
Abstract
Molecular docking and molecular dynamics simulations were employed to screen plant-derived compounds from Aralia armata for potential inhibition of tumor necrosis factor-alpha (PDB ID: 2AZ5). Aralia armata has a history of use in traditional medicine and contains bioactive compounds with anti-inflammatory potential, such as triterpene glycosides, as reported in previous studies. The docking protocol was validated using root mean square deviation, yielding a value of 1.827 Å, confirming its reliability. The binding affinity analysis revealed that compounds calenduloside E (
Keywords
Introduction
Tumor necrosis factor-alpha (TNF-α) is a key pro-inflammatory cytokine that plays a vital role in various biological processes, including cell proliferation, differentiation, apoptosis, immune regulation, and inflammation induction. 1 Dysregulation of TNF-α has been implicated in the pathogenesis of numerous inflammatory and autoimmune diseases, such as rheumatoid arthritis (RA), psoriasis, and ankylosing spondylitis.2,3 Due to its pivotal role in inflammation, TNF-α has become a major therapeutic target in drug development, leading to the introduction of TNF-α inhibitors such as infliximab, etanercept, adalimumab, and golimumab.4,5 These biologics have significantly transformed the treatment of autoimmune diseases. However, biologic TNF-α inhibitors are often associated with high production costs and require parenteral administration, which can limit patient accessibility and compliance.6,7 In contrast, small-molecule TNF-α inhibitors are being actively explored as a cost-effective alternative, offering the advantages of oral bioavailability, lower manufacturing costs, and ease of administration. TNF-α is a 17-kDa monomeric protein that forms a biologically active non-covalent homotrimer. Its activity is mediated through interactions with two receptors: TNFR1 and TNFR2. TNFR1 primarily triggers inflammatory signaling pathways, such as the mitogen-activated protein kinase (MAPK) cascade, extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK), which contribute to the progression of autoimmune diseases, including RA and hepatitis.8–10 In addition, TNFR1 signaling activates the nuclear factor-kappa B (NF-κB) pathway, leading to the transcription of pro-inflammatory cytokines and survival proteins. 11 In contrast, TNFR2, which is mainly expressed on immune and endothelial cells, plays a crucial role in immune modulation, tissue regeneration, and vascular homeostasis. 12 Beyond its involvement in autoimmune diseases, TNF-α is also a critical mediator of the inflammatory response during infections. 13 It is rapidly produced by macrophages, dendritic cells, and T cells in response to pathogen-associated molecular patterns (PAMPs) recognized by Toll-like receptors (TLRs). 14 TNF-α enhances immune defense by increasing vascular permeability, promoting leukocyte recruitment, and stimulating the production of other pro-inflammatory mediators such as interleukin-1 (IL-1) and interleukin-6 (IL-6). 15 However, excessive TNF-α production can lead to systemic inflammation, as seen in conditions such as sepsis and cytokine storm syndromes. Understanding the molecular pathways governing TNF-α activity highlights its dual role in host defense and pathology, reinforcing the importance of targeted therapeutic interventions.
Aralia armata (Wall.) Seem., a member of the Araliaceae family, is a well-known medicinal plant in Vietnam traditionally used to treat hepatitis, arthritis, stomachache, malaria, and snake bites (Figure 1). 16 The main components of A. armata’s leaves and roots have been identified as oleanane-type triterpene glycosides as a result of phytochemical investigations.17,18 These compounds have demonstrated a variety of bioactivities, including antibacterial, antifungal, antioxidant, anti-inflammatory, anticancer, and anti-HIV effects.19–25 However, the underlying mechanisms of action of these compounds, particularly in relation to inflammation-associated diseases, remain poorly understood. This suggests the potential of bioactive compounds from A. armata as inhibitors of key biological targets involved in inflammatory pathways, paving the way for the identification of promising anti-inflammatory drug candidates.

Aralia armata (Wall.) Seem.
Furthermore, computational approaches such as molecular docking combined with molecular dynamics (MD) simulations have proven effective in the preliminary screening of natural compounds, allowing the identification of potential inhibitors of the key target protein. These methods offer a cost-effective and efficient strategy to accelerate the discovery of bioactive molecules for further biological evaluation.26,27
Therefore, in this study, we conducted in silico investigations on compounds derived from A. armata to support the discovery of potential TNF-α inhibitors for the treatment of inflammatory diseases.
Materials and methods
Molecular docking
The structures of the compounds derived from A. armata were obtained from previous reports and drawn using ChemSketch software.17,18,28–31 Then, their structures were energy-optimized through MMFF94s using Avogadro software and prepared for docking using AutoDockTools software, saved in PDBQT format.32,33 The three-dimensional (3D) protein structure of TNF-α was downloaded from the RCSB Protein Data Bank (PDB ID: 2AZ5). 34 Water molecules and co-crystallized ligands were removed using PyMOL software. In addition, polar hydrogen atoms were added to the protein structure, Kollman charges were calculated, and the structure was saved in PDBQT format using AutoDockTools software. The docking process was performed using AutoDock Vina v1.2.3 program with the exhaustiveness parameter set according to previous studies.35,36 The grid box parameters were set within the active site of the receptor, specifically with center coordinates at x = −19.2 Å, y = 74.5 Å, z = 33.8 Å, a grid size of 24 Å × 24 Å × 24 Å, and a grid spacing of 1 Å. The binding affinity (∆G) of the compounds was ranked, and the most potent compounds were selected. The best docking poses of these compounds were visualized in two-dimensional (2D) and three-dimensional (3D) interaction images using Discovery Studio Visualizer software.
MD
To gain deeper insights into the configurational dynamics of the TNFα system upon ligand binding, we conducted all-atom MD simulations of protein-ligand complexes for 100 ns using GROMACS v.2023.3. 37 First, the topology files for the ligand structures were generated using AmberTools and the AnteChamber PYthon Parser interfacE (ACPYPE).38,39 The initial protein topology was parameterized with the AMBER99SB-ILDN force field. 40 Each protein–ligand complex was solvated in a triclinic box filled with TIP3P water molecules, ensuring a minimum 4.0-nm buffer between the solute and the box boundaries. The system was then neutralized by adding counterions. Energy minimization was carried out using the steepest descent algorithm, followed by sequential equilibration under NVT and NPT ensembles at 300 K and 1 bar for 100 ps. Temperature and pressure were controlled using the Berendsen thermostat and the Parrinello–Rahman barostat, respectively. Finally, MD simulations were performed for 100 ns with a 2-fs integration timestep. All trajectory coordinates were recorded in the MD simulation files, and the results were visualized and analyzed using Excel software. Furthermore, the binding free energy was calculated using the MMGBSA approach with the gmx_MMPBSA program. 41
ADMET prediction
The online web server pkCSM (https://biosig.lab.uq.edu.au/pkcsm/prediction) was used to predict the pharmacokinetic properties (absorption, distribution, metabolism, excretion, and toxicity) of the selected compounds. 42 The predicted models include water solubility, Caco-2 permeability, intestinal absorption (human), skin permeability, P-glycoprotein substrate, P-glycoprotein I inhibitor, P-glycoprotein II inhibitor, VDss (human), fraction unbound (human), BBB permeability, CNS permeability, CYP2D6 substrate, CYP3A4 substrate, CYP1A2 inhibitor, CYP2C19 inhibitor, CYP2C9 inhibitor, CYP2D6 inhibitor, CYP3A4 inhibitor, total clearance, renal OCT2 substrate, AMES toxicity, maximum tolerated dose (human), hERG I inhibitor, hERG II inhibitor, oral rat acute toxicity (LD50), oral rat chronic toxicity (LOAEL), hepatotoxicity, skin sensitization, Tetrahymena pyriformis toxicity, and minnow toxicity.
Results and discussion
Molecular docking simulation plays a crucial role in screening plant-derived natural compounds to identify promising candidates for in vitro and in vivo experiments. 43 This computational approach predicts the binding affinity between compounds and target proteins, significantly accelerating the screening process while reducing time and cost compared to traditional biological assays.44,45 In this study, a set of compounds from A. armata was docked into the active site of the TNF-α protein (PDB ID: 2AZ5). Prior to screening, docking protocol validation was conducted to ensure reliability. The accuracy of the protocol was assessed using the root mean square deviation (RMSD) value, which measures the difference between the redocked co-crystallized ligand structure and its experimentally determined conformation. An RMSD value below 2.0 Å typically indicates high reliability, demonstrating that the method can accurately reproduce the ligand’s actual binding position.35,46 Conversely, a high RMSD suggests the need for adjustments, such as refining simulation parameters, selecting an optimized docking algorithm, or improving input structure quality. The RMSD value obtained in this study was 1.827 Å, which is below 2.0 Å, with minimal deviation as shown in Figure 2, confirming the reliability of the docking protocol. Therefore, this validated approach was applied to the subsequent screening process.

Superimposition of the co-crystallized ligand (cyan) and the redocked ligand (magentas) in the TNF-α protein with an RMSD value of 1.827 Å.
The binding affinities and molecular interactions of A. armata compounds with TNF-α reveal significant variations in binding strength and interaction patterns among the studied compounds. The binding affinities ranged from −4.741 to −10.27 kcal/mol, as shown in Table S1. Notably, compound
Binding Affinities and Molecular Interactions of Top-Hit Compounds With Amino Acid Residues Within the Active Site Pocket of the TNF-α Protein (PDB ID: 2AZ5).

These compounds demonstrated strong interactions with TNF-α, suggesting a higher inhibitory potential compared to the others, making them prime candidates for further molecular interaction analysis (Figure 3). Compounds
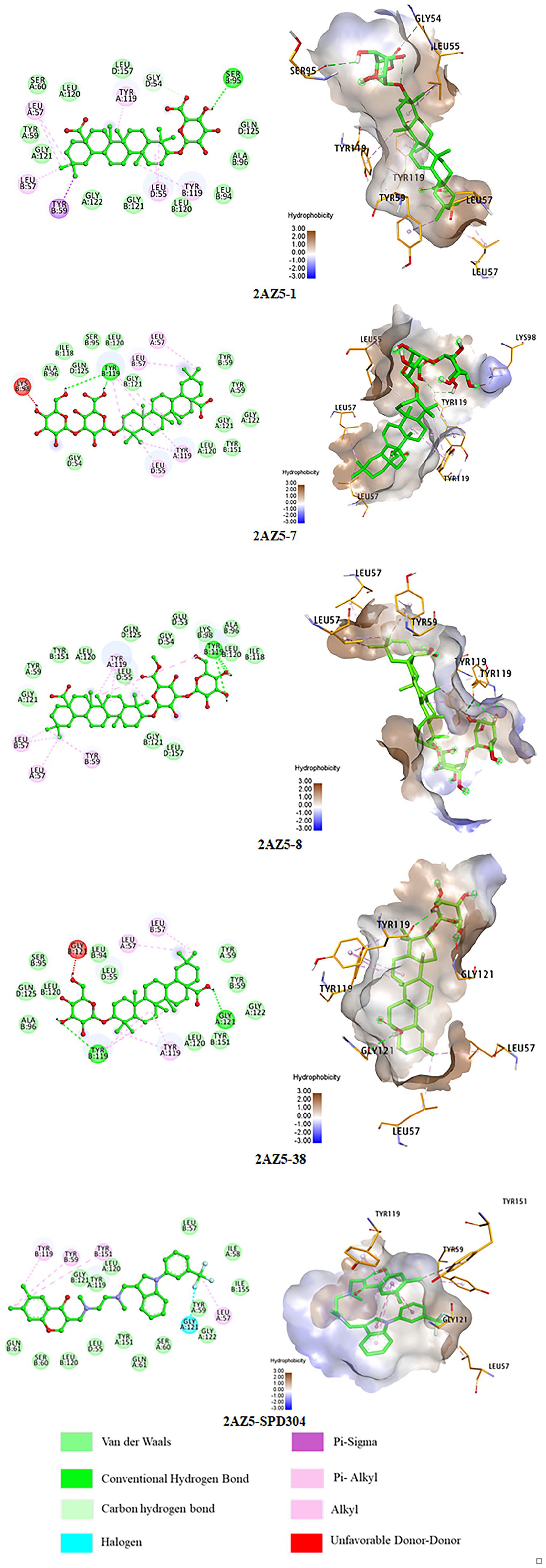
2D and 3D interactions of top-hit compounds within the active site pocket of the TNF-α protein (PDB ID: 2AZ5).
MD
In MD simulations, root mean square deviation (RMSD) is a key parameter used to assess the structural stability of a system over time. A low and stable RMSD value indicates that the system maintains its structural integrity, while a high or fluctuating RMSD suggests significant conformational changes.47,48 This stability assessment is evident in Figure 4, where the RMSD values of the protein backbone in complexes 2AZ5-

Root mean square deviation (RMSD) analysis of the backbone (left) and ligand (right) atoms of TNF-α complexes.
While RMSD provides insights into the structural stability of protein–ligand complexes, a more comprehensive understanding of their binding strength requires energetic analysis. To achieve this, the MMGBSA (Molecular Mechanics/Generalized Born Surface Area) method is employed to estimate the binding free energy between small molecules and proteins.49,50 Compared to conventional docking methods, MMGBSA offers superior accuracy by accounting for solvent effects and system flexibility. While docking primarily relies on a rigid model of the protein–ligand complex and evaluates only static interactions, MMGBSA recalculates binding energy across multiple conformational states derived from MD simulations. This dynamic evaluation provides a more realistic representation of binding interactions in biological environments, making MMGBSA particularly valuable for systems with significant conformational changes or strong solvent interactions.49,50 The data in Table 2 present the binding free energy of various compounds with TNF-α, a crucial target in pharmaceutical research. The total binding free energy (ΔGtotal) is derived from several energy components, including van der Waals interactions (EVDWAALS), electrostatic interactions (EEL), polar solvation energy (EGB), and surface energy (ESURF). A more negative ΔGtotal indicates a stronger binding affinity between the compound and the protein. The results show that compound
The Bind Free Energy (in kcal/mol) Analysis of TNF-α and Selected Compounds Complexes, Determined via a MMGBSA Method.

ADMET profiles
Based on the ADMET data of the four selected compo-unds (Table 3), their absorption, distribution, metabolism, excretion, and toxicity can be assessed to determine their pharmacokinetic properties and safety profiles. All four compounds exhibit low water solubility (−2.96 to −3.249), which may negatively impact their oral bioavailability. Their permeability across the Caco-2 membrane is also poor (−0.109 to −0.404), suggesting limited passive diffusion through the intestinal epithelium. However, intestinal absorption varies significantly, with compound
ADMET Profiles of Selected Compounds.

Conclusion
This study utilized molecular docking, MD simulations, and MMGBSA binding energy calculations to identify promising TNF-α inhibitors from A. armata compounds. The docking protocol was validated with an RMSD value of 1.827 Å, ensuring accuracy in predicting binding interactions. Among the screened compounds, compound
Supplemental Material
sj-docx-1-chl-10.1177_17475198251339509 – Supplemental material for Phytochemicals from Aralia armata as potential tumor necrosis factor-alpha inhibitors: A computational study
Supplemental material, sj-docx-1-chl-10.1177_17475198251339509 for Phytochemicals from Aralia armata as potential tumor necrosis factor-alpha inhibitors: A computational study by Vu Thi Thu Le, Luc Quang Tan, Do Khac Hung and Nguyen Xuan Ha in Journal of Chemical Research
Footnotes
Ethical Considerations
Ethical approval is not applicable to the article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported under the project with the code: B2024-TNA-28 by the Ministry of Education and Training, Vietnam.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Statement of human and animal rights
This article does not contain any studies with human or animal subjects.
Statement of informed consent
There are no human subjects in this article, and informed consent is not applicable.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.