
Abstract
Molecular docking and molecular dynamics (MD) simulations were employed to evaluate Baeckea frutescens–derived compounds as potential MAPK3 inhibitors. The docking protocol was validated with an RMSD of 0.620373 Å, confirming its reliability. Docking results revealed binding affinities ranging from –5.279 to –11.77 kcal/mol, with 12 compounds exhibiting stronger affinities than –10 kcal/mol including frutescone B (42), baeckein A (69), baeckein B (70), baeckein C (71), baeckein D (72), baeckein E (73), baeckein I (77), 6-C-methylquercetin 4′-O-β-D-glucopyranoside (87), myricetin 3-O-(5′′-O-galloyl)-α-L-arabinofuranoside (90), quercetin-3-O-α-L-rhamnoside (93), 7-O-(4′,6′-digalloyl)-β-D-glucopyranosyl-5-hydroxy-2-methylchromone (108), and 6-β-C-(2′-galloylglucopyranosyl)-5,7-dihydroxy2-isopropylchromone (117). Compounds 69, 70, and 72 displayed the highest affinities, engaging in hydrogen bonding, π-sigma, and π-alkyl interactions, stabilizing the MAPK3 complex. MD simulations further confirmed their stability, with RMSD values lower than the reference system. MMGBSA (Molecular Mechanics Generalized Born Surface Area) analysis identified compound 87 as the strongest binder (ΔGbind = −60.43 kcal/mol), followed by compounds 72 and 73. ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) predictions suggested compound 42 as the most promising candidate for oral administration, while compounds 71–73 showed poor intestinal absorption but limited CNS penetration. Most compounds exhibited no genotoxicity or hepatotoxicity, though hERG II inhibition indicated potential cardiac risks. These findings provide insights into the potential of B. frutescens–derived compounds as MAPK3 inhibitors and guide future optimization for enhanced stability and pharmacokinetic properties.

Introduction
Baeckea frutescens L., a shrub belonging to the genus Baeckea in the Myrtaceae family, is primarily distributed across Vietnam, China, Thailand, India, Burma, the Malay Peninsula, Sumatra, Borneo, the Philippines, and New Guinea.1,2 This plant, known as “Jungrahab” in Indonesia and “Chổi sể” in Vietnam, has been traditionally used in folk medicine for treating malaria, fever, influenza, headaches, dysentery, and abdominal pain.3 –5 Its leaves, in particular, are commonly utilized for alleviating headaches, rheumatism, and fever. Recent studies have unveiled a wide range of biological activities exhibited by B. frutescens, including cytotoxic, anti-inflammatory, mosquito larvicidal, antimicrobial, antioxidant, anti-rheumatoid arthritis, and skin-protective effects.6 –11 To date, approximately 128 phytochemicals have been isolated and identified from various parts of B. frutescens. These include phloroglucinols (14 compounds), phloroglucinol-based meroterpenoids (46 compounds), sesquiterpenoids (4 compounds), triterpenoids (5 compounds), flavonoids (34 compounds), chromones (19 compounds), and other compounds (7 compounds). 12 Several compounds derived from this plant have demonstrated potent cytotoxic effects. Notably, frutescones A-G inhibited the proliferation of Caco-2 and A549 cancer cells, with IC50 values ranging from 7.96 to 41.33 µM. 13 Baeckenone F moderately suppressed the growth of A549, PSN-1, and MDA-MB-231 cancer cells, exhibiting IC50 values of 33.3–39.3 µM. 14 Baeckenones J-K demonstrated strong inhibitory effects against A549 and PSN-1 cancer cells, with IC50 values between 11.8 and 19.2 µM. 4
Despite its well-documented pharmacological properties, the potential of B. frutescens phytochemicals against MAPK3 has not been explored. This represents a critical gap in research, as MAPK3 is a key regulator of cancer cell proliferation, survival, and metastasis. 15 Given that several compounds from B. frutescens have exhibited cytotoxic effects against various cancer cell lines, it is logical to investigate their interaction with MAPK3, a well-established target in oncology. Traditional medicinal uses of B. frutescens for conditions linked to inflammation and infection further support its relevance in targeting MAPK3, as this kinase is also implicated in inflammatory responses and oncogenic signaling. Bridging this gap will not only contribute to the field of natural product research but also provide insights into novel therapeutic strategies for cancer treatment.
Mitogen-activated protein kinase 3 (MAPK3), also known as extracellular signal-regulated kinase 1 (ERK1), is a key component of the ERK/MAPK signaling pathway, playing a central role in transducing extracellular signals into intracellular responses.16,17 MAPK3 is involved in regulating critical biological processes such as apoptosis, cell proliferation, and migration. 18 Overexpression and/or hyperactivation of MAPK3 has been closely linked to metastasis and drug resistance in various malignancies. 19 In addition, MAPK3 is part of a highly conserved family of serine/threonine protein kinases that are crucial for cell differentiation, proliferation, and survival. The MAPK signaling cascade, where MAPK3 serves as a pivotal component, interacts with several other oncogenic pathways, such as JAK/STAT and PI3K, thereby promoting tumor growth and therapy resistance.20 –24 Numerous studies have identified MAPK3 as an oncogenic driver in several malignancies, including liver, lung, thyroid, and gastric cancers. 19 Consequently, MAPK3 has emerged as a critical target for cancer therapy, with Type I and Type II kinase inhibitors targeting its ATP-binding site and allosteric pockets, paving the way for novel anticancer treatment strategies. 25
Recent studies have explored the potential of natural products as inhibitors of MAPK3 and related kinases. For example, computational screening of natural anthraquinones, emodin-8-glucoside, aloe-emodin 8-glucoside, pulmatin, rhodoptilometrin, and hypericin has identified several promising inhibitors of MAPK3. 26 In silico screening of cinnamic acid derivatives through molecular docking and molecular dynamics (MD) simulations has demonstrated that compounds such as cynarin, chlorogenic acid, rosmarinic acid, caffeic acid 3-glucoside, and cinnamyl caffeate possess strong binding affinities toward the MAPK3 active site, with binding free energies (ΔGbinding) consistently lower than −10 kcal/mol. 25 However, to our knowledge, this is the first comprehensive in silico screening of B. frutescens–derived compounds against MAPK3.
Given its diverse bioactive compounds and promising pharmacological potential, B. frutescens represents a valuable resource for drug discovery, particularly in cancer research targeting MAPK3. Therefore, our study focuses on applying computational approaches, including molecular docking, MD, and ADMET predictions, to aid in the discovery of MAPK3 inhibitors for the treatment of cancer diseases.
Materials and methods
Molecular docking
The compound information was gathered from previously published literature, and their chemical structures were drawn using Marvin Sketch. 12 These structures were then energy-optimized using Avogadro with the MMFF94s force field to achieve a stable conformation. 13 The human ERK1 protein in complex with SCH772984 (PDB ID: 4QTB) was retrieved from the RCSB Protein Data Bank (https://www.rcsb.org/structure/4QTB) with a resolution of 1.4 Å. 27 The protein structure was refined by adding polar hydrogen atoms and removing co-crystallized molecules using PyMOL to prepare it for molecular docking studies. The ligand and protein structures were then formatted for docking by converting them to PDBQT format using AutoDockTools software. The docking grid box was centered at x = 35 Å, y = 56.7 Å, z = 50.4 Å, with a grid size of 24 × 24 × 24 and a spacing of 1 Å, ensuring optimal coverage of the binding site. For molecular docking, AutoDock Vina v1.2.3 was employed with an exhaustiveness value of 400 to enhance the accuracy of the binding predictions.28,29 Compounds with docking scores lower than −10 kcal/mol were considered significant hits, serving as a cutoff criterion for selecting promising inhibitors. After docking, nine binding poses were generated for each compound. The top-ranked pose based on the Vina score was initially considered; however, the final selection of the best binding conformation was carried out by carefully analyzing the docking score and the binding orientation and key molecular interactions with the target protein. Poses demonstrating significant hydrogen bonding, hydrophobic interactions, and favorable positioning within the active site were prioritized, even if their docking score was slightly lower than the highest-ranked pose. This approach ensured that the selected conformations were not solely based on scoring but also on their potential to establish meaningful and stable interactions with the protein target. The final selected conformations were analyzed and visually represented using Discovery Studio Visualizer software to provide deeper insights into their interactions with the target protein.
Molecular dynamics
MD simulation is a cutting-edge computational approach that captures the intricate motions and interactions of biomolecular systems at the atomic scale, providing deep insights into their structural dynamics and behavior over time. The most promising selected compounds undergo extensive 100 ns MD simulations using the GROMACS v2023.3 program.30,31 The AMBER99SB-ILDN force field is applied to prepare the protein topology, 32 ensuring accuracy in molecular interactions. For ligand topology preparation, the docked ligand structures are optimized using density functional theory (DFT) calculations with the B3LYP/6-31g(d, p) basis set in the Gaussian 09 program. The obtained energy-minimized structures are then used to generate ligand topology via AmberTools v23 and ACPYPE. 33 The MD simulations were conducted for the top 12 selected compounds (42, 69, 70, 71, 72, 73, 77, 80, 87, 90, 93, 108, 117) in complex with MAPK3 to evaluate their dynamic stability. The protein-ligand complexes are placed in a triclinic simulation box with a 1 nm buffer in all directions. The system is solvated using explicit TIP3P water molecules, and counterions (Na+ or Cl−) are added to neutralize the overall charge, ensuring a physiologically relevant environment. Energy minimization is conducted using the steepest descent algorithm to remove steric clashes and stabilize the system. The equilibrations proceed through sequential NVT and NPT ensembles, each run for 100 ps at 310 K and 1 bar, allowing the system to adapt to the simulation conditions. Finally, the MD simulation is carried out, with coordinates saved every 10 ps for analysis. Each protein-ligand complex was simulated once to assess their binding stability over time. Following the production MD simulations, the resulting trajectories are analyzed in detail to assess structural stability, flexibility, and intermolecular interactions. The results are visualized using XMGRACE software, providing insightful graphical representations of MD over time. To further evaluate the binding free energy of the complexes, MM/GBSA calculations were performed using the gmx_MMPBSA program. Binding free energy (ΔGbind) values were obtained as an average over extracted frames from the 100 ns traject of the simulation. The analysis considered both polar and non-polar contributions but did not explicitly include entropic effects. This approach provides a quantitative estimation of the binding strength between the selected ligands and MAPK3, offering additional insights into their potential as inhibitors.
ADMET prediction
To predict the ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of potential compounds, this study employs pkCSM, a powerful and widely used online tool for pharmacokinetic predictions. 34 The process begins with preparing the molecular structures in SMILES format using OpenBabel software, ensuring accuracy through double-checking before submission. 35 These structures are then uploaded to the pkCSM platform, which automatically analyzes and predicts key pharmacokinetic properties, including absorption (water solubility, Caco2 permeability, human intestinal absorption, skin permeability, P-glycoprotein substrate, P-glycoprotein I inhibitor, P-glycoprotein II inhibitor), distribution (VDss, fraction unbound, BBB permeability, CNS permeability), metabolism (CYP2D6, CYP3A4 substrates, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 inhibitors), excretion (Total Clearance, Renal OCT2 substrate), and toxicity (AMES toxicity, maximum tolerated dose (human), hERG I inhibitor, hERG II inhibitor, oral rat acute toxicity (LD50), oral rat chronic toxicity (LOAEL), hepatotoxicity, skin sensitisation, Tetrahymena pyriformis toxicity, minnow toxicity). Summarizing these parameters in the methodology provides a clearer overview of the ADMET evaluation process. The predicted data provide valuable insights into the drug-like behavior of each compound, helping to assess their feasibility as potential drug candidates. By evaluating these properties, the study aims to identify promising molecules with favorable pharmacokinetics, paving the way for further optimization and development in drug discovery.
Results and discussion
Molecular docking results
Molecular docking is an essential technique in pharmacological research and drug discovery, particularly for screening natural compounds as potential inhibitors. This computational method predicts how ligands interact with a target protein, providing crucial insights into their binding affinity and mode of action.36,37 In this study, molecular docking was utilized to evaluate compounds derived from B. frutescens against MAPK3, a key kinase involved in cancer development. Before conducting molecular docking, the protocol was validated by re-docking the co-crystallized ligand SCH772984 into its binding site within MAPK3 (PDB: 4QTB). The root-mean-square deviation (RMSD) between the initial coordinates of SCH772984 and its redocked coordinates was calculated to be 0.620373 Å (Figure 1). Since an RMSD below 2 Å indicates a reliable docking method, this result confirms the validity of our docking protocol.38,39 This step is crucial because it ensures that the docking approach can accurately reproduce the experimental binding mode of a known ligand, thereby providing confidence in the subsequent docking results.
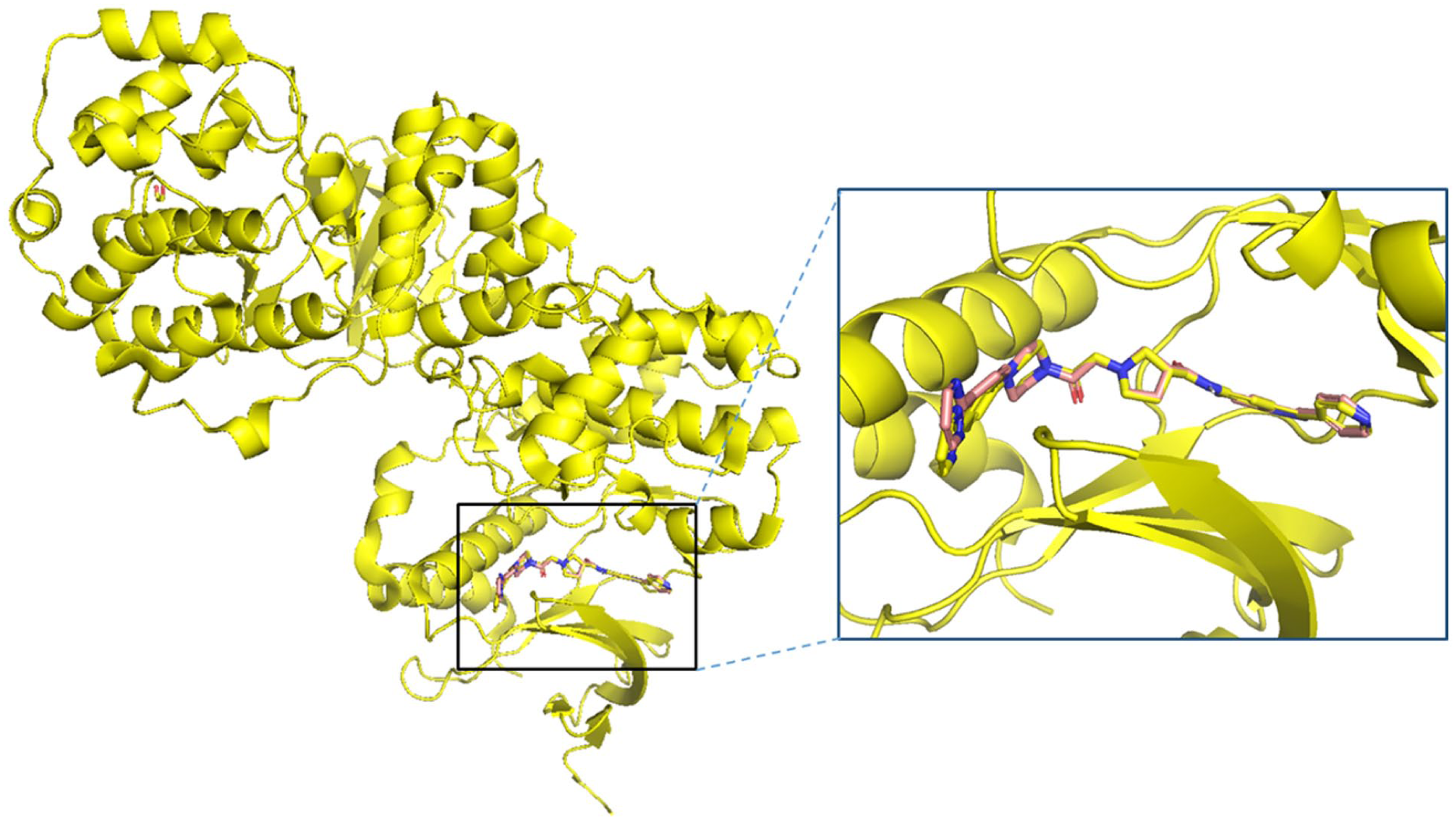
Superimposition of the co-crystallized ligand (yellow) and the redocked ligand (wheat) complexed with the PDE4B protein with an RMSD value of 0.620373 Å.
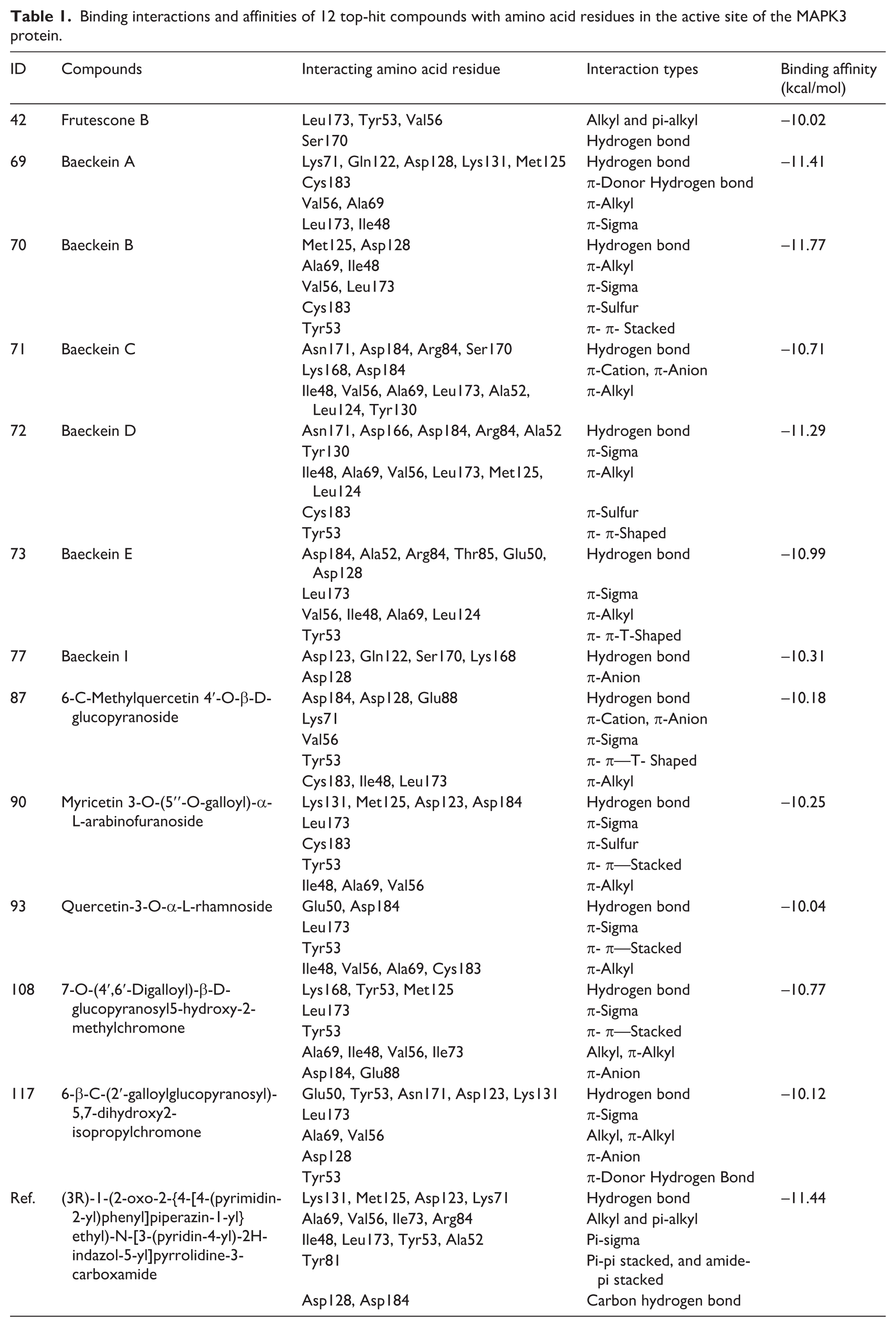
Following validation, a database of B. frutescens–derived compounds was docked into the ATP-binding cleft of the MAPK3 protein. The results showed binding affinities ranging from –5.279 to –11.77 kcal/mol, with 12 compounds exhibiting affinities stronger than –10 kcal/mol (Table 1), making them promising candidates for further analysis. These selected compounds displayed diverse molecular interactions, including hydrogen bonding, π-alkyl, π-sigma, π-π stacking, and π-sulfur interactions, all of which contributed to strong binding potential with the MAPK3 protein (Figure 2, Table 1). Compared to the reference compound, several tested compounds demonstrated notable binding characteristics. The reference compound formed strong hydrogen bonds with Lys131, Met125, Asp123, and Lys71, along with alkyl and π-alkyl interactions with Ala69, Val56, and Ile73, stabilizing the ligand-protein complex. In addition, π-sigma interactions with Ile48, Leu173, and Tyr53, as well as π-π stacking with Tyr81, further reinforced the molecular stability. Among the tested compounds, baeckein A (compound 69), baeckein B (compound 70), and baeckein D (compound 72) exhibited the strongest binding affinities of –11.41 kcal/mol, –11.77 kcal/mol, and –11.29 kcal/mol, respectively, comparable to or even stronger than the reference compound (–11.44 kcal/mol). All three compounds are flavonoids (Supplemental Figure S1), consistent with previous reports that flavonoids such as kaempferol 3-rutinoside-4′-glucoside, kaempferol 3-rutinoside-7-sophoroside, rutin, and vicenin-2 can strongly bind to the MAPK3 catalytic site and act as potent inhibitors. 40
Binding interactions and affinities of 12 top-hit compounds with amino acid residues in the active site of the MAPK3 protein.

Two-dimensional interactions between 12 top-hit compounds and amino acid residues in the active site of MAPK3 protein (PDB ID 4QTB).
Among them, compound 70 displayed the highest binding affinity (−11.77 kcal/mol), surpassing the reference, which underscores its potential as a potent MAPK3 inhibitor. This flavonoid engaged hydrogen bonding and π-sigma interactions with critical amino acid residues such as Met125 and Leu173, respectively while π-alkyl interactions with Ile48 and Ala69 contributed to enhanced stability. Particularly, compound 70 also exhibited π-sulfur and π-π stacking interactions, further strengthening the ligand-protein complex in a manner similar to the reference compound. In addition, compound 73 also showed a promising binding affinity, further reinforcing the potential of these selected hits. Other studied compounds, including 42, 71, 73, 77, 87, 90, 93, 108, and 117, showed weaker affinities than the reference compound but still demonstrated significant interactions with key amino acid residues within the ATP-binding region and the activation loop such as Leu173, Tyr53, Val56, Ala69, Ile48, Asp128, Lys131, Met125, and Asp184. Although their binding affinities were lower, their interactions suggest notable potential as MAPK3 inhibitors.
Importantly, when compared to known MAPK3 inhibitors from the literature, the predicted binding affinities of these phytochemicals remain highly competitive. For instance, the reference compound SCH772984 docked at –11.44 kcal/mol, which aligns with reported IC50 values in the low nanomolar range. 27 In contrast, other known MAPK3 inhibitors such as purvalanol and ulixertinib exhibited binding affinities of –8.53 kcal/mol and –9.12 kcal/mol, respectively.26,40 Therefore, the top compounds identified here, particularly compound 70 (–11.77 kcal/mol), demonstrate comparable or superior docking performance. This suggests that these B. frutescens–derived compounds, especially the flavonoids, possess real-world binding potential analogous to established MAPK3 inhibitors.
Overall, the molecular docking results indicate that several B. frutescens–derived compounds exhibit strong binding potential with MAPK3, resembling the interaction patterns of the reference compound. These findings highlight their potential for further development as MAPK3 inhibitors in cancer treatment.
Molecular dynamics
To further investigate the binding stability and dynamic behavior of these promising compounds, MD simulations were performed on the selected protein-ligand complexes. Based on the results of a 100 ns MD simulation, the stability of protein-ligand complexes can be assessed by analyzing RMSD (Root Mean Square Deviation) values, as illustrated in Figure 3. 41 Overall, most of the protein backbone in the studied systems demonstrate good stability compared to the reference system 4QTB-ref (average RMSD: 0.1815 nm) and the apo-protein, except for a few cases exhibiting significant fluctuations. Specifically, the 4QTB-90 and 4QTB-108 systems’ protein backbone shows considerable RMSD variations throughout the simulation. In the case of 4QTB-90, the RMSD value increases sharply after 50 ns, rising from approximately 0.2 nm to 0.35 nm, indicating a significant structural change in the protein backbone. Meanwhile, the 4QTB-108 system exhibits an RMSD increase of around 85 ns, suggesting potential structurally large fluctuations within the protein-ligand complex. Although some systems display strong fluctuations, most others maintain good stability, with average RMSD values ranging from 0.1577 nm to 0.2372 nm. The low RMSD values (below 0.2 nm for most systems) throughout the 100 ns trajectory indicate that the ligands remain stably bound and do not induce significant structural perturbations in the protein. This stability suggests that the docking poses were realistic and that the MAPK3 binding pocket can accommodate these ligands without undergoing major conformational rearrangements. Notably, systems such as 4QTB-71, 4QTB-77 (both at 0.1577 nm), 4QTB-70 (0.1733 nm), and 4QTB-72 (0.1730 nm) exhibit lower average RMSD values than the reference system and are similar to apo-protein, indicating high structural stability of the protein backbone. In addition, RMSD values provide insights into the stability of ligands within the 4QTB complexes. RMSD reflects the positional changes of ligands relative to their initial structures throughout the simulation, offering valuable information about complex stability. As observed in Figure 3, ligands 69, 70, 72, 73, 87, and 117 exhibit good stability within the 4QTB complex. This is evident as their average RMSD values remain lower than or comparable to that of the reference system 4QTB-ref (0.0996 nm). In particular, ligand 70 has the lowest average RMSD (0.032 nm), followed by ligand 69 (0.0448 nm) and ligand 87 (0.0730 nm), indicating the highest stability among the studied ligands. Conversely, some ligands show significant RMSD fluctuations, reflecting structural changes during the simulation. For instance, the 4QTB-42 system experiences an RMSD increase after 30 ns but stabilizes afterward, with an average RMSD of 0.1295 nm. The 4QTB-71 system exhibits substantial variations, with fluctuations ranging from 0.1 nm to 0.3 nm and an average value of 0.1892 nm, indicating considerable ligand movement within the complex. Notably, the 4QTB-77 system experiences an RMSD increase after 40 ns but stabilizes from 60 ns onward, with an average RMSD of 0.1641 nm. The 4QTB-90 system shows a sharp RMSD increase after 95 ns, while the 4QTB-93 system undergoes significant fluctuations between 55–75 ns before stabilizing. Particularly, the 4QTB-108 system exhibits the highest average RMSD (0.2733 nm); although it maintains stability throughout the simulation, its initial fluctuation exceeding 0.2 nm suggests considerable structural adjustments before reaching equilibrium.
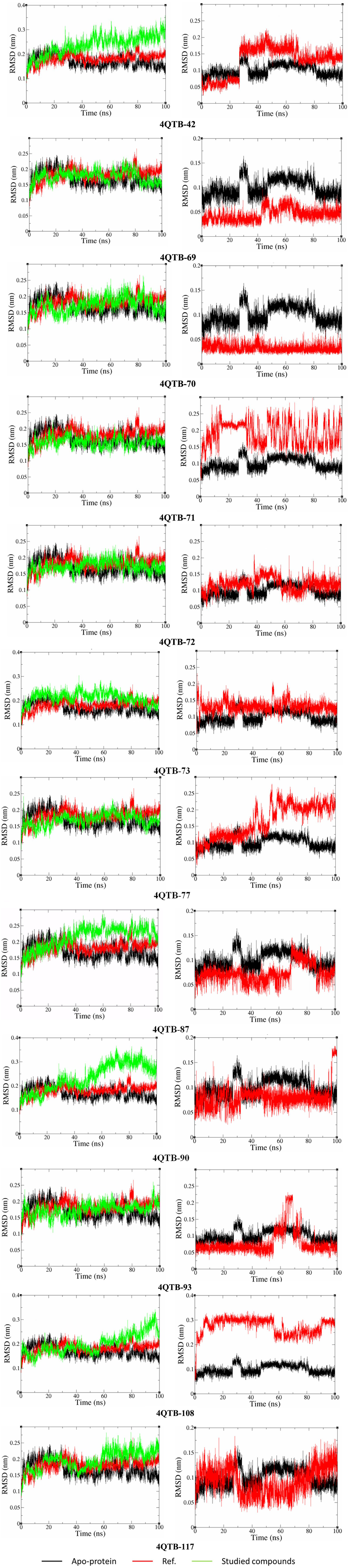
RMSD analysis of the protein backbone (left) and ligand (right) in MAPK3 complexes.
In summary, compared to the reference system, ligands 69, 70, 72, 73, 87, and 117 exhibit strong stability, whereas ligands 42, 71, 77, 90, 93, and 108 show significant RMSD fluctuations. These findings provide crucial insights into ligand dynamics within the protein complex, guiding further studies on the stability of the complexes and the interaction potential of ligands with the MAPK3 protein.
Furthermore, preliminary analysis of root mean square fluctuation (RMSF) values revealed no major deviations in critical functional regions such as the ATP-binding cleft and the activation segment (Supplemental Figure S2). In particular, the amino acid residues Leu173, Tyr53, Val56, Ala69, Ile48, Asp128, Lys131, Met125, and Asp184 remained structurally stable when bound to top-performing ligands, further supporting the integrity of the complexes. Although not all dynamic changes led to destabilization, these observations suggest differential ligand-induced flexibility.
Binding free energy calculation using the MMGBSA method
The MMGBSA method was employed to calculate the binding free energy (ΔGtotal) between the selected compounds and the MAPK3 protein, incorporating various energy components such as Van der Waals interactions (VDWAALS), electrostatic interactions in vacuum (EEL), polar solvation energy (EGB), surface energy (ESURF), gas-phase free energy (ΔGgas), and solvation free energy (ΔGsolv).42,43 Among the analyzed compounds, VDWAALS energy ranged from –39.26 to –76.65 kcal/mol, indicating varying strengths of Van der Waals interactions. Notably, compound 73 exhibited the lowest VDWAALS value (–76.65 kcal/mol), suggesting the strongest surface interaction with MAPK3. Meanwhile, electrostatic energy (EEL) varied from –0.40 to –85.26 kcal/mol, with compound 72 displaying the lowest value (-85.26 kcal/mol), underscoring the significant role of electrostatic interactions in binding. The polar solvation energy (EGB) had positive values ranging from 19.49 to 100.32 kcal/mol, reflecting the instability of compounds in solvent. Compound 72 exhibited the highest EGB value (100.32 kcal/mol), indicating that strong solvation effects could weaken ligand-protein binding in aqueous environments. In addition, surface energy (ESURF) contributed modestly to binding, ranging from –5.16 to –10.37 kcal/mol. Overall, the binding free energy (ΔGtotal) of the compounds ranged from –27.15 to –60.43 kcal/mol (Table 2). Compound 87 demonstrated the strongest binding free energy with ΔGtotal = –60.43 kcal/mol, while compound 42 exhibited the binding free energy at –27.15 kcal/mol. Generally, a more negative corresponds to stronger binding. Van der Waals and electrostatic interactions were the two primary factors driving complex stability. However, excessive solvation energy, particularly EGB, could reduce binding affinity. Although compound 73 had the strongest Van der Waals interactions, solvation effects prevented it from being the most stable binder. Therefore, optimizing ligand binding necessitates balancing Van der Waals forces, electrostatic interactions, and solvation effects. Beyond these factors, gas-phase free energy (ΔGgas) and solvation free energy (ΔGsolv) also play crucial roles in determining ligand-protein complex stability. ΔGgas, which combines Van der Waals and electrostatic interactions in vacuum, reflects the intrinsic binding strength between the ligand and protein. Its values ranged from –41.48 to –149.78 kcal/mol, with compound 72 exhibiting the lowest value (–149.78 kcal/mol), indicating strong binding driven by both Van der Waals and electrostatic forces. Conversely, solvation free energy (ΔGsolv) counteracts by representing the influence of the solvent environment on binding stability. had positive values ranging from 14.33 to 90.48 kcal/mol, with compound 72 displaying the highest value (90.48 kcal/mol), suggesting that high solvation energy could negatively impact ligand stability in aqueous conditions. A key observation is the distinction between compounds with similar values but different binding mechanisms. For instance, although compound 87 had the lowest (–60.43 kcal/mol), compound 73 exhibited stronger Van der Waals interactions yet was not the most stable ligand. Furthermore, compounds 72, 73, and 87 displayed better binding free energy than the reference compound (–58.63 kcal/mol). Interestingly, although compound 87 had a slightly less favorable docking score than compounds 69 and 70, MM/GBSA rescoring identified it as the most promising binder. This discrepancy highlights the value of dynamic simulations in capturing stabilizing interactions beyond static docking. During the 100 ns MD simulation, compound 87 maintained persistent hydrogen bonds with critical residues such as Glu88, Asp184, and Asp128, and additionally formed a stable hydrogen bond with Met125 an interaction not observed in the initial docking pose (Supplemental Figure S3). These hydrogen bonds likely contributed to the enhanced binding free energy. Moreover, key hydrophobic and polar interactions with Lys71, Val56, Leu173, Ile48, Cys183, and Tyr53 were preserved throughout the simulation, further supporting the stability of the complex (Supplemental Figure S3). This confirms that binding stability cannot be assessed based on a single factor; instead, a comprehensive evaluation of Van der Waals forces, electrostatic interactions, and solvation effects is necessary. In conclusion, compound 87 emerged as the most promising ligand due to its strongest binding free energy, indicating superior affinity toward MAPK3. Compound 72 also demonstrated strong binding potential, particularly in terms of electrostatic interactions, though its high solvation energy could limit stability in aqueous environments. Meanwhile, compound 73 exhibited the most robust Van der Waals interactions, yet solvation effects prevented it from being the strongest binder overall. These findings highlight the importance of balancing intermolecular forces and solvation effects when designing optimized MAPK3 inhibitors. Future research should focus on modifying these promising compounds to enhance binding stability while minimizing unfavorable solvation energy, thereby improving their potential as therapeutic agents.
The binding free energy (in kcal/mol) for the systems studied, calculated using MM–GBSA methods.

ADMET profiles
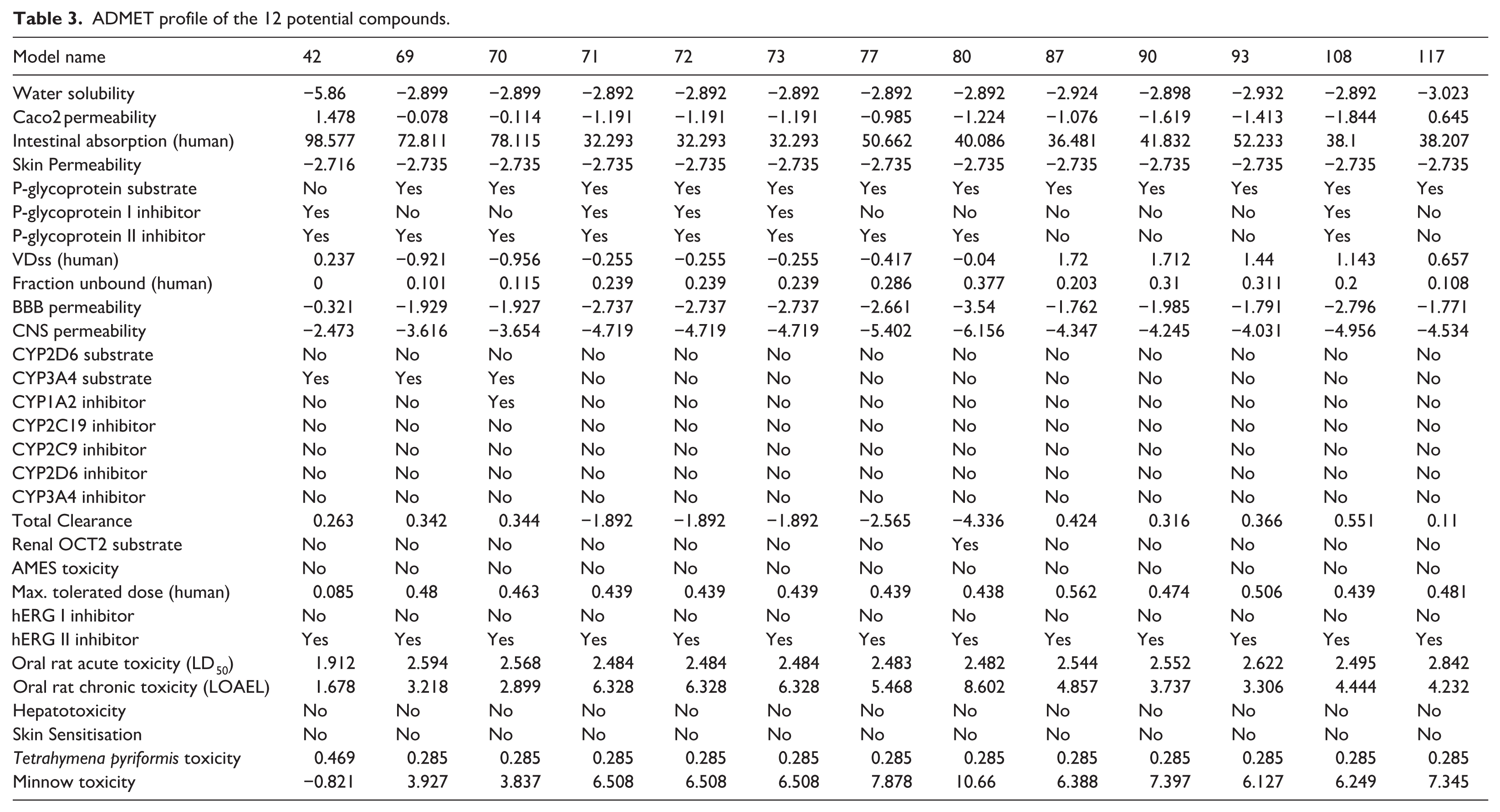
The potential compounds continue to be evaluated for their ADMET pharmacokinetic properties using pkCSM, with results presented in Table 3. 34 The solubility of these compounds in water ranges from –5.86 to –2.892. Regarding intestinal permeability (Caco-2 permeability), only compound 42 has a positive value (1.478), indicating good permeability, whereas most other compounds have negative values, especially compounds 71, 72, and 73, with low values (–1.191), suggesting poor intestinal absorption. This observation aligns with human intestinal absorption data, where compound 42 shows the highest absorption rate (98.577%), while compounds 71–73 exhibit significantly lower absorption (32.293%). This could be problematic for oral bioavailability, as poor absorption may limit their effectiveness when administered orally. Formulation strategies or structural modifications might be necessary to improve their permeability, or these compounds could be considered more suitable for intravenous administration, where intestinal absorption is less of a concern. In terms of skin permeability, all compounds have very low values (–2.735), indicating poor absorption through the skin, which reduces the risk of toxicity from dermal exposure. Similarly, blood-brain barrier (BBB) permeability is also quite low, particularly for compounds 71–73, with values ranging from –2.737 to –3.54, suggesting limited BBB penetration and reduced suitability for central nervous system (CNS)-related applications. CNS permeability is also very low (below –4.0), further confirming this characteristic. This lack of BBB penetration could make these compounds unsuitable for CNS-targeted therapies unless their structures are modified to enhance BBB permeability. Regarding their impact on CYP metabolic enzymes, none of the compounds act as substrates for CYP2D6, suggesting they are not metabolized via this pathway. However, three compounds (42, 69, 70) are substrates of CYP3A4, indicating a potentially faster metabolism. In terms of CYP enzyme inhibition, only compound 70 inhibits CYP1A2, while none of the compounds inhibit CYP2C9, CYP2C19, CYP2D6, or CYP3A4, thereby reducing the risk of drug-drug interactions through hepatic enzyme inhibition. The total clearance of these compounds varies significantly, with some showing negative clearance values (e.g. compound 80 with –4.336), suggesting prolonged retention in the body, whereas compounds 42, 69, and 70 exhibit positive clearance values, indicating faster elimination. Compounds with negative clearance values may need to be carefully monitored for accumulation and potential toxicity over time. In terms of toxicity, most compounds do not exhibit genotoxicity (AMES toxicity), hepatotoxicity, or skin sensitization. However, the majority show a risk of hERG II inhibition, which may be associated with potential cardiac arrhythmias. This is a significant concern for safety, and further testing would be needed to assess the likelihood of these compounds causing cardiac issues in vivo. Acute toxicity in mice (LD50) ranges from 1.912 to 2.842, indicating moderate toxicity levels, whereas chronic toxicity (LOAEL) varies significantly among compounds, with compound 80 having the highest value (8.602), suggesting a higher dose is required to cause long-term adverse effects. These findings indicate that while most compounds appear to have manageable acute toxicity, long-term exposure to compounds like 80 could pose greater risks, warranting further investigation. Regarding environmental toxicity, some compounds exhibit significant ecological risks, particularly compound 80, which has a Minnow toxicity value of 10.66, indicating a strong potential impact on aquatic ecosystems. This highlights the need to consider environmental safety when advancing compounds like 80 in development. In conclusion, compound 42 stands out as a promising candidate for oral drug development due to its high intestinal absorption and favorable clearance rate. Conversely, compounds 71–73 show poor intestinal absorption and limited BBB permeability, making them more suitable for non-CNS applications. In terms of safety, most compounds do not exhibit genotoxicity or hepatotoxicity, but the risk of cardiac effects due to hERG II inhibition should be considered. Environmental toxicity is also an important factor to evaluate, especially for compounds with high Minnow toxicity values. These in silico ADMET results are preliminary and require further validation through experimental ADMET and toxicity studies.
ADMET profile of the 12 potential compounds.
Conclusion
The study provides valuable insights into the potential of B. frutescens–derived compounds as MAPK3 inhibitors. Molecular docking and MD simulation results revealed that several compounds demonstrate significant binding potential, with strong interactions with key amino acid residues of MAPK3. Compound 87, in particular, exhibited the most favorable binding free energy (−60.43 kcal/mol), making it the most promising candidate for further exploration. However, binding stability is influenced by a balance between Van der Waals interactions, electrostatic forces, and solvation effects, suggesting that careful optimization of these factors is essential for improving ligand stability. In terms of pharmacokinetic properties, compound 42 stands out due to its high intestinal absorption and favorable clearance rate, making it a strong candidate for oral drug development. On the other hand, compounds 71–73 showed poor intestinal absorption and limited blood-brain barrier penetration, making them better suited for non-CNS applications. Despite the promising binding potential of several compounds, the risk of cardiac toxicity (hERG II inhibition) and environmental toxicity should be considered during the drug development process. In conclusion, this study identified several promising MAPK3 inhibitors from B. frutescens, with lead candidates based on binding affinity and pharmacokinetic potential. By integrating molecular docking, MD simulations, and ADMET analysis, this research provides a comprehensive framework for selecting and optimizing drug-like candidates. These findings emphasize the importance of balancing potency, stability, and pharmacokinetics in early-stage drug discovery. Future research should focus on experimental validation, structural refinements to enhance efficacy and safety, and further exploration of their therapeutic applications, including synthesizing or isolating the top candidates for in vitro and in vivo testing to confirm their potential as MAPK3 inhibitors.
Supplemental Material
sj-docx-1-chl-10.1177_17475198261417807 – Supplemental material for In silico molecular docking and molecular dynamics simulation of Baeckea frutescens phytochemicals as mitogen-activated protein kinase 3 (MAPK3) potential inhibitors
Supplemental material, sj-docx-1-chl-10.1177_17475198261417807 for In silico molecular docking and molecular dynamics simulation of Baeckea frutescens phytochemicals as mitogen-activated protein kinase 3 (MAPK3) potential inhibitors by Hoang Thi Tue Trang, Ngu Thi Tra Giang, Nguyen Thi Thuy Tram, Nguyen Xuan Ha and Cao Hong Le in Journal of Chemical Research
Footnotes
Ethical considerations
This paper does not contain any studies with human or animal participants.
Consent to participate
This paper has no human participants, and informed consent is not required.
Consent for publication
The authors declare that they have no known competing financial interests or personal relationships that may affect the work reported in this paper.
Author contributions
All authors contributed to the article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Data will be made available on request from the corresponding author.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.