacterial, immunosuppressive, 8 antioxidant, and skin-protective effects, 9 Currently recognized as one of the four principal natural coumarins, coumestrol has emerged as a promising scaffold for designing novel therapeutics targeting diverse diseases. Natural products, owing to their vast chemical and biological diversity, constitute a valuable resource for drug discovery. In a screening campaign against phosphodiesterase type 2 (PDE2) using the L6000-Natural Compound Library, coumestrol was identified as a significant PDE2 inhibitor, with an IC₅₀ value of 7.71 ± 1.17 µM.
Several inhibitors targeting phosphodiesterase type 2 (PDE2) have been developed, including EHNA, 10 BAY 60-7550, 11 PF-05180999, and ND-7001. However, no PDE2 inhibitor has yet received clinical approval or entered the market. This is primarily due to inherent limitations of the existing small-molecule candidates, which have consequently hindered their progress beyond preclinical research or prevented them from advancing through later stages of clinical trials.
Results and discussion
Docking results
Previous investigations into the structure–activity relationships (SAR) of phosphodiesterases (PDEs), based on their crystal structures, have identified common features that govern the interactions between different PDEs and their inhibitors. 12 In 2013, Jian Zhu and colleagues reported the X-ray crystal structure of PDE2 in complex with the highly selective nanomolar inhibitor BAY60-7550. The catalytic domain, represented by the structure 4HTX, consistently contains several conserved regions upon inhibitor binding. Ligands that interact with these regions often exhibit high inhibitory potency. These key interaction sites are primarily categorized as the Q-pocket, characterized by the glutamine switch mechanism and a hydrophobic clamp, and the H-pocket, defined by clusters of hydrophobic amino acid residues. 13 The Q-pocket predominantly involves the residues Gln812, Gln859, and Phe862, which constitute the core components of the glutamine switch mechanism. The H-pocket comprises the residues Leu770, Leu809, Ile866, Ile870, His773, and Leu774, all of which play significant functional roles. The crystal structure of the PDE2A catalytic site was elucidated by Iffland et al. 14 PDE2A interacts with cyclic nucleotides via residue Gln859, which exhibits conformational flexibility. This flexibility allows Gln859 to rotate freely and form a critical hydrogen bond either with the exocyclic carbonyl oxygen of cGMP or the exocyclic amino group of cAMP. This interaction targets the purine ring of the respective cyclic nucleotide, thereby conferring dual substrate selectivity for both cGMP and cAMP to PDE2A.
Computer-aided drug design (CADD) offers valuable structural insights by enabling the visualization of three-dimensional (3D) receptor structures and receptor–ligand complexes. CADD methods are broadly classified into two main approaches: structure-based drug design (SBDD), which directly targets the relevant biological macromolecule, and ligand-based drug design (LBDD), used when the target’s crystal structure is unavailable or the molecular target remains unknown. SBDD includes techniques such as molecular docking and virtual screening, whereas LBDD encompasses quantitative structure–activity relationship (QSAR) modeling and pharmacophore modeling. In this study, the CDOCKER protocol within the Discovery Studio software suite was used to analyze the interaction patterns between the phosphodiesterase type 2 (PDE2) crystal structure (PDB ID: 4HTX) and its ligands, thereby elucidating key protein–ligand interactions. The designed ligands were subsequently prioritized based on their predicted interaction profiles and calculated binding energies.
Molecular docking of the reference inhibitor BAY 60-7550 with the catalytic domain of PDE2 (PDB ID: 4HTX) was conducted using the Discovery Studio software package. The simulation produced a CDOCKER_INTERACTION_ENERGY of 72.5 kcal/mol. The resulting ligand pose, which illustrates key binding-site interactions, is shown in Figure 1.

2D and 3D results of BAY60-7550 docking with 4HTX crystal.
Crystallographic analysis indicates that inhibitors engage the PDE2A active site not only via the glutamine switch mechanism but also through interactions with hydrophobic residues within the H-pocket, such as Leu770 and Leu809. Specifically, the reference inhibitor BAY 60-7550 predominantly occupies the Q-pocket. The carbonyl oxygen atom of its imidazotriazine ring participates in the glutamine switch mechanism, forming hydrogen bonds with residues Gln859 and Gln812. Concurrently, the imidazotriazine ring engages in parallel π-π stacking interactions with Phe862, a key component of the hydrophobic clamp. Within the H-pocket, the propylphenyl moiety of BAY 60-7550 establishes significant hydrophobic contacts, namely alkyl and π-alkyl interactions, with residues Leu770 and Leu809. Analysis of the docking pose of BAY 60-7550 with the target protein 4HTX and its comparison with previously reported structural data 15 confirmed the accuracy and reliability of our docking methodology. Subsequently, molecular docking of coumestrol with the 4HTX structure was performed, yielding a CDOCKER INTERACTION ENERGY value of 35.33 kcal/mol. The resulting binding pose of coumestrol is shown in Figure 2.

2D and 3D results of coumestrol docking with 4HTX crystal.
Analysis of the docking pose indicates that the carbonyl group in the coumarin ring of coumestrol forms hydrogen bonds with Gln859 and Gln812, key residues of the glutamine switch mechanism in the Q-pocket. Concurrently, the coumarin ring participates in π–π stacking interactions with Phe862, a component of the hydrophobic clamp. These observed interactions align well with those identified for the reference inhibitor BAY 60-7550, thereby satisfying the key structural requirements for potent PDE2 inhibition. Furthermore, coumestrol forms additional stabilizing contacts within the binding site—in the Q-pocket: π-alkyl interactions with Ile826 and Met847, and π–π stacking with Phe830; in the M-pocket: a hydrogen bond with Asp808; in the H-pocket: a π-alkyl interaction with Leu809 and π–π stacking with Tyr655.
Coumestrol was modified at the 3-hydroxyl and 9-hydroxyl positions using the Grow Fragment method in Discovery Studio (DS) software. The Grow Fragment procedure involves three steps: generating replacement fragments from the ligand structure; refining potential fragments within the protein active site; and identifying novel scaffolds with desirable properties. The resulting compounds were subsequently redocked using CDOCKER. Based on their interactions with PDE2, binding energy, and synthetic feasibility, 42 compounds (21 of which share identical substituents) were selected for further study. Specific docking results are provided in Table 1.
CDOCKER Interaction Energy (kcal/mol) for screening compounds.

Synthesis of coumestrol derivatives
Synthesis of 3-Hydroxyl coumestrol derivatives
3-Bromophenol (compound 10) undergoes nucleophilic addition with glyoxylic acid (compound 9) under alkaline (NaOH) conditions to afford 2-(2-bromo-4-hydroxyphenyl)-2-hydroxyacetic acid (compound 8). Compound 8 is then treated in an HCl-acidic and SnCl₂-reducing environment to yield 2-bromo-4-hydroxyphenylacetic acid (compound 7). Next, the carboxylic acid group of compound 7 is esterified with methanol to obtain methyl 2-bromo-4-hydroxyphenylacetate (compound A6). The phenolic hydroxyl group at position 4 then undergoes Williamson ether synthesis with halide RX, producing a series of etherified products (compound A5 series). Under conditions involving copper(II) salts, alkalinity, and elevated temperature, these products undergo hydrolysis, in which the bromine at position 2 is replaced by a phenolic hydroxyl group, and the methoxycarbonyl-protected phenylacetic acid moiety is hydrolyzed to regenerate the phenylacetic acid group. Acidification then affords a series of 4-etherified 2-hydroxyphenylacetic acid compounds (compound A4 series). These compounds undergo intramolecular lactonization in a POCl₃-acidic environment to form a series of benzofuran-2-ones (compound A3 series). The benzofuran-2-one compounds subsequently undergo reflux condensation with 2,4-dihydroxybenzaldehyde under Et₃N-basic conditions, producing 4-substituted 7-hydroxy-3-(2-hydroxyphenyl)-2H-chromen-2-ones, where substitution occurs at the phenolic hydroxyl position 4 on the benzene ring at position 3 (compound A2 series). Finally, in the presence of DDQ as an oxidant in toluene, oxidative ring-closure occurs between the active hydrogen of the phenolic hydroxyl group at position 3 of the benzene ring and the active hydrogen on the olefinic carbon of the coumarin ring, forming a furan ring and yielding a series of 3-hydroxycoumestrol derivatives (compound A1 series).
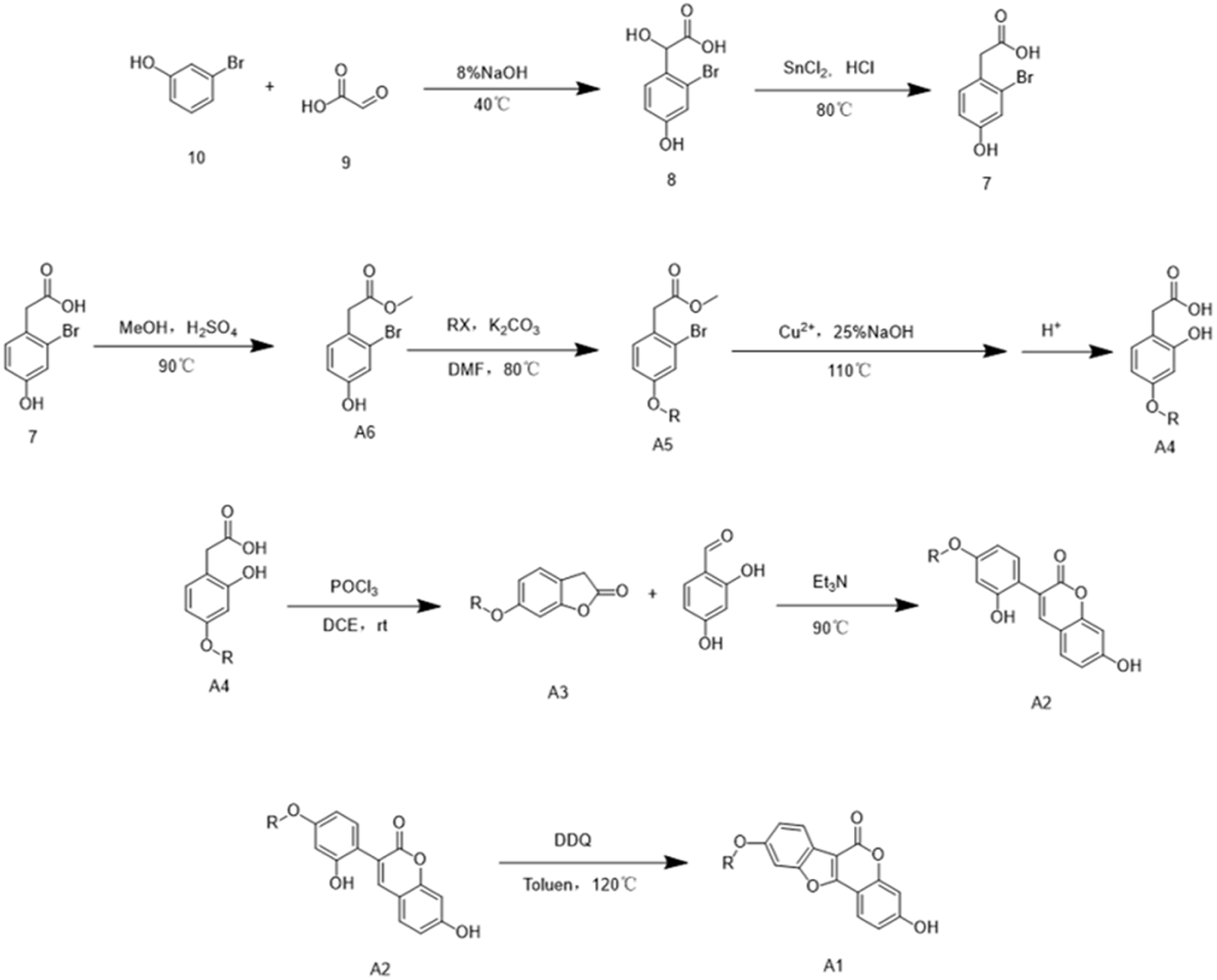
Synthesis of 9-hydroxy coumestrol derivatives
The synthetic route for 9-hydroxy coumestrol derivatives follows the same steps as for the 3-hydroxy analogues up to the formation of 2-bromo-4-hydroxyphenylacetic acid (compound 7). Compound 7 is then hydrolyzed under conditions of copper(II) salts, alkalinity, and elevated temperature to afford 2,4-dihydroxyphenylacetic acid (compound B6). Meanwhile, 2,4-dihydroxybenzaldehyde (compound B5) undergoes Williamson ether synthesis with halide RX to yield a series of 4-substituted 2-hydroxybenzaldehyde compounds (compound B4 series). A Perkin condensation between 2,4-dihydroxyphenylacetic acid and the compound B4 series, carried out with Et₃N, acetic anhydride, and heating, produces acetylated 3-phenylcoumarin compounds (compound B3 series). After removal of the acetyl protecting group from the compound B3 series, the resulting intermediates (compound B2 series) undergo DBU(1,8-diazabicyclo[5.4.0]undec-7-ene)-catalyzed cyclization, followed by acidification, to give the target 9-hydroxy coumestrol derivatives (compound B1 series).

In Vitro PDE2 inhibitory activity assay of coumestrol derivatives
The reagents used in this assay include cNMP, ATP, Kinase-Glo® reagent (luciferase), inactive protein kinase A, and PDE2 protein. The principle of the assay is as follows: cNMP binds to inactive protein kinase A, inducing a conformational change that releases the catalytic subunit. The released catalytic subunit reacts with ATP, consuming free ATP and thus lowering its concentration. Free ATP can react with the Kinase-Glo® reagent to generate a luminescent signal—when free ATP levels are low, the luminescent signal is weak. In the reaction system, PDE2 hydrolyzes cNMP, thereby controlling its concentration and indirectly regulating the intensity of the luminescent signal. If a PDE2 inhibitor (PDE2I) is added, it suppresses the hydrolytic activity of PDE2, leading to an accumulation of cNMP, a further decrease in free ATP, and a corresponding reduction in luminescence. The higher the potency of the PDE2I, the weaker the luminescent signal produced. By measuring the luminescence intensity, the inhibitory activity of the compound against PDE2 can be evaluated.
The synthesized compounds were initially screened at a concentration of 50 µM. Those exhibiting >50% inhibition were further tested at seven concentrations: 50, 25, 12.5, 6.25, 3.12, 1.56, and 0.78 µM. IC₅₀ values derived from these measurements are presented in Table 2.
IC50 of coumestrol derivatives on PDE2.

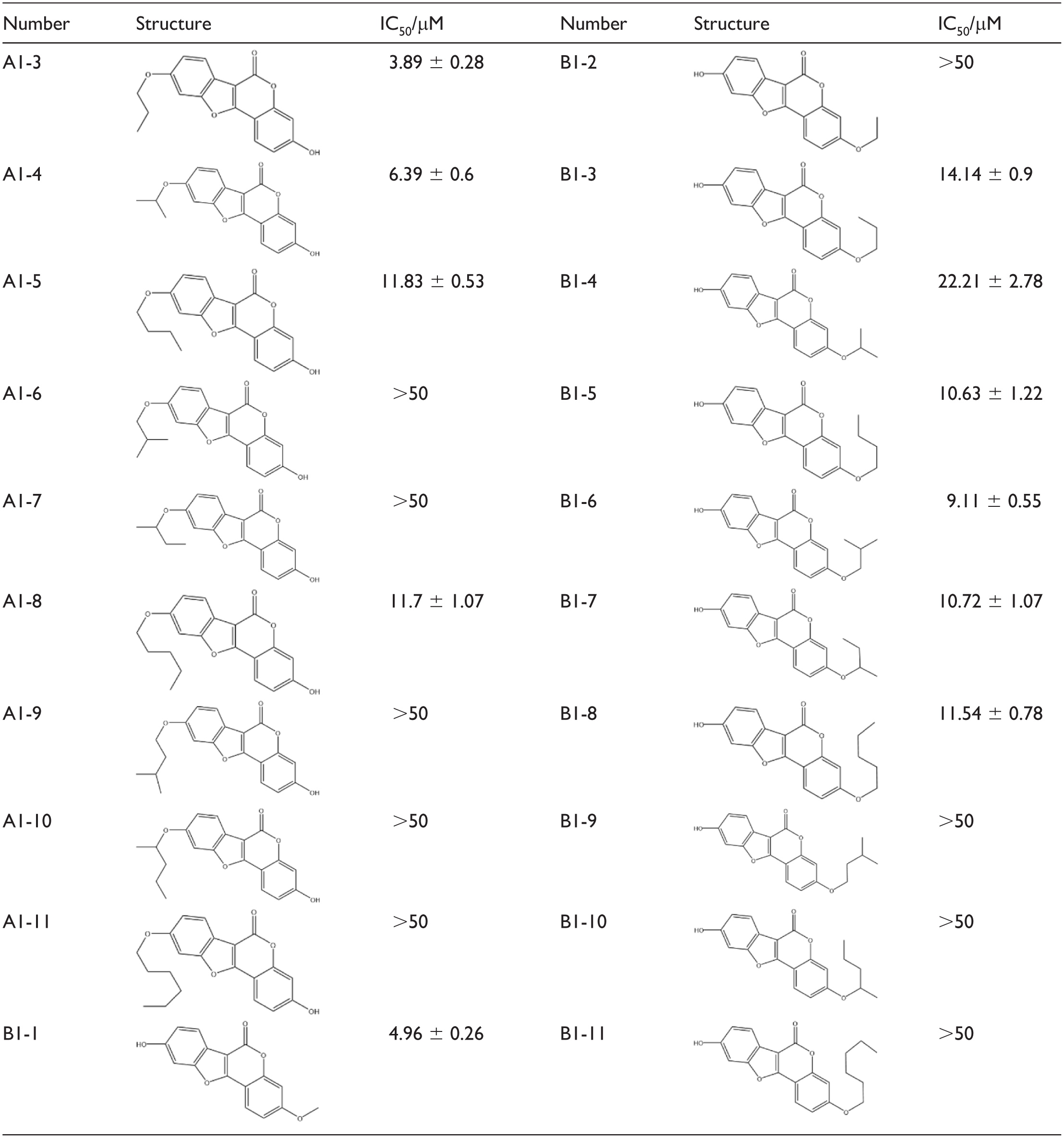
The data above summarize the PDE2 inhibitory activity of coumestrol derivatives. Most alkyl-substituted derivatives show activity comparable to that of coumestrol (IC₅₀ = 7.71 ± 1.17 µM). Among the 20 synthesized derivatives, only A1-3, A1-4, and B1-1—which contain fewer carbon atoms—exhibit improved potency. Other active derivatives did not show significant enhancement in inhibition. Furthermore, derivatives retaining the hydroxyl group at position 9 generally display higher activity than those retaining it at position 3.
Drug discovery strategies encompass multiple approaches, including serendipitous discovery, isolation of bioactive compounds from natural products, identification from drug metabolites, and screening of synthetic intermediates. During the synthesis of coumestrol derivatives, an intermediate with a 3-(2-hydroxyphenyl)coumarin structure was obtained. Based on our laboratory’s established expertise and accumulated findings in PDE2 inhibitor development,15–19 and given the well-documented, diverse pharmacological activities of coumarin derivatives, this synthetic 3-(2-hydroxyphenyl)coumarin intermediate was subsequently evaluated for its phosphodiesterase type 2 (PDE2) inhibitory activity.
The IC₅₀ values for the inhibition of PDE2 by 3-(2-hydroxyphenyl)coumarin derivatives are summarized in Table 3.
IC50 of 3-(2-hydroxyphenyl) coumarin derivatives on PDE2.
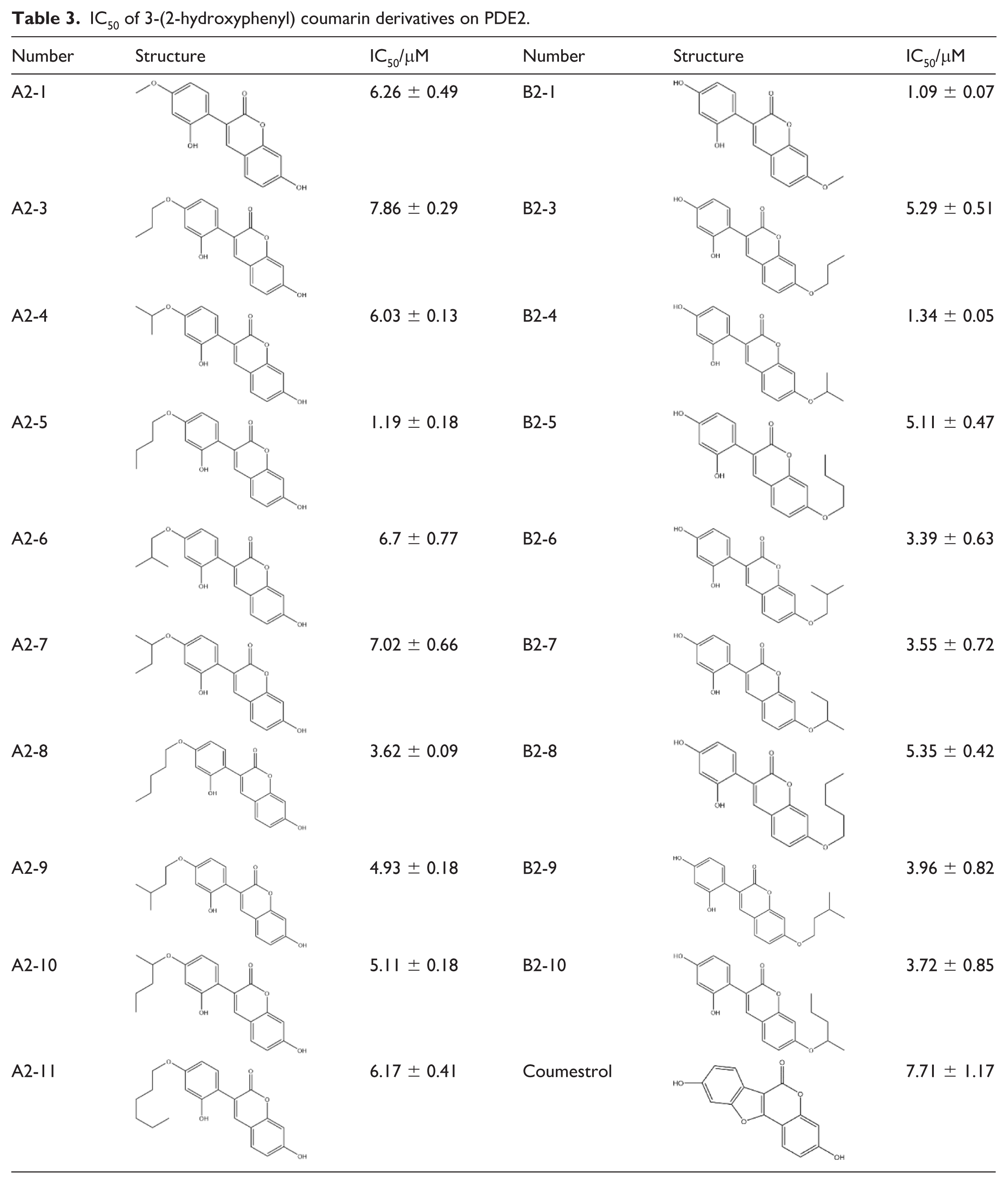

Compared with coumestrol (IC₅₀ = 7.71 ± 1.17 µM), 18 of the derivatives showed stronger PDE2 inhibitory potency, with the exceptions of compounds A2-2, B2-2, and B2-11 (IC₅₀ > 50 µM). Compound A2-3 exhibited activity comparable to that of coumestrol. Among the derivatives, the A series displayed the highest overall potency, with the most active member having an IC₅₀ of 1.19 ± 0.18 µM. Within the B series, compounds B2-1 and B2-4 were the most potent inhibitors, with IC₅₀ values of 1.09 ± 0.07 µM and 1.34 ± 0.05 µM, respectively.
Analysis of the coumestrol derivatives indicated that alkyl substitution at either the 3- or 9-position did not significantly enhance PDE2 inhibitory potency relative to the parent coumestrol. Moreover, IC₅₀ values revealed a progressive decline in activity as the carbon chain length of the alkyl substituent increased. Introducing branching at the terminus or proximal positions of the chain—compared to linear analogs with the same number of carbons—did not improve activity and in some cases even reduced potency. In contrast, the 3-(2-hydroxyphenyl)coumarin derivatives as a group exhibited stronger PDE2 inhibitory activity than the coumestrol derivatives. Analysis of their IC₅₀ values identified distinct structure–activity trends based on the substitution site. Substitution at the 4-position of the pendant phenyl ring: activity followed a parabolic trend with alkyl chain length, initially increasing and then decreasing as the number of carbons rose. Within this series, linear chains consistently conferred higher potency than their branched counterparts of equal carbon number. Within this series, linear chains consistently conferred greater potency than their branched counterparts of equivalent carbon number. Substitution at the 3-position of the coumarin core: activity showed a slight decrease with longer alkyl chains. Notably, for substituents of the same carbon number, branched analogs displayed greater potency than linear analogs.
ADMET properties prediction of coumestrol and B2-1
The ADMET model can be used to calculate absorption, distribution, metabolism, and excretion properties, particularly for predicting human intestinal absorption, blood-brain barrier permeability, cytochrome P450 2D6 inhibition, hepatotoxicity, plasma protein binding, and other characteristics. It provides theoretical data to support further development and research.
The absorption of drugs in the intestine plays a crucial role, as absorption is the process by which drugs move from the site of administration into the bloodstream. This primarily depends on the metabolism and barrier effects of intestinal enzymes and mucosal cells on the drugs. According to the data in Tables 4 and 5, the intestinal absorption values of coumestrol and B2-1 are both 0, indicating favorable absorption.
ADMET-related data of coumestrol and B2-1.

Passive intestinal absorption of coumestrol and B2-1.

Cytochrome P450 enzymes belong to a family of multifunctional enzymes. A decrease in cytochrome P450 enzyme activity can directly or indirectly impair the liver’s detoxification capacity. CYP2D6 is a critical component of the cytochrome P450 family and participates in the metabolism of various substrates in the liver. According to the prediction results in Table 4, the CYP450 2D6 enzyme inhibition value for coumestrol is −4.194, while that for B2-1 is −1.698. Based on Table 6, neither coumestrol nor B2-1 inhibits CYP2D6, and thus, they do not affect liver detoxification.
Coumestrol and B2-1 bind to human cytochrome P4502D6 enzyme.

In Table 4, the hepatotoxicity value for coumestrol is 7.384, while that for B2-1 is 2.188. Following structural modification, the hepatotoxicity of B2-1 is significantly reduced.
Plasma protein binding is a model used to predict whether a compound can bind extensively (⩾90% binding) to carrier proteins in the blood. The plasma protein binding of drug molecules can influence drug efficacy, as only the unbound fraction exhibits pharmacological activity. The results show that the ADMET_AlogP98 values for plasma protein binding of coumestrol and B2-1 are 3.061 and 2.878, respectively. In Table 7, neither compound binds to plasma proteins, suggesting that both may possess drug activity.
Coumestrol and B2-1 binding to plasma proteins.

Conclusions
Among the 20 synthesized coumestrol derivatives and 22 intermediate compounds evaluated, compound B2-1 displayed the most potent PDE2 inhibitory activity, with an IC₅₀ of 1.09 ± 0.07 µM, and the PDE2 inhibitory activities of compounds A2-5 and B2-4 were respectively 1.19±0.18, 1.34±0.05 µM, Overall, three compounds—A2-5, B2-1, and B2-4—showed sub-micromolar IC₅₀ values close to 1 µM. Notably, 21 compounds exhibited greater potency than the parent compound coumestrol (IC₅₀ = 7.71 ± 1.17 µM). These results clearly identify B2-1 as the most promising inhibitor in this series. Although its activity is lower than that of the reference PDE2 inhibitor BAY 60-7550, it represents a substantial improvement over coumestrol. ADMET was employed to predict the intestinal absorption, hepatotoxicity, and protein-binding capacity of coumestrol and B2-1. The intestinal absorption values were both 0, indicating favorable intestinal absorption. The protein-binding capacities were both below 90%, reflecting good drug activity. In the hepatotoxicity prediction, the value for B2-1 was 2.188, significantly lower than that of coumestrol (7.384), suggesting a substantial reduction in hepatotoxicity for B2-1. The ADMET prediction results demonstrate that B2-1 exhibits superior properties compared to coumestrol.
Based on our work and existing studies on PDE2 inhibitors,20,21 most compounds with good inhibitory activity feature a bicyclic conjugated core, retain ring structures capable of hydrogen bonding, and contain flexible functional side chains.
This core was designed to engage key interactions within the PDE2 active site. π-π Stacking with the Hydrophobic Clamp: the planar, electron-rich bicyclic core is positioned to form optimal π–π stacking interactions with the hydrophobic clamp formed by Phe862 in the Q-pocket. Hydrogen Bonding for the Glutamine Switch: Functional groups integrated into the core scaffold can act as hydrogen‑bond donors or acceptors to interact with the key residues Gln812 and Gln859. Hydrophobic Interactions in the H‑pocket: Diverse hydrophobic substituents are introduced at suitable positions flanking the core scaffold to maximize hydrophobic contacts within sub‑pockets of the H‑pocket, thereby enhancing binding affinity and inhibitory potency against PDE2.
Methods
Synthesis
All chemicals, reagents, and solvents utilized in this investigation are of analytical grade and were obtained from the following suppliers: Alfa, Meyer, and Aladdin. The 1H NMR spectra were obtained using a Bruker drx spectrometer at 300 and 400 MHz. Similarly, the Bruker BioSpin GmbH spectrometer records the 13C NMR spectrum at 101 MHz. The coupling constant is represented in Hertz. The melting point was determined with an X4-A microscope. TLC was carried out on a pre- coated silica GF-254 plate (25 × 75 mm, Shanghai Pinjia Chemical Co. Ltd.) and samples were viewed under ultraviolet light. All raw materials and reagents come from commercial providers.
Molecular docking method
Molecular docking simulations were performed using the CDOCKER protocol within Discovery Studio to characterize the protein-ligand interaction patterns between the PDE2 catalytic domain (PDB: 4HTX) and its cognate ligands. The resulting binding poses were systematically evaluated based on criteria encompassing key intermolecular forces and calculated binding energies to prioritize compounds with optimal target engagement.
Synthesis method
Synthesis of 2-bromo-4-hydroxyphenylacetic acid(7)
A solution of glyoxylic acid monohydrate (0.12 mol, 22.2 g, 50 wt% in water) and an aqueous NaOH solution (0.18 mol, 7.2 g, 8 wt%) were added simultaneously via dropwise addition over 30 min to a mechanically stirred suspension of 3-bromophenol (0.12 mol, 20.64 g) in a three-necked flask equipped with a magnetic stirrer. The reaction mixture was maintained at 40 °C in an oil bath. Upon completion of the addition, the reaction was allowed to proceed at 40 °C for 10 h, with progress monitored by thin-layer chromatography (TLC). The resulting mixture was acidified to pH 1−2 with concentrated hydrochloric acid and subsequently neutralized to pH 7–8 with a saturated sodium bicarbonate solution. Unreacted 3-bromophenol was extracted with ethyl acetate (3 × 100 mL), and the combined organic extracts were concentrated under reduced pressure to recover the starting material. The aqueous phase was re-acidified to pH 1–2 with concentrated HCl and extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried over anhydrous Na₂SO₄ and concentrated in vacuo to afford 2-bromo-4-hydroxymandelic acid as a pale yellow viscous liquid (24.6 g, 0.10 mol). 2-Bromo-4-hydroxymandelic acid (0.10 mol, 24.6 g) was dissolved in concentrated hydrochloric acid (50 mL) in a round-bottom flask. Tin(II) chloride dihydrate (SnCl₂·2H₂O, 0.12 mol, 22.8 g) was added portionwise, and the reaction mixture was heated at 80 °C in an oil bath for 5 h under reflux. Formation of a white precipitate was observed during the reaction. After confirmation of reaction completion by TLC, the mixture was carefully poured into preheated water (300 mL, 80 °C) for recrystallization. The volume was reduced to approximately 120 mL by evaporation, and the solution was cooled to 2 °C in a refrigerator. The resulting crystals were collected by vacuum filtration, washed with ice-cold water, and dried under reduced pressure to yield 2-bromo-4-hydroxyphenylacetic acid as white needle-like crystals.
Synthesis of methyl 2-(2-bromo-4-hydroxyphenyl)acetate(A6)
Dissolve 2-bromo-4-hydroxyphenylacetic acid (0.07 mol, 16.1 g) in methanol (60 mL), add 2 mL sulfuric acid, and reflux at 90 °C for 2 h while monitoring the reaction progress via TLC. After the reaction is complete, neutralize the sulfuric acid with saturated sodium bicarbonate solution to adjust the pH to 7–8. Extract the mixture with ethyl acetate. Remove the ethyl acetate by rotary evaporation to obtain a yellow oily liquid, identified as methyl 2-bromo-4-hydroxyphenylacetate.
Synthesis of 2-hydroxyphenylacetic acid derivatives(A4)
To a mixture of methyl 2-bromo-4-hydroxyphenylacetate (12.3 mmol, 3.0 g) and anhydrous potassium carbonate (18.45 mmol, 2.55 g) in 40 mL anhydrous DMF, add halide RX (18.45 mmol) dropwise. Stir the system in a 80 °C oil bath. Monitor the reaction via TLC. On completion, dilute with water and extract with ethyl acetate. Wash the organic layer with saturated brine thrice. Remove ethyl acetate by rotary evaporation to get a crude etherified methyl 2-bromophenylacetate. For purification, take the crude product and add copper(II) bis(8-hydroxyquinoline) and a 25% NaOH solution in a molar ratio of 1:0.15:15. Heat in a 110 °C oil bath for 10 h. Monitor the reaction via TLC. After completion, filter off the copper salt and acidify the filtrate to pH 1–2 with concentrated HCl. Extract with ethyl acetate and remove the solvent. Purify the oily residue by column chromatography (PE:EA:AcOH = 3:1:1%). Finally, obtain white to yellow solids, the etherified 2-hydroxyphenylacetic acid derivatives.
Synthesis of benzofuranone derivatives(A3) and 7-hydroxy-3-(2-hydroxyphenyl)-2H-chromen-2-one(A2)
Add 4.2 mmol of a 2-hydroxyphenylacetic acid derivative to a 100 mL flask. Dissolve in 22.7 mL of 1,2-dichloroethane (DCE), then add 3.4 mL POCl₃. Stir at room temperature for 4 h. Monitor by TLC. Quench with saturated NaHCO₃ after completion. Extract with ethyl acetate, concentrate, and purify by column chromatography (PE:EA = 20:1) to obtain compound A3. Add 5.7 mmol of benzofuran-2-one (compound A3) to a flask with 25 mL Et₃N. Slowly add 5.7 mmol (0.79 g) of 2,4-dihydroxybenzaldehyde. Stir in a 90 °C oil bath for 3 h. Monitor by TLC. Remove excess Et₃N, dissolve the residue in MeOH, acidify with 1 M HCl to pH 2~3. Extract with ethyl acetate, concentrate, and purify by column chromatography (PE:EA = 3:1) to obtain compound.
Synthesis of 3-hydroxy coumestrol derivatives(A1)
Dissolve 3-phenylcoumarin derivative (4.3 mmol) in 104 mL toluene in a flask. Add DDQ (8.6 mmol, 2.0 g) slowly with stirring, then heat to 120 °C and react for 6 h. Monitor by TLC. After completion, remove toluene by rotary evaporation and purify the residue by column chromatography (PE:EA = 3:1) to obtain 3-hydroxy coumestrol derivatives as white solids.
Synthesis of 2,4-dihydroxyphenylacetic acid(B6)
To a round-bottom flask containing 2-bromo-4-hydroxyphenylacetic acid (0.07 mol, 16.1 g) and a stirrer, add copper(II) bis(8-hydroxyquinoline) and a 25% NaOH solution in a molar ratio of 1:0.15:15. Stir and heat the mixture in a 110 °C oil bath for 10 h. Monitor the reaction via TLC. Once the reaction is complete, filter off the solid copper salt. Acidify the filtrate to pH 1–2 with concentrated HCl and extract with ethyl acetate. Remove the solvent to obtain a reddish-brown oil, which crystallizes on cooling. This crude 2,4-dihydroxyphenylacetic acid can be used directly in the next step without further purification.
Synthesis of 2-hydroxybenzaldehyde derivatives(B4)
To a round-bottom flask containing 2,4-dihydroxybenzaldehyde (0.022 mol, 3.0 g) and a stirrer, dissolve in 30 mL DMF. Add NaHCO₃ (0.033 mol, 2.8 g), then slowly add halide RX (0.033 mol). Reflux in a 110 °C oil bath for 8 h while monitoring via TLC. After completion, dilute with water and extract with ethyl acetate. Wash the organic layer with saturated brine three times. Purify the residue by column chromatography (PE:EA = 200~500:1) to obtain colorless, fragrant oily 2-hydroxybenzaldehyde derivatives.
Synthesis of 3-phenyldiacetate coumarin derivatives(B3)
To a 100 mL flask containing 2-hydroxybenzaldehyde derivative (9.8 mmol) and 2,4-dihydroxyphenylacetic acid (9.8 mmol), add Et₃N (19.6 mmol, 1.98 g) and acetic anhydride (29.4 mmol, 3.0 g). Stir the mixture in a 110 °C oil bath with a reflux condenser for 6 h. Monitor the reaction via TLC. After completion, add water and extract with ethyl acetate. Purify the residue by column chromatography (PE:EA = 3:1) to obtain crude 3-phenylcoumarin diacetate.
Synthesis of 3-(2,4-dihydroxyphenyl)-2H chromene-2-one derivatives(B2) and 9-hydroxy coumestrol derivatives (B1)
Place 6.3 mmol of crude 3-phenylcoumarin diacetate in a flask with a stirrer. Add 25 mL of 20% HCl/EtOH and reflux at 80 °C for 2 h. Monitor by TLC. After completion, filter, rinse the solid with cold ethyl acetate, and dry to get a yellow solid product. This is the 7-hydroxyphenyl substituted 3-(2,4-dihydroxyphenyl)-2H-chromen-2-one. Add 2 mmol of compound B2 to a flask. Include 10 mmol of DBU and 20 mL of water. Stir uncovered at 40 °C for 12 h. Monitor by TLC. After completion, add concentrated HCl to adjust the pH to 1 and stir for an additional 2 h. Extract with ethyl acetate, concentrate, and purify by column chromatography (PEs:EA = 4:1) to obtain 9-hydroxy coumestrol derivatives as white solids.
PDE2 enzyme inhibitory activity assay
The PDE-GloTM Phosphodiesterase Assay kit was used to detect the inhibitory activity of the compounds against phosphodiesterase II (PDE2) with BAY60-7550 as the positive control. Escherichia coli BL21 Codon Plus (DE3) and DH5α were purchased from Stratagene. The recombinant plasmid pET15b-PDE2A (580–941) was provided by Professor Ke Hengming’s laboratory at the University of North Carolina. The specific determination methods are as follows: Add 1 μL of the compound to be tested with the diluent and 1.5 μL of PDE2 protein diluent to the white hole plate, shock at room temperature for 30 min, then add 2.5 μL of cNMP, 2.5 μL of termination buffer, and 2.5 μL of detection solution successively. For each addition of reagent, centrifuge for 1 min and then shake at room temperature for 20 min. Finally, Kinase Reagent buffer was added for light avoidance reaction for 10 min. After the reaction was complete, the IC50 was calculated using GraphPad Prism and the multifunctional enzymograph. During the experiment, positive control (no PDE2 protein) and negative control (no inhibitor) were set up and supplemented with 1× Reaction Buffer in equal amounts. Three parallel experiments were required for each group.
Supplemental Material
sj-docx-1-chl-10.1177_17475198261424070 – Supplemental material for Design, synthesis, and biological evaluation of coumestrol-based inhibitors targeting phosphodiesterase type 2
Supplemental material, sj-docx-1-chl-10.1177_17475198261424070 for Design, synthesis, and biological evaluation of coumestrol-based inhibitors targeting phosphodiesterase type 2 by Guoqiang Song, Tongtong Zhou, Qiulin Hu, Ming Ni, Xiaoqing Feng and Long Tang in Journal of Chemical Research
Footnotes
Ethical considerations
This study did not involve any ethical concerns requiring approval from an institutional review board.
Consent to participate
Not applicable, as this study did not involve human participants.
Consent for publication
Not applicable.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data supporting the findings of this study are available upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.