
Abstract
In our quest to discover effective inhibitors against severe acute respiratory syndrome coronavirus 2 helicase, a diverse set of more than 300 naturally occurring antiviral metabolites was investigated. Employing advanced computational techniques, we initiated the selection process by analyzing and comparing the co-crystallized ligand (
Keywords

Introduction
As of 23 July 2023, the World Health Organization (WHO) reported an alarming total of 768,237,788 confirmed cases of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection worldwide. Tragically, among these cases, 6,951,677 individuals have lost their lives. 1 These distressing figures highlight the urgent need for extensive efforts from scientists in the quest for a cure, underscoring the crucial role of research and collaborative efforts in combatting this global health crisis.
The use of the computer as a tool in drug discovery (computer-aided drug discovery (CADD)) has been an essential approach in pharmaceutical and medicinal research since the 1970s and before. Progressively, it drew more attention to being a trustworthy technique that can expect the potential of a compound to be biologically active, saving time, and money. 2 There are two main types of CADD techniques. The first is the structure-based that starts with the target protein and measures the ability of various ligands to bind with that protein through the calculation of binding energies. 3 Contrastingly, the ligand-based method uses ligands (mostly a group of molecules) checking their possible activities against an identified ligand through pharmacophore, structural similarity, or quantitative structure–activity relationship (QSAR) models.4,5 Regarding coronavirus-19 (COVID-19), the CADD approach was applied in several reports.6–8
Throughout history, nature has been a generous source of remedies, offering humans a vast array of cures for various diseases and infections. From the earliest records of human existence, our ancestors relied on their surroundings to find plants, herbs, and other natural substances that could alleviate ailments and restore health.9,10
In our team’s efforts to counteract the impact of COVID-19, a sophisticated multistage in silico CADD strategy was implemented. The objective was to discern natural inhibitors and propose the most efficacious one tailored to specific COVID-19 enzymes. Among a cohort of 310 antiviral natural metabolites, promising inhibitors for the main protease,11,12 nsp10, 13 and the papain-like protease 14 have been pointed. Furthermore, among 3009 Food and Drug Administration (FDA)-approved drugs, the most potential inhibitors against the RNA-dependent RNA polymerase 15 and the SARS-CoV-2 nsp16-nsp10 2′-o-methyltransferase 16 have been indicated. Also, the exploration of 5956 traditional Chinese medicine to identify potential inhibitors for the SARS-CoV-2 helicase enzyme was conducted. 17
Viral helicases are a sort of enzyme that is responsible for the unpack of the duplex oligonucleotides (RNA or DNA) into their constituent single strands besides other significant functions in the replication process. 18 Consequently, the inhibition of that vital enzyme could be a crucial step in the fight against the target virus.
The aim of this study is to single out a natural compound to be an anti-COVID-19 candidate through the inhibition of SARS-CoV-2 helicase protein. The compounds were subjected to various in silico filtration processes. We herein report different advanced ligand and structure-based computational studies for a set of 310 reported antiviral natural compounds (Fig.S.1. and Table S.1. in the Supplementary data) to determine the most convenient SARS-CoV-2 helicase inhibitor. The used compounds were selected based on deep research on the scientific websites for natural metabolites that exhibited antiviral activities. The selected compounds belonged to diverse classes, such as alkaloids, peptides, flavonoids, anthraquinones, steroids, and carbohydrates.
The rationale of this work comprises seven successive computational steps to reach a promising candidate against SARS-CoV-2 helicase. First, a set of 310 naturally occurring compounds was examined for their structural similarity with the co-crystallized ligand of SARS-CoV-2 helicase. Then, the most similar 30 compounds were subjected to the pharmacophore study to select the most related compound to the co-crystallized ligand of SARS-CoV-2 helicase. Next, a set of 13 compounds that showed good fit values with the generated pharmacophore was docked against the crystal structure of SARS-CoV-2 helicase. Seven compounds that showed good fitting and binding mode at the active pocket of SARS-CoV-2 helicase were subjected to in silico physicochemical profiling assessment according to Lipinski’s and Veber’s rules. The tested compound that obeyed Lipinski’s and Veber’s rules (seven compounds) was subjected to ADMET study to reach the most convenient compounds for druggability and drug likeness. After that, the most convenient four compounds were evaluated for their expected toxicity potential. One compound showed an accepted range of toxicity and was subjected to an MD simulation experiment to assess its stability in the active pocket of SARS-CoV-2 helicase (Figure 1).

Rationale of the discovery of a promising candidate against SARS-CoV-2 helicase.
Results and discussion
Molecular similarity
The molecular similarity is an in silico ligand-based type of calculation that is used to explore the similarity between two molecules following the scientific principle that the structurally similar molecules are predicted to have related biological properties. 19 Molecular similarity targets the molecular properties of the explored compounds, such as the LogP and molecular weight. In addition, the molecular similarity mostly depends on the distances between atoms in space, structural and physicochemical descriptors. 20
Although molecular similarity can also be defined using molecular properties, such as molecular weight, and LogP, molecular similarity is most often defined in terms of a distance measure in a structural or physicochemical descriptor space. There many different distance measures available. In this work, we used Euclidean distance method that is available in Discovery Studio.
In this research, a molecular similarity study has been preceded for 310 naturally isolated compounds that showed antiviral activities before (Supplementary data) against the co-crystallized ligand of SARS-CoV-2 helicase protein (

The similarity analysis between the tested molecules and (VXG). Green ball = reference molecule, red balls = similar compounds, blue balls = not similar compounds. (a) Compounds 1–50, (b) compounds 51–100, (c) compounds 101–150, (d) compounds 151–200, (e) compounds 201–250, and (f) compounds 251–310.
In total, 30 compounds (Figure 3 and Table 1) exhibited similar values of molecular properties with (VXG).

The most structurally similar 30 compounds with (VXG).
The most structurally similar 30 compounds with (VXG) and their molecular properties.
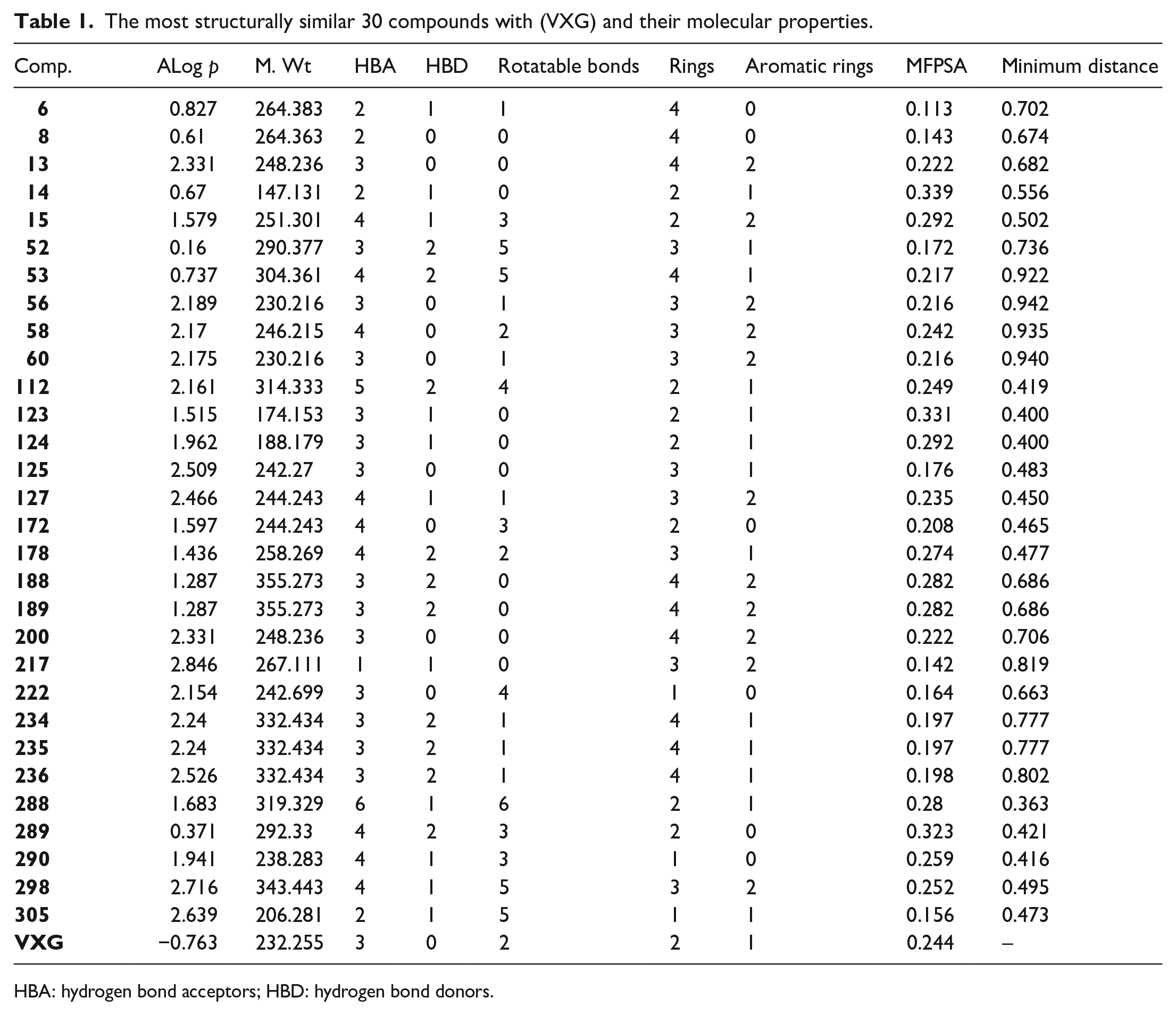
HBA: hydrogen bond acceptors; HBD: hydrogen bond donors.
Pharmacophore study
Generation of 3D-pharmacophore model
The main objective of the pharmacophore-based drug design is to generate a 3D pharmacophore model based on the known receptor–ligand pharmacophore generation approach. 21 In this approach, various features would be expected from the study of the binding of the ligand with the target protein.22,23 These features are essential for the activity. Consequently, any compound that contains these features is expected to be active. 24 The resulted pharmacophore model was generated based on receptor–ligand interactions. At first, a set of features from the binding ligand was identified. The feature types that considered during generation process are HBA, HBD, hydrophobic groups, positive and negative ionizable groups, and aromatic rings. The features that matched the interactions between the binding ligand and the protein were used to build the pharmacophore models. Excluded volumes were added based on the locations of atoms on the protein. Second, the pharmacophore models were ranked based on a measure of sensitivity and specificity and the top models are returned. The pharmacophore models were validated based on a genetic function approximation (GFA) model. 25 The resulted 3D pharmacophore model consisted of three features: two HBA and one hydrophobic center (Figure 4). The generated model was used to examine the tested compounds as possible SARS-CoV-2 helicase inhibitors.

(a) The generated hypothetical 3D-pharmacophore geometry with four features; two HBA (red) and one hydrophobic (yellow). (b) Mapping of the co-crystallized ligand on the generated pharmacophore (Fit value = 36.08). (c) The calculated distances between the different features.
Activity prediction
The test set of the selected 30 compounds and the co-crystallized ligand (VXG) were examined against the generated 3D-pharmacophore model. In this process, fit values (that quantitatively represent the existence of that essential features) between the tested compound and the generated 3D-pharmacophore were generated.26,27
In total, 13 compounds (
Essential features, fit values, and relative fit of the tested compounds and the co-crystallized ligand (VXG) based on the generated pharmacophore model.

 Hydrogen bond acceptor
Hydrogen bond acceptor  Hydrophobic center
Hydrophobic center
Relative Fit = Fit value of a compound/Fit value of co-crystallized ligand (VXG).

Mapping of the tested compounds on the generated pharmacophore. (a) Compound
The specified compounds are metabolites of natural sources with proved antiviral potentialities. In detail, isatin or tribulin (
Atropine (
The furanocoumarin, isopimpinellin (
Mycophenolic acid (
Docking studies
The molecular docking process was conducted for the most favored 13 compounds against SARS-CoV-2 helicase (PDB codes: 5RMM) using MOE 19.0901 software as described in previous reports.54–59 In this test, a flexible docking procedure was applied for both the co-crystallized ligand and the tested molecules at the same pocket of the co-crystallized ligand with the same dimensions.
Validation of docking process
A redocking process of (VXG) in the active site of SARS-CoV-2 helicase has been conducted and showed a root-mean-square deviation (RMSD) value of 0.66 Å indicating a valid docking algorithm (Figure 6).

Superimposition of the docked and original poses of the co-crystallized ligand in SARS-CoV-2 helicase active site.
Molecular docking of the Crystal ligand
The binding mode of (
Docking score of 13 compounds against SARS-CoV-2 helicase (PDB ID: 5RMM).

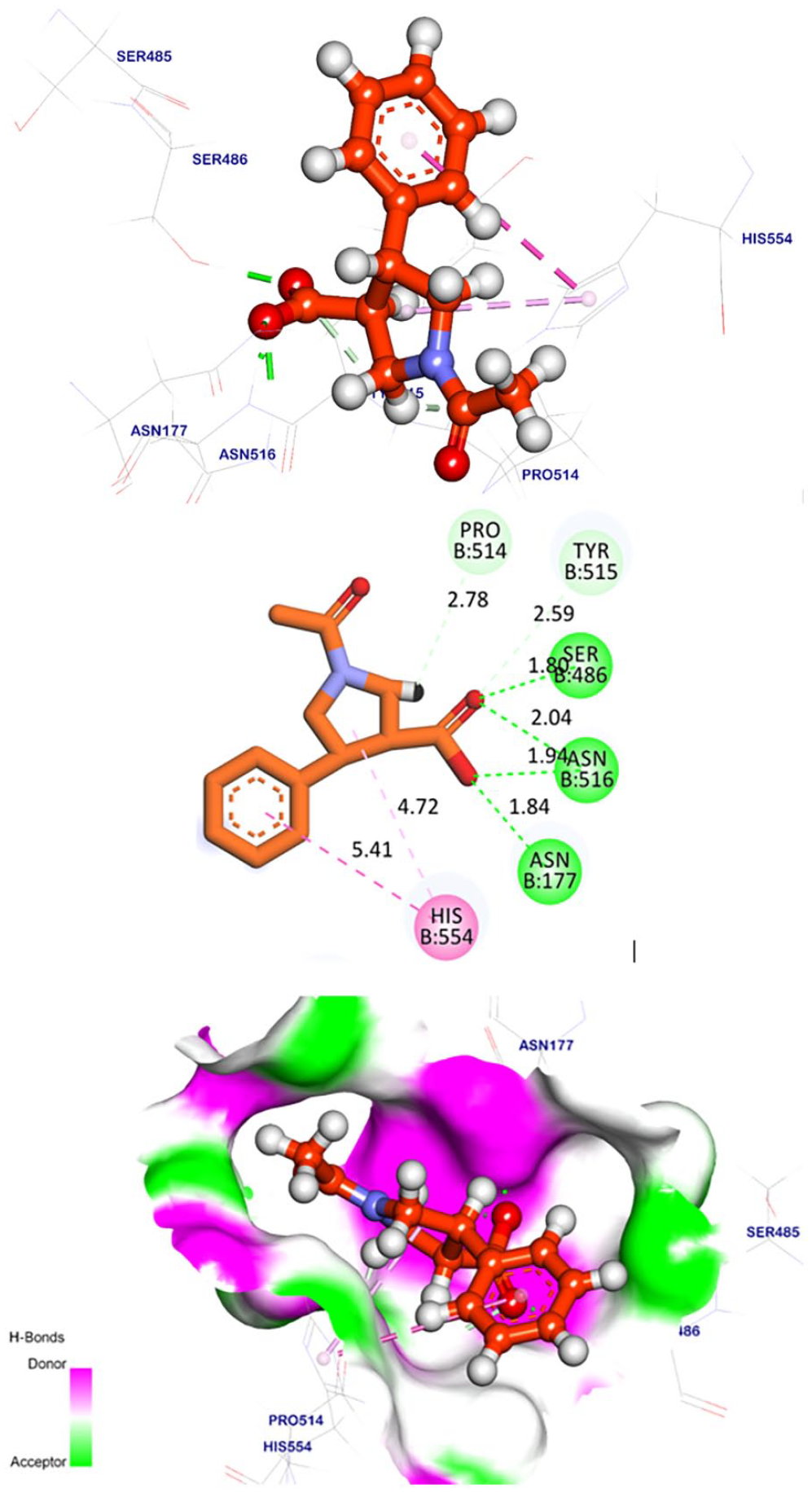
Molecular docking of the selected compounds
Compound
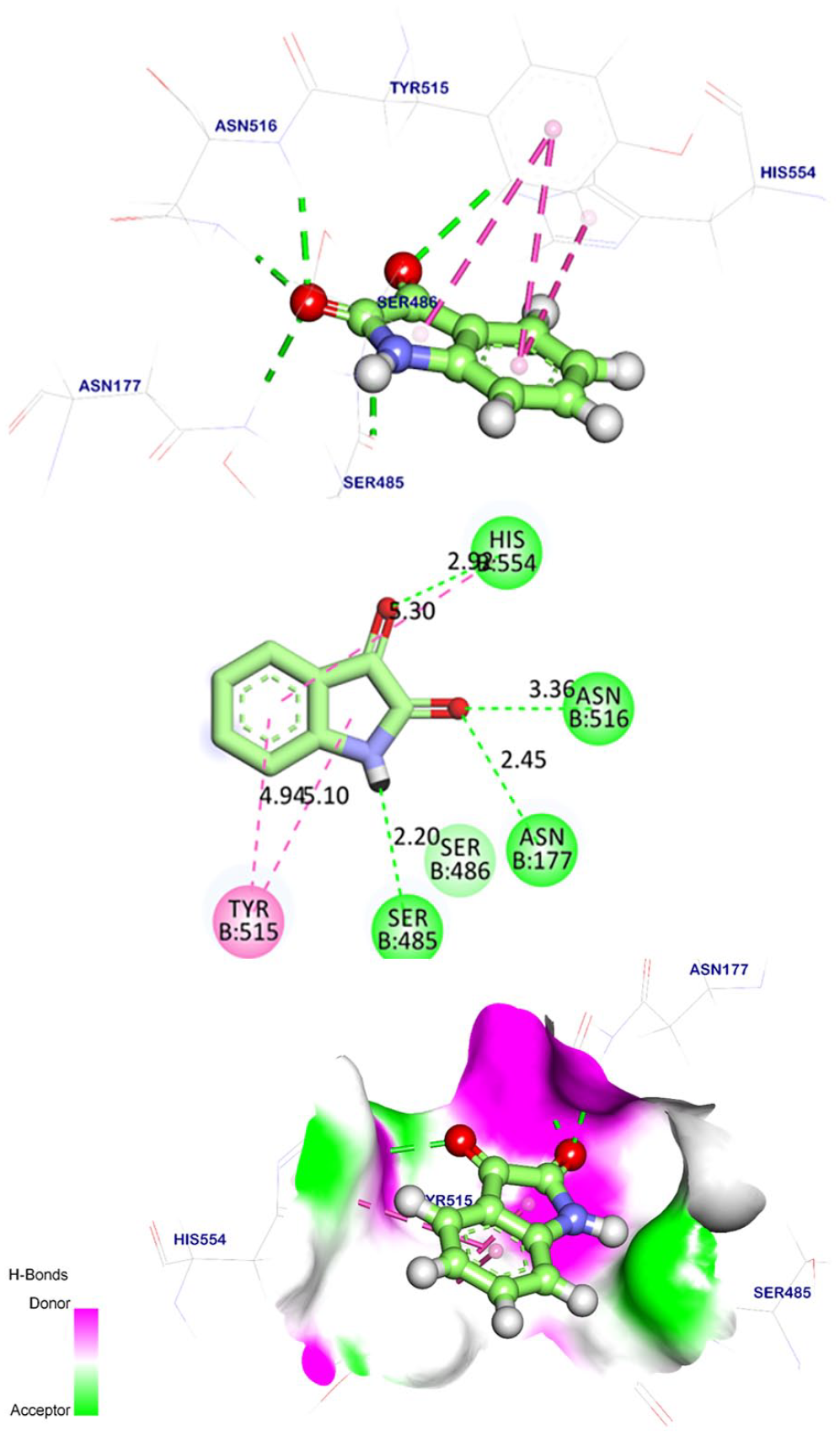
Compound
The binding mode of candidate

Compound
The binding mode of compound
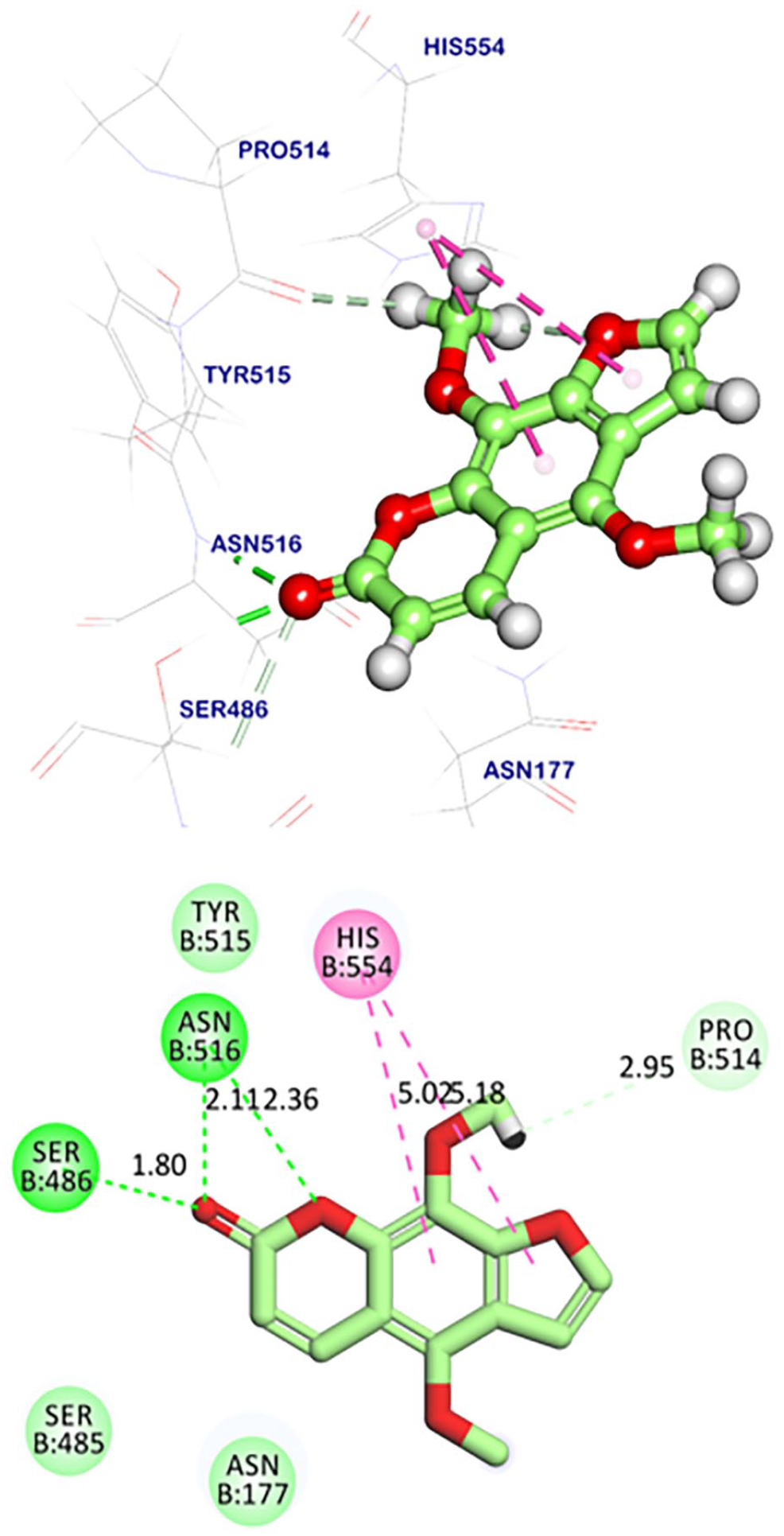
Compound
Compound

Compound
Regarding compound
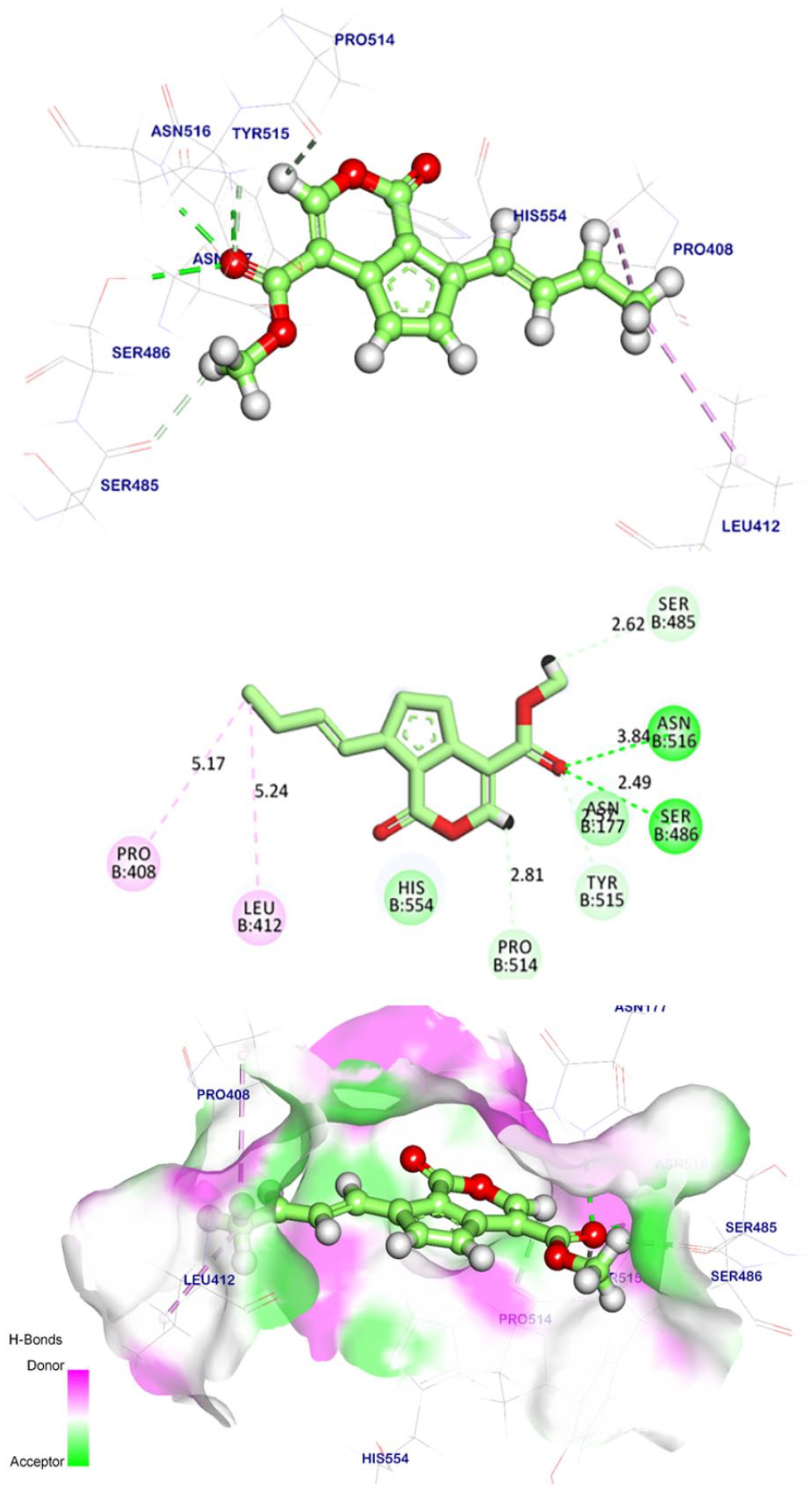
Compound
With regard to the binding mode of compound

Compound
Concerning compound

Compound
Pharmacokinetic profiling study
Seven compounds (
Lipinski’s rule describes the oral absorption of a drug if the molecule fulfills at least three of the four principles (HBD ⩽ 5; HBA ⩽ 10; molecular weight < 500; LogP < 5). In addition, Veber’s rule deals with the bioavailability of a compound if the molecule fulfills its two principles: rotatable bonds < 10 and polar surface area ⩽ 140 Å. Compounds with 10 or fewer have a high probability of good oral bioavailability.60,61 As shown in Table 4, all tested molecules and reference ligand
Pharmacokinetic profiling of the tested molecules according to Lipinski’s and Veber’s rules.

HBA: hydrogen bond acceptors; HBD: hydrogen bond donors.
Computational ADMET studies
Six ADMET parameters were assessed using Discovery studio software using amenamevir (reference drug) as described in the literature.62–66 The results were summarized in Table 5 and Figure 15. The results revealed that blood penetration levels of all compounds are ranging from medium to low, indicating less possibility for CNS side effects. Additionally, all compounds had good or optimal solubility levels. Furthermore, all the tested molecules showed good absorption levels and appeared to be non-inhibitors for cytochrome P450. Concerning hepatotoxicity, compounds
Predicted ADMET for the elected metabolites and the reference compound.

Blood-brain barrier (BBB) penetration level, 4 = very low and 0 = very high.
4 is optimal while 1 is very low.
0 is good, and 3 is very poor.
CYP2D6, the inhibition of cytochrome P2D6, T = inhibitor, F = non inhibitor.
Hepatotoxicity, T = toxic, F = non-toxic.
Plasma protein-binding (PBB), binding with plasma protein, F: < 90%, T: > 90%
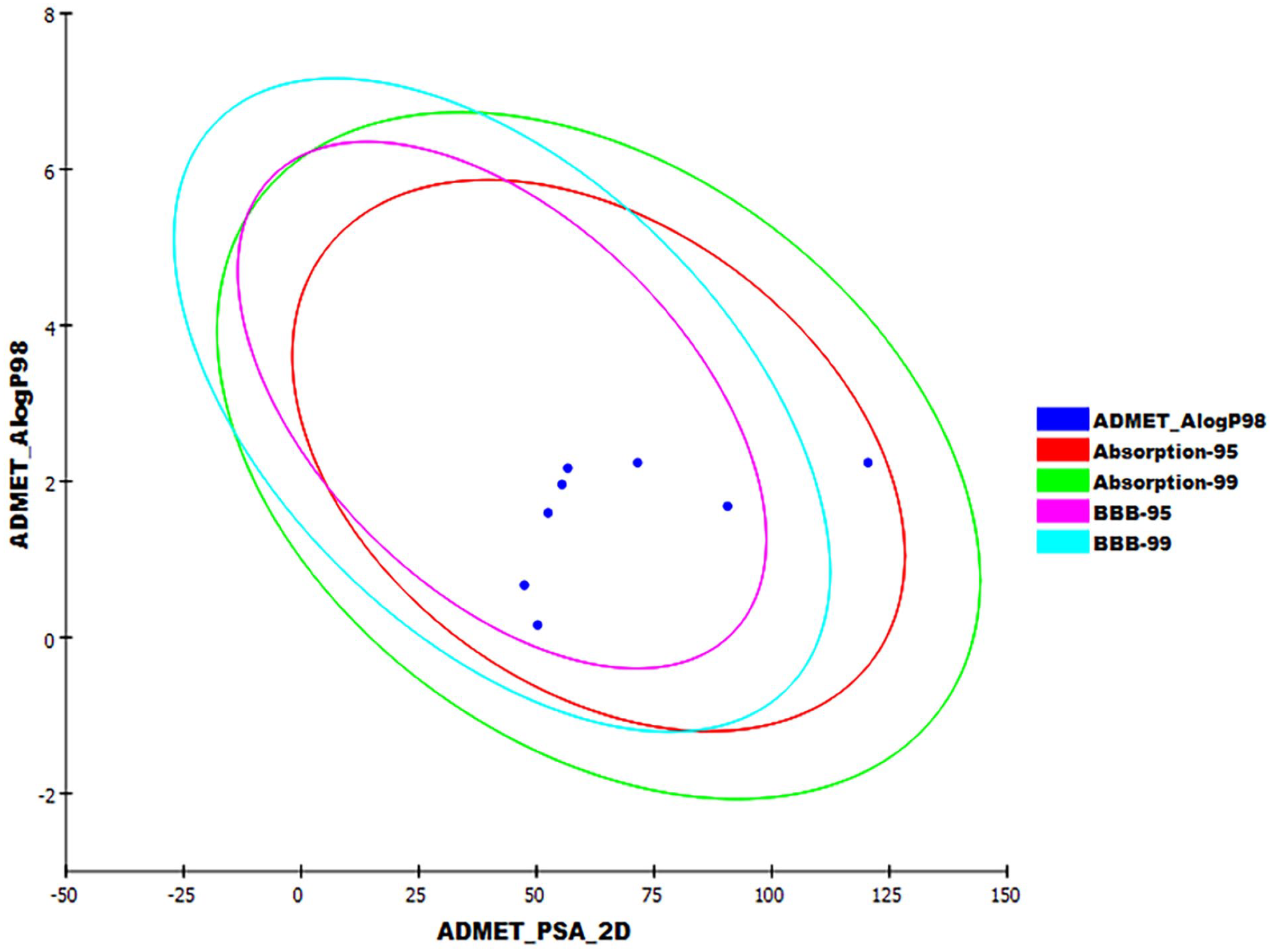
The expected ADMET study.
Computational toxicity studies
Virtual toxicity studies against different toxicity models have been carried out using Discovery studio software as described before 67 using amenamevir as a reference molecule. The results were summarized in Table 6 and detailed in the Supplementary data.
Toxicity properties of compounds.

TD: tolerated dose; MTD: maximum tolerated dose
Unit: mg kg−1 day−1.
Unit: g kg−1.
I = irritant, N = non-irritant.
Compound
However, compounds
MD simulations studies
Molecular dynamics (MD) simulations can provide plentiful information regarding the dynamical and structural changes that happen on a protein after binding to a ligand. Also, it affords generous energetic information about the protein–ligand complex. 68 The obtained results could be very useful to understand the interactions between the ligand and the target protein on an atomic resolution. 69
The conformational changes and variations, and the atomic dynamic movements of backbone atoms in the mycophenolic acid–SARS-CoV-2 helicase complex were computed by RMSD to verify the stability of the mycophenolic acid–SARS-CoV-2 helicase on both apo and ligand bonded states. It was noticed that mycophenolic acid, SARS-CoV-2 helicase, and the mycophenolic acid–SARS-CoV-2 helicase complex exhibited very low RMSD values without major fluctuations demonstrating the great stability (Figure 16(a)). The flexibility of SARS-CoV-2 helicase, during the 100 ns of the experiment, was computed in the term of RMSF to investigate closely the region of proteins that have been fluctuated during the MD simulation. The root-mean-square fluctuation (RMSF) denoted that the binding of mycophenolic acid makes the SARS-CoV-2 helicase slightly flexible in 150–250 residue areas (Figure 16(b)). To study the compactness of mycophenolic acid–SARS-CoV-2 helicase complex, the radius of gyration (Rg) was investigated. Figure 16(c) designated the decrease of the Rg values of the mycophenolic acid–SARS-CoV-2 helicase complex than the starting period. Such results marking the high degree of compactness of the mycophenolic acid–SARS-CoV-2 helicase complex. Aiming at the analysis the conformational changes occurred during the mycophenolic acid–SARS-CoV-2 helicase interaction, the solvent accessible surface area (SASA) was measured over 100 ns. Interestingly, the SARS-CoV-2 helicase displayed a decrease in the surface area giving relatively low SASA value at the end of experiment than the starting period (Figure 16(d)). Finally, hydrogen bonding in the mycophenolic acid–SARS-CoV-2 helicase was computed. Advantageously, the results verified the high number of conformations of SARS-CoV-2 helicase formed up to eight hydrogen bonds (Figure 16(e)). Such interesting results authenticate the stability in with the mycophenolic acid–SARS-CoV-2 helicase complex.

MD simulations results; (a) RMSD values of mycophenolic acid, SARS-CoV-2 helicase complex and mycophenolic acid–SARS-CoV-2 helicase complex. (b) RMSF, (c) Rg, (d) SASA of SARS-CoV-2 helicase, and (e) hydrogen bonding mycophenolic acid–SARS-CoV-2 helicase complex in the MD run.
Molecular Mechanics Poisson–Boltzmann Surface Area results
The binding-free energy of mycophenolic acid–SARS-CoV-2 helicase complex was computed over the last 20 ns of the MD run with a 100-ps interval using Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA) method. Mycophenolic acid displayed a low binding-free energy value of −294 kJ mol−1 with SARS-CoV-2 helicase (Figure 17(a)). Furthermore, the total binding-free energy of the mycophenolic acid–SARS-CoV-2 helicase complex was disintegrated into per amino acid residue share energy. The output of that experiment gave us further details about the primary (essential) residues that share favorably in the binding of mycophenolic acid to the SARS-CoV-2 helicase. ARG-178, LYS-202, LYS-414, and ARG-560 residues of SARS-CoV-2 helicase were found to be the higher contributors with free energies that more than −20 kJ mol−1 (Figure 17(b)).

MM/PBSA results of mycophenolic acid–SARS-CoV-2 helicase complex; (a) free binding energy over the 100 ns, and (b) crucial amino acids that were contributed in the mycophenolic acid–SARS-CoV-2 helicase complex.
In 1969, mycophenolic acid exhibited significant in vitro antiviral activities against DNA and RNA viruses using agar diffusion plaque reduction and tube culture methods. The inhibition power was noticed against vaccinia virus, adenovirus, herpes, and measles.
70
Mycophenolic acid protected the virus-infected cells with 100% at a concentration of 10 μg mL−1.
71
In another experiment, it was active against orthopoxviruses through the inhibition of the viral cellular inosine monophosphate dehydrogenase.
72
As well, it inhibited the viral RNA replication of the dengue virus.
73
In addition, mycophenolic acid inhibited in vitro and in vivo hepatitis C virus (HCV) infection and augmented interferon-stimulated gene expression.
74
Besides, the antiviral activity of
Method
Molecular similarity detection
Discovery studio 4.0 software was used for 310 natural metabolites against
Pharmacophore studies
Discovery studio 4.0 software was used for the most similar 30 metabolites as (see method part in Supplementary data).
Docking studies
Docking studies were done for the selected 13 metabolites and
ADMET analysis
Discovery studio 4.0 84 was used for the selected seven metabolites (see method part in Supplementary data).
Toxicity studies
Discovery studio 4.0 85 was used for the selected four metabolites (see method part in Supplementary data).
Molecular dynamics simulation
The mycophenolic acid–SARS-CoV-2 helicase system was prepared using the web-based CHARMM-GUI86–88 interface using CHARMM36 force field 89 and NAMD 2.13 90 package. The TIP3P explicit solvation model was used (see supporting data).
MM/PBSA studies
The g_mmpbsa package of GROMACS 91 was used to calculate the MM/PBSA of mycophenolic acid–SARS-CoV-2 helicase system (see supporting data).
Conclusion
This research used a set of 310 reported antiviral natural compounds as an examination set to single out the most convenient SARS-CoV-2 helicase inhibitor. The chemical structural similarity test against the co-crystallized ligand assigned the most similar 30 compounds. Then, the study of the pharmacophoric features selected the most suitable 13 compounds. Next, the molecular docking against the SARS-CoV-2 helicase protein (PDB ID: 5RMM) filtered seven candidates. Furthermore, the ADMET and toxicity studies elected three metabolites. The Density functional theory (DFT) study suggested mycophenolic acid (
Supplemental Material
sj-pdf-1-chl-10.1177_17475198231221253 – Supplemental material for Computer-aided drug discovery of natural antiviral metabolites as potential SARS-CoV-2 helicase inhibitors
Supplemental material, sj-pdf-1-chl-10.1177_17475198231221253 for Computer-aided drug discovery of natural antiviral metabolites as potential SARS-CoV-2 helicase inhibitors by Eslam B Elkaeed, Ibrahim H Eissa, Abdulrahman M Saleh, Bshra A Alsfouk and Ahmed M Metwaly in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R142), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors extend their appreciation to the Research Center at AlMaarefa University for funding this work.
Supplemental material
Supplemental material for this paper is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.