
Abstract
Phosphodiesterase 4B is an important enzyme belonging to the phosphodiesterase family, playing a role in regulating the levels of cyclic AMP in cells. Phosphodiesterase 4B degrades cyclic AMP, a crucial signaling molecule involved in numerous biological processes, including inflammation regulation. Recently, the search for potential inhibitors with fewer side effects and high biological activity in valuable medicinal plants has drawn the attention of current scientists. Various in silico methods have been applied to reduce costs and time for experimental studies. In this study, an in silico screening involving a set of 131 natural compounds sourced from Prinsepia utilis species was conducted. These compounds were docked into the active site of the phosphodiesterase 4B protein. As a result, 10 compounds exhibited the most potential inhibitory activity against phosphodiesterase 4B, including 2α-O-trans-p-coumaroyl-3β,19α-dihydroxy-urs-12-en-28-oic acid, 2α-O-cis-p-coumaroyl-3β,19α-dihydroxy-urs-12-en-28-oic acid, cyanidin-3-O-rutinoside, delphinidin-3-O-rutinoside, peonidin-3-O-rutinoside, rutin, isorhamnetin-3-O-rutinoside, kaempferol 3-O-α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside-7-O-β-D-glucopyranoside, kaempferol 3-O-α-L-rhamnopyranosyl-(1→6) [α-L-rhamnopyranosyl-(1→2)]-β-D-glucopyranoside, quercetin 3-O-α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside were identified through molecular docking simulations. Subsequently, molecular dynamics simulations were performed on these complexes, revealing significant findings regarding their stability. Furthermore, MM–GBSA calculations indicated that the potential compounds had stronger binding free energies than the reference inhibitor. Finally, the selected compounds were subjected to toxicity prediction, showing noteworthy results with large LD50 values and safe toxicity levels. Therefore, these compounds could be potential candidates for further experimental studies as phosphodiesterase 4B inhibitors.
Introduction
Inflammation is a multifaceted process that is often studied from various angles, with infectious and autoimmune inflammations being the most researched. It can occur in different parts of the body and lead to serious health issues. 1 One enzyme of interest in the context of inflammation is phosphodiesterase 4B (PDE4B). PDE4B, a member of the phosphodiesterase (PDE) family, degrades cyclic nucleotides such as cAMP and cGMP, thus reducing these crucial second messenger signals within cells. cAMP is renowned for its anti-inflammatory properties and is widely used in pharmacology to treat inflammatory diseases.2,3 Recent studies have identified cAMP as a key coordinator in resolving inflammation. 4 In addition, cAMP regulates cellular metabolism by activating protein kinase A (PKA) and targeting proteins directly activated by cAMP, impacting numerous essential cellular functions across all cell types. Research has shown that knocking down PDE4B effectively inhibits lipopolysaccharide (LPS)-induced activation of NF-κB and inflammatory responses in various cell types. Deleting PDE4B also reduces the LPS-induced production of reactive oxygen species (ROS). 4 PDE4B plays a role in releasing inflammatory mediators from immune cells, thereby amplifying the inflammatory response. Therefore, inhibiting PDE4B activity could be a potential strategy for controlling inflammation. Recently, the application of molecular docking and molecular dynamics (MD) simulations in discovering PDE4B inhibitors has shown considerable success.5 –7
Prinsepia utilis Royle, a member of the Prinsepia genus within the Rosaceae family, holds significant medicinal value and is often widely used in traditional medicine for treating various ailments like joint pain and inflammation. 8 So far, around 131 compounds have been identified from different parts of P. utilis, spanning terpenoids, flavonoids, lignans, and sterols.9 –11 Alongside chemical exploration, researchers have focused on assessing its diverse biological activities, including antioxidant, hypoglycemic, α-glucosidase inhibitory, cytotoxic, anti-inflammatory, immunosuppressive, antibacterial, and lipase inhibitory properties. 9 Particularly noteworthy are its potential anti-inflammatory effects. Thakur et al. conducted an in vivo study on the methanolic extract of P. utilis flowers using a rat pedal edema model induced by carrageenan. The results revealed significant anti-inflammatory activity at both 100 mg/kg body weight (64.38% inhibition) and 200 mg/kg body weight (65.75% inhibition). 12 P. utilis seed oil has shown promise as a natural anti-inflammatory and analgesic, evident from in vitro trypsin inhibition assays and serum bovine albumin denaturation tests. These tests demonstrated a dose-dependent response with IC50 values of 63.57 and 518.14 μg/mL, respectively. Moreover, in vivo experiments showcased effective anti-inflammatory activity, with significant inhibition lasting up to 4 h against carrageenan- and formalin-induced mouse paw edema at a maximum experimental dose of 200 mg/kg body weight. 13 In addition, P. utilis water extract exhibited inhibitory effects on allergic contact dermatitis symptoms in mice induced by fluorescein isothiocyanate (FITC), by repairing tissue barriers and reducing Th2-type allergic inflammation. 14
Despite these promising findings, information regarding the anti-inflammatory properties of this species remains limited. Hence, current research endeavors have undertaken in silico studies on compounds from P. utilis to aid in the discovery of phosphodiesterase-4 inhibitory agents for treating inflammatory diseases.
Materials and methods
Molecular docking
The chemical structures of all compounds derived from P. utilis were drawn using Marvin JS software and then energy-minimized in the Avogadro software with the MMFF94s force field.9,15 The chemical structures of the ligands were further converted to PDBQT format using AutoDockTools software. The crystal structure of the human phosphodiesterase 4B (PDE4B) protein was downloaded from the RCSB Protein Data Bank (https://www.rcsb.org/structure/4KP6) with PDB ID: 4KP6 and a resolution of 1.50 Å. 16 The protein molecules were prepared before docking using Chimera and AutoDockTools software. The preparation process involved removing water molecules and co-crystallized ligands, then adding polar hydrogens, Kollman partial charges, and converting to PDBQT format. AutoDock Vina was used for docking the molecules between the ligand structure and the target protein.17,18 A grid box was selected to cover the binding site of the co-crystallized ligand at the center coordinates x = −41.8 Å, y = 91.2 Å, z = 114.4 Å, the box size of 24 × 24 × 24, and the spacing of 1. All parameters during the docking experiment were set to default, except for the exhaustiveness value which was set to 400. The protocol validation for this docking process was conducted through re-docking. PyMOL and Discovery Studio Visualizer software were used to visualize the interaction modes of ligand-protein.
Molecular dynamics (MD)
To monitor the movement of each atom in a system over time and have information about their relative positions, MD simulations were applied using the GROMACS v2023.1 software package. 19 The AMBER99SB-ILDN force field was used to parameterize the protein PDE4B (PDB ID: 4KP6). The energy of the docked ligand structure was calculated using the B3LYP/6-31G** function set with the Gaussian09 program, followed by generating ligand topology parameters through a combination of the GAFF and GLYCAM_06j-1 force fields, utilizing ACPYPE and AmberTools 22.20,21 The protein–ligand complex system was solvated with the TIP3P water model placed in a triclinic box and neutralized by adding counter ions (Na+). To ensure system stability, the steepest descent algorithm was used for energy minimization with a maximum force of 1000 kJ.mol-1.nm-1 and performed with 50,000 steps. Then, the systems were equilibrated through two steps: the NVT ensemble and the NPT ensemble at 100 ps to maintain the temperature and pressure at 310 K and 1 atm. The production MD were run for 200 ns to relax the system, and trajectories were saved every 2.0 ps. Post-simulation analyses including RMSD and RMSF were plotted using the Matplotlib tool in Python.
MM–GBSA calculations
The binding free energy of the protein complex system in a 200 ns MD simulation was estimated using the gmx_MMPBSA v1.4.3 program, employing the MM/GBSA (Molecular Mechanics/Generalized Born Surface Area) approach. 22 These calculations were performed using the single trajectory (ST) method, with the AMBERFF99SB-ILDN and GAFF2 force fields applied to the protein and ligand, respectively. A total of 20,000 snapshots from each 200 ns MD simulation trajectory were utilized for the binding free energy calculations. The MM/GBSA binding energy was determined by summing the contributions of various interactions, represented as follows

Here, ΔEMM = ΔEcovalent + ΔEele + ΔEvdW = (ΔEbond + ΔEangle + ΔEdihedral) + ΔEele + ΔEvdW and ΔGsol = ΔGGB + ΔGSA.
In this context, Gcomplex is the Gibbs free energy of the PDE4B complex with the studied compound, Greceptor is the Gibbs free energy of the PDE4B protein, and Gligand is the Gibbs free energy of the unbound studied compound. ΔH represents the enthalpy of binding, while −TΔS corresponds to the entropy changes upon ligand binding. ΔEMM (or ΔGGas) denotes the change in molecular mechanic energy in the gas phase, encompassing changes in internal energy ΔEint (bond, angle, and dihedral energies), electrostatic energy ΔEele, and van der Waals energy ΔEvdW . The solvation free energy ΔGso includes the polar contribution (electrostatic solvation energy calculated via the GB model) and the nonpolar contribution ΔEsurf or ΔGSA (estimated using the solvent-accessible surface area) between the solute and the continuum solvent.22,23
Toxicity prediction
ProTox 3.0 (https://tox-new.charite.de/protox3/) is a powerful and efficient tool for predicting the toxicity of potential compounds. Utilizing this tool not only saves time and costs but also enhances accuracy and efficiency in toxicity research. This significantly contributes to the development of safe and effective compounds in the pharmaceutical and chemical fields. 24 Therefore, ProTox 3.0 online tools were employed to study the toxicity properties of the selected compounds for the current research.
Results
Molecular docking
Molecular docking simulation is an important computational method in the search for potential inhibitors from screening natural compound datasets of medicinal plant species.25,26 In this study, a database of compounds sourced from P. utilis was subjected to molecular docking against the target protein PDE4B (PDB ID: 4KP6). Before docking, protocol confirmation is necessary through the redocking process. The obtained results are presented in Figure 1, where the overlaid ligand after redocking with the initial ligand shows deviation within an acceptable range as the calculated RMSD value is 1.25556 Å (<2Å).27,28 The redocked ligand 2-ethyl-2-{[4-(methylamino)-6-(1H-1,2,4-triazol-1-yl)-1,3,5-triazin-2-yl]amino}butanenitrile (1S1), a known PDE-4B inhibitor with an experimental in vitro IC50 value of 0.81 nM, was chosen as the positive control in this study. 16 After successful protocol confirmation, the compounds were docked, and the results are presented in Tables 1 and Supplemental Table S1. It can be observed that the binding affinities of the studied compounds range from −1.55 to −11.22 kcal/mol, whereas the control compound 1S1 has an affinity of −7.25 kcal/mol. As observed in Supplemental Table S1, the top 10 potential compounds exhibited affinities lower than −10 kcal/mol. These compounds were selected for further analysis of their interaction mode and binding capability with the target protein PDE4B as presented in Table 1 and Figure 2.

Superimposition of the co-crystallized ligand (cyan) and the redocked ligand (yellow) in the PDE4B protein with an RMSD value of 1.25556 Å.
The binding affinities of the top-hit compounds and their interactions with the PDE4B protein.

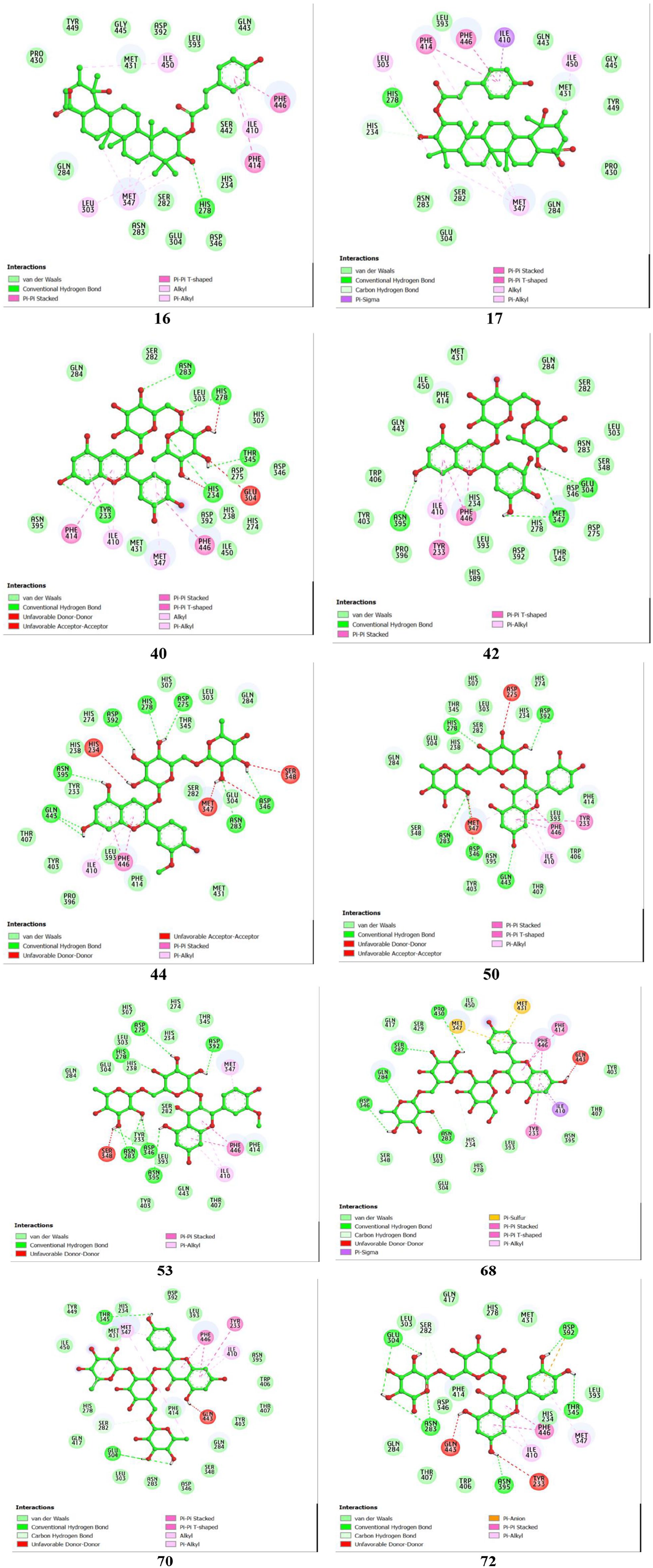
2D interactions between amino acid residues of PDE4B protein (PDB ID 4KP6) and top-hit compounds.
Five natural rutinosides—specifically cyanidin-3-O-rutinoside (compound 40), delphinidin-3-O-rutinoside (compound 42), peonidin-3-O-rutinoside (compound 44), rutin (compound 50), and isorhamnetin-3-O-rutinoside (compound 53)—demonstrate strong binding affinities with ΔG values of −10.29, −10.24, −11.22, −10.52, and −10.24 kcal/mol, respectively. Among these, compound 44 forms the highest number of hydrogen bonds with seven amino acid residues—Gln443, Asn395, Asp392, His278, Asp275, Asp346, and Asn283—resulting in the strongest binding affinity (ΔG = −11.22 kcal/mol). The predominance of hydrogen bonds significantly enhances the stability and binding affinity of the protein–ligand complexes. Compound 40 forms hydrogen bonds with Tyr233, Asn283, His278, Thr345, and His234. Compound 42 forms hydrogen bonds with Asn395, Met347, and Glu304. Compound 50 establishes hydrogen bonds with residues His278, Asp392, Asn283, Asp346, and Gln443. Compound 53 forms hydrogen bonds with residues Asn395, Asp346, Asn283, Asp275, Asp392, and His278. Hydrophobic interactions, including π-alkyl, π-π T-shaped, π-π stacked, π-sulfur, and π-σ interactions, are also observed in the complexes of these compounds with the PDE4B protein. Detailed interactions of compounds 40, 42, 44, 50, and 53 with the amino acid residues of the PDE4B protein are presented in Table 1.
Two kaempferol derivatives, compounds 68 and 70, and one quercetin derivative, compound 72, show similar binding affinities of −10.2 kcal/mol, −10.13 kcal/mol, and −10.1 kcal/mol, respectively. Compound 68 forms hydrogen bonds with Asp346, Asn283, Gln284, Ser282, and Pro430, while compound 70 forms bonds with Thr345 and Glu304. Compound 72 establishes hydrogen bonds with five residues—Glu304, Asn283, Asn395, Thr345, and Asp392. Other interactions of compounds 68, 70, and 72 with the amino acid residues of the PDE4B protein are detailed in Table 1.
Overall, the compounds interact with key amino acid residues in PDE4B’s catalytic domain such as a metal binding M pocket (His234, Asp346, Met347, Asp392), solvent-filled side pocket (Phe414, Ser282), and Q switch and P clamp pocket (Ile410, Asn395, Phe446, and Gln443) and, indicating their potential as PDE4B inhibitors. 29
MD simulation
To evaluate the stability and interactions of the protein–ligand complexes with PDE4B for the most promising compounds (ΔG ⩽10 kcal/mol), we analyzed the results of MD simulations. The RMSD values for the protein backbone and ligands are presented in Figure 3 and Supplemental Figure S1. Throughout the 200 ns simulation, the RMSD values of the protein in the complexes showed minor changes, staying below 0.3 nm with average values ranging from 0.1557 to 0.2375 nm, compared with 0.1671 nm for the apo-protein. The RMSD values for the proteins in the PDE4B-17, PDE4B-42, PDE4B-50, and PDE4B-70 complexes remained stable, showing minimal fluctuations around fixed points with no significant deviations. For the PDE4B-16 complex, a slight increase in protein backbone RMSD was observed, rising from approximately 0.15 nm at 55 ns to 0.23 nm at 60 ns, after which it stabilized for the remainder of the simulation. A similar trend was seen in the PDE4B-44 complex, where the RMSD increased modestly from 0.13 nm at 55 ns to 0.22 nm at 60 ns before reaching stability. Notably, the PDE4B-44 complex displayed minor structural adjustments during the early phases but achieved stability after 30 ns. The PDE4B-53 complex experienced fluctuations in protein backbone RMSD between 75 and 110 ns, after which it stabilized. In contrast, the PDE4B-68 complex showed an unusual spike in RMSD at 125 ns, remaining unstable through the end of the simulation. Meanwhile, the protein backbone RMSD of the PDE4B-72 complex remained unstable until around 100 ns, when it gradually began to stabilize.

Root mean square deviation (RMSD) analysis of the backbone (right) and ligand (left) atoms of PDE4B complexes.
Regarding ligand structure, the average RMSD values ranged from 0.1817 to 0.3627 nm. Among them, the ligand in the PDE4B-17 complex proved to be the most stable, with an average RMSD of just 0.1817 nm. In the PDE4B-16 complex, the ligand displayed a slight increase of approximately 0.1 nm at 20 ns but maintained stability afterward. The RMSD values for the ligands in the PDE4B-17, PDE4B-40, PDE4B-42, and PDE4B-44 complexes fluctuated around fixed points, showing no significant shifts. The ligand in the PDE4B-53 complex underwent considerable changes during the first 25 ns, yet its RMSD eventually stabilized for the remainder of the simulation. These observations highlight the resilience of the bound complexes, which retained their structural integrity despite internal thermal and dynamic fluctuations. In conclusion, the MD simulations strongly suggest that these protein–ligand complexes are stable, with the ligands firmly and consistently bound to the proteins.
The root mean square fluctuation (RMSF) measures the motion and flexibility of amino acid residues in a protein. Higher RMSF values indicate greater movement and flexibility, particularly at active sites, while extremely low RMSF values signify a stable and rigid active site. The RMSF profiles of the PDE4B complexes with the studied compounds, shown in Figure 4, highlight these dynamics. In the regions of amino acids 280-300 and 430-450, the PDE4B complexes with compounds 16, 17, 40, 42, 53, 70, and 72 exhibit lower RMSF values compared with the apo-protein structure, indicating stable binding in these areas. Conversely, the PDE4B-44 complex shows increased flexibility at both regions, and the PDE4B-50 and PDE4B-68 complexes exhibit significant flexibility at the 430-450 region. Fluctuations in other regions align with those of the apo-PDE4B protein. Notably, the regions 280-300 and 430-450 contain key residues involved in binding interactions, as reported in the “Molecular docking” section, demonstrating the significant potential for effective PDE4B inhibition by the compounds.

Root mean square fluctuation (RMSF) analysis of the backbone atoms of PDE4B and its complexes.
MM–GBSA binding free energy calculation
The MM–PBSA analysis was conducted to evaluate the binding affinities of the studied compounds and the reference inhibitor to the target protein PDE4B. The nonbonding energy, the more negative, indicates stronger ligand–protein interactions. The energies corresponding to the ligand–target complexes are presented in Table 2. The results indicate that the PDE4B complexes with the studied compounds have binding free energies ranging from −79.54 to −45.09 kcal/mol. It can be observed that compared with the reference inhibitor 1S1, all the compounds exhibit stronger binding free energies. This suggests that the most potent plant compounds show stronger binding affinities and increased stability with PDE4B.
The binding free energy in kcal.mol−1 for the studied systems, determined via MM–GBSA calculations.

Toxicity profiles
Toxicity prediction is a crucial step in drug development to protect human health by ensuring that new compounds are not harmful. This process saves time and costs, increases research efficiency, and minimizes risks for subsequent experimental studies. It also enhances research ethics by reducing the use of animals in testing.30,31 Thus, the ProTox 3.0 online tool was utilized in the current study to predict the toxicity of potential compounds through parameters such as LD50, hepatotoxicity, neurotoxicity, nephrotoxicity, respiratory toxicity, and cardiotoxicity. 24 The results, recorded in Table 3, show that the predicted compounds have high LD50 values (⩾5000 mg/kg) and low toxicity levels, indicating safety for oral administration. Specifically, compounds 16 and 17 were categorized with a toxicity level of 6, which is considered safe, while compounds 40, 42, 44, 50, 53, 68, 70, and 72 were classified at toxicity level 5. In addition, organ-specific toxicity analyses were conducted, revealing that hepatotoxicity and neurotoxicity of the compounds were predicted to be inactive. However, these compounds exhibited respiratory toxicity, suggesting they should not be used via inhalation. Compounds 16 and 17 were found to be active for cardiotoxicity but inactive for nephrotoxicity. Conversely, compounds 40, 42, 50, 53, 68, 70, and 72 showed the opposite predictions. Compound 44 was noted to be active for both cardiotoxicity and nephrotoxicity. These findings provide a foundation for the further experimental evaluation of the surveyed compounds in future studies.
The oral toxicity prediction of the top-hit compounds.

Conclusion
In this study, a virtual screening method involving molecular docking and MD was conducted to identify novel PDE4B inhibitors and provide insights into the underlying mechanisms of action of the identified potential compounds. As a result, the top 10 potential compounds, including 2α-O-trans-p-coumaroyl-3β,19α-dihydroxy-urs-12-en-28-oic acid, 2α-O-cis-p-coumaroyl-3β,19α-dihydroxy-urs- 12-en-28-oic acid, cyanidin-3-O-rutinoside, delphinidin-3-O-rutinoside, peonidin-3-O-rutinoside, rutin, isorhamnetin-3-O-rutinoside, kaempferol 3-O-α-L- rhamnopyranosyl-(1→6)-β-D- glucopyranoside-7-O-β-D-glucopyranoside, Kaempferol 3-O-α-L- rhamnopyranosyl- (1→6) [α-L- rhamnopyranosyl- (1→2)]-β-D- glucopyranoside, quercetin 3-O-α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside, demonstrated strong binding affinities with PDE4B (ΔG ⩽10 kcal/mol). Furthermore, MD simulations were performed to confirm the binding affinities and stability of these compounds in the active site of PDE4B. The binding free energies via MM–GBSA calculations showed that the surveyed compounds had stronger binding free energies than the reference inhibitor. In addition, regarding toxicity, the predicted compounds were all indicated to have safe toxicity levels; however, there were some considerations for use in subsequent stages. Overall, these results are significant for researchers in developing novel PDE4B receptor inhibitors for treating chronic inflammation.
Supplemental Material
sj-docx-1-chl-10.1177_17475198241305879 – Supplemental material for In silico molecular docking and molecular dynamics of Prinsepia utilis phytochemicals as potential inhibitors of phosphodiesterase 4B
Supplemental material, sj-docx-1-chl-10.1177_17475198241305879 for In silico molecular docking and molecular dynamics of Prinsepia utilis phytochemicals as potential inhibitors of phosphodiesterase 4B by Cao Hoang Minh Chau, Ngu Thi Tra Giang, Nguyen Thi Thuy Tram, Le Thi My Chau, Nguyen Xuan Ha and Phan Thi Thuy in Journal of Chemical Research
Footnotes
Author contributions
P.T.T. and N.X.H. supervised the project. P.T.T., N.X.H., and C.H.M.C. designed the project. C.H.M.C., N.T.T.G., and L.T.M.C. calculated and analyzed the data. P.T.T. and N.X.H. wrote the paper. All authors discussed and analyzed the results of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval is not applicable for the article.
Statement of human and animal rights
This article does not contain any studies with human or animal subjects.
Statement of informed consent
There are no human subjects in this article, and informed consent is not applicable.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.