
Abstract
Imidazole derivatives such as miconazole and econazole have shown promising antibacterial and antifungal activities. Newer generation 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (BP1-BP5) were synthesized by the reaction between oxazolone derivatives and 4,5-diphenyl-1H-imidazol-2-amine. The synthesized molecules were characterized by different spectroscopic techniques. Then the synthesized molecules were evaluated against four bacterial and two fungal strains. Molecular docking of synthesized molecules with antibacterial and antifungal targets showed that BP1 and BP5 are the most effective docked structures. MD simulation expressed the structural integrity. DFT analysis showed that BP5 is the soft molecule, and BP3 showed maximum electrophilicity. BP1 and BP5 showed good antibacterial activity against B. subtilis, S. aureus, E. coli, and S. typhi. MIC and MBC analysis revealed that B. subtilis and S. aureus are the most sensitive bacterial strains toward synthetic derivatives. BP5, BP1, and BP3 showed good antifungal activities compared to the standard nystatin. These studies confirmed that the synthesized 1′H-[1,2′-biimidazol]-5(4H)-one derivatives are effective as antibacterial and antifungal agents with validated computational insights.
Keywords

Introduction
Microbial infection is a broad category of viruses, bacteria, fungi, and other parasites that induce pathological conditions, which can cause adverse effects and sometimes be life-threatening if left untreated.1,2 There are millions of bacteria present in the environment which impact a fatal adverse effect on humans. Bacterial colonization of the host system is the initial process of bacterial infections of the host, followed by adhesion, invasion, and replication.3,4 Bacterial infections affect various body parts, such as the skin, respiratory tract, digestive system, and urinary tract. 5 Bacteria such as Streptococcus pneumoniae and Haemophilus influenzae affect the respiratory system (lungs, airways), leading to conditions like sinusitis, pneumonia, and bronchitis. Staphylococcus aureus and Streptococcus pyogenes are responsible for skin infections, including impetigo, cellulitis, and abscesses. Staphylococcus aureus, commonly found on human skin, causes infections if it enters the body through cuts or wounds.6,7 These infections range from minor skin issues like boils and abscesses to more serious conditions such as pneumonia, sepsis (bloodstream infections), and bone infections (osteomyelitis and septic arthritis).8,9 Some strains, like MRSA (Methicillin-resistant Staphylococcus aureus), are resistant to antibiotics. 10 Urinary tract infections, gastrointestinal infections, and bloodstream infections are often caused by Escherichia coli. Other than these bacteria, Salmonella typhi (S. typhi), a harmful bacterium, causes typhoid fever. It is transmitted through contaminated food or water and invades the bloodstream, resulting in elevated body temperature, abdominal pain, weakness, and other symptoms. 11 These infections can cause serious and sometimes life-threatening illnesses.12,13 Penicillin, cephalosporins, macrolides, tetracyclines, fluoroquinolones, sulfonamides, and glycopeptides are the classes of therapeutic agents that are used to treat bacterial infections. 14 Whereas, fungal infection is caused by the fungal spores present in animal feces, chicken coops, and rotting vegetation. It affects various parts of the body, such as the skin, nails, internal organs, and mucous membranes. Inhalation of the fungal spores can lead to an internal fungal infection. Athlete’s foot, ringworm, and Seborrheic dermatitis are some examples of fungal infections. Aspergillus niger is known for its dual nature, offering immense industrial value while posing health risks to certain medical conditions. 15 Sometimes it does not affect healthy beings, but it infects the individual with a weakened immune system. It can cause aspergillosis, allergic bronchopulmonary aspergillosis, aspergillomas (fungal balls) in pre-existing lung cavities (caused by sarcoidosis, tuberculosis). Aspergillus niger invades the bloodstream (invasive aspergillosis) and affects other organs, such as the brain, heart, or kidneys. There is another harmful fungus that belongs to the Aspergillus genus, namely, Aspergillus flavus. It is a type of filamentous fungus that thrives in tropical and subtropical climates. Along with allergic reactions, it induces invasive aspergillosis with severe respiratory issues and spread to other organs, resulting in a serious condition. One of the significant issues associated with Aspergillus flavus is its ability to produce aflatoxins. It is a group of carcinogenic toxic compounds that can contaminate food supplies and can pose severe health risks, including liver damage and cancer. Among the therapeutic categories, imidazole (a five-membered aromatic ring containing two nitrogen atoms with electron-rich characteristics) derivatives play the most versatile role in inhibiting microbial growth. Two adjacent nitrogen atoms present within the five-membered ring contain the ability to form hydrogen bonds. 16 The basicity contribution and hydrogen bond formation ability enable imidazole and its derivatives to interact effectively with proteins.17,18 Imidazole derivatives such as miconazole (imidazole linked with 2,4-dichlorophenyl oxy), econazole (imidazole linked with 2,4-dichlorobenzene and 4-chlorobenzyloxy), isoconazole (imidazole linked with 2,4-dichlorobenzene and 2,6-dichlorobenzyloxy), sulconazole (imidazole linked with 2,4-dichlorobenzene and 2,6-dichlorobenzylsulfanyl), ketoconazole (imidazole linked with 2,4-dichlorobenzene, dioxolan, and piperizine), clotrimazole (imidazole with diphenyl and 2-chlorophenyl), and metronidazole (imidazole with nitro group at 5 position and hydroxyethyl group) have proven their antimicrobial activity, antiparasitic activity to its ability to disrupt the cell membranes, inhibition of ergosterol synthesis, interfere with DNA and RNA synthesis in microbes. CHEMBL5276104 (amino imidazole with 1,2,3-triazole), CHEMBL5424433 (5-nitro imidazole with 1,2,4-oxadiazole), and HY-144255 (imidazole with pyrimidine, oxazole, and sulphonamide) showed good antibiofilm activity against Pseudomonas aeruginosa, antibacterial activity against Clostridioides difficile, and antibacterial activity against multidrug-resistant Klebsiella pneumoniae and Acinetobacter baumannii, respectively (Figure 1).19,20 In the structure of miconazole, econazole, isoconazole, sulconazole, ketoconazole, and clotrimazole, monochloro or dichlorobenzene plays an important role in antimicrobial activity. In the same manner, the structure of clotrimazole, CHEMBL5424433, and HY-144255, imidazole rings are directly linked with phenyl, 1,2,4-oxadiazole, and pyrimidine ring systems. These attachments play a pivotal role in antimicrobial activity. By understanding these structural features of antimicrobial imidazole derivatives, novel 1′H-[1,2′-biimidazol]-5(4H)-one (BP1-BP5) derivatives were developed as antibacterial and antifungal agents using insights from molecular docking, molecular dynamics simulation, and other computational approaches.

Imidazole derivatives with antimicrobial activity and background of design.
Results and discussion
Synthesis
Scheme 1 depicts the synthesis of 4,5-diphenyl-1H-Imidazol-2-amine by the reactions of benzoin and guanidine hydrochloride. The melting point of 4,5-diphenyl-1H-Imidazol-2-amine was recorded at 138°C. In the next step, different aromatic aldehydes (benzaldehyde/salisaldehyde/4-chlorobenzaldehyde/2/2-chlorobenzaldehyde/cinnamaldehyde) are reacted with hippuric acid to form the corresponding oxazolone derivative. Melting points of (4E)-4-benzylidene-2-phenyl-1,3-oxazol-5(4H)-one (4E)-4-[(2-hydroxyphenyl)methylidene]-2-phenyl-1,3-oxazol-5(4H)-one, (4E)-4-[(4-chlorophenyl)methylidene]-2-phenyl-1,3-oxazol-5(4H)-one, ((4E)-4-[(2-chlorophenyl)methylidene]-2-phenyl-1,3-oxazol-5(4H)-one, and ((4Z)-2-phenyl-4-[(2Z)-3-phenylprop-2-en-1-ylidene]-1,3-oxazol-5(4H)-one were 160±0.4°C, 170±0.4°C, 140±0.4°C, 152±0.4°C, and 170±0.4°C, respectively (Scheme 2). In the final step, different oxazolone derivatives were reacted with 4,5-diphenyl-1H-Imidazol-2-amine to form 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (Scheme 3). All the compounds were characterized by different spectroscopic and elemental analyses. Melting points of all the synthesized imidazole derivatives were 112°C, 95°C, 125°C, 130°C, and 125°C, respectively. Sharp melting points are indicative of the purity of the structures. Progression of reactions is monitored by TLC with n-hexane and ethyl acetate (7:3) ratio as the solvent system. Single spots are observed in the TLC plate reader. The FTIR analysis of the final compounds confirmed the presence of functional associated groups. In case of BP1, FTIR peaks are observed at 3149.82 cm−1, 1594.92 cm−1, 755.84 cm−1, 1672.77 cm−1, and 3370.28 cm−1, which are associated with CH str, C=C str, CH out of plane bending, C=O str, and NH str, respectively. In case of BP2, FTIR peaks are observed at 2929.53 cm−1, 1594.07 cm−1, 759.19 cm−1, 1675.73 cm−1, 3381.14 cm−1, and 1385.50 cm−1, which are associated with CH str, C=C str, CH out of plane bending, C=O str, NH str, and OH deformation, respectively. In case of BP3, FTIR peaks are observed at 3195 cm−1, 757.6 cm−1, 1695.2 cm−1, 694.4 cm−1, and 833.4 cm−1, which are associated with CH str, CH out of plane bending, C=O str, NH wagging, and C-Cl deformation, respectively. In case of BP4, FTIR peaks are observed at 1594.45 cm−1, 765.23 cm−1, 1674.73 cm−1, 3375.65 cm−1, and 833.85 cm−1, which are associated with C=C str, CH out of plane bending, C=O str, NH str, and C-Cl deformation, respectively. In case of BP5, FTIR peaks are observed at 3153.33 cm−1, 1592.13 cm−1, 755.36 cm−1, 1672.67 cm−1, and 3375.28 cm−1, which are associated with CH str, C=C str, CH out of plane bending, C=O str, and NH str, respectively. 1HNMR and 13CNMR spectral data of the synthesized compounds confirmed the position and nature of protons present in the structures. 1HNMR spectral data of BP1 observed a singlet at 8.042 ppm associated with the NH group of imidazole, and a multiplet near (6.1.00–7.993) ppm for the phenyl ring, and 13CNMR spectral data observed a singlet at 166.509 ppm for the ketone group and a multiplet within (127.263–139.755) ppm for the phenyl ring. 1HNMR spectral data of BP2 observed a singlet at 8.857 ppm for the NH group of imidazole, 6.085 ppm for the hydroxyl group, a multiplet near (7.226–8.029) ppm for the phenyl ring, and 13CNMR spectral data observed a singlet at 166.556 ppm for ketone group and multiplet within (127.278–139.763) ppm for phenyl ring. 1HNMR spectral data of BP3 observed a singlet at 8.857 ppm for the NH group of imidazole, 6.085 ppm for the hydroxyl group, a multiplet near (7.226–8.029) ppm for the phenyl ring, and 13CNMR spectral data observed a singlet at 171.414 ppm for ketone group and multiplet within (127.275–139.762) ppm for phenyl ring. 1HNMR spectral data of BP4 observed a singlet at 8.870 ppm for the NH group of imidazole, 7.332 ppm for the methylene group, a multiplet near (7.096–7.982) ppm for the phenyl ring, and 13CNMR spectral data observed a singlet at 171.405 ppm for ketone group and multiplet within (127.272–139.758) ppm for phenyl ring. 1HNMR spectral data of BP5 observed a singlet at 8.886 ppm for NH group of imidazole, multiplet (6.086–7.226) ppm for (=CH-CH=CH) group, multiplet near (7.250–7.979) ppm for phenyl ring, and 13CNMR spectral data observed a singlet at 171.430 ppm for ketone group and multiplet within (127.289–139.771) ppm for phenyl ring. Mass spectrometric data of (BP1-BP5) confirmed the presence of molecular ion peaks. Furthermore, elemental (CHN) analysis data confirmed the formation of compounds. Calculated and analyzed percentage CHN analysis data of BP1 showed the presence of carbon 79.73% and 78.92%; hydrogen 4.71% and 4.25%; and nitrogen 12.00% and 11.84%, respectively. Calculated and analyzed percentage CHN analysis data of BP2 showed the presence of carbon 77.09% and 76.82%; hydrogen 4.55% and 4.12%; and nitrogen 11.60% and 11.12%, respectively. Calculated and analyzed percentage CHN analysis data of BP3 showed the presence of carbon 74.25% and 73.77%; hydrogen 4.19% and 4.02%; and nitrogen 11.17 % and 10.67%, respectively. Calculated and analyzed percentage CHN analysis data of BP4 showed the presence of carbon 74.25% and 73.21%; hydrogen 4.19% and 3.85%; and nitrogen 11.17% and 10.21%, respectively. Calculated and analyzed percentage CHN analysis data of BP5 showed the presence of carbon 80.39% and 79.29%; hydrogen 4.87% and 4.14%; and nitrogen 11.36% and 10.61%, respectively. All spectral images are incorporated in the Supplementary file.

Synthesis of 4,5-diphenyl-1H-imidazol-2-amine (III) from benzaldehyde.

Synthesis of oxazolone derivatives (IX-XIII).
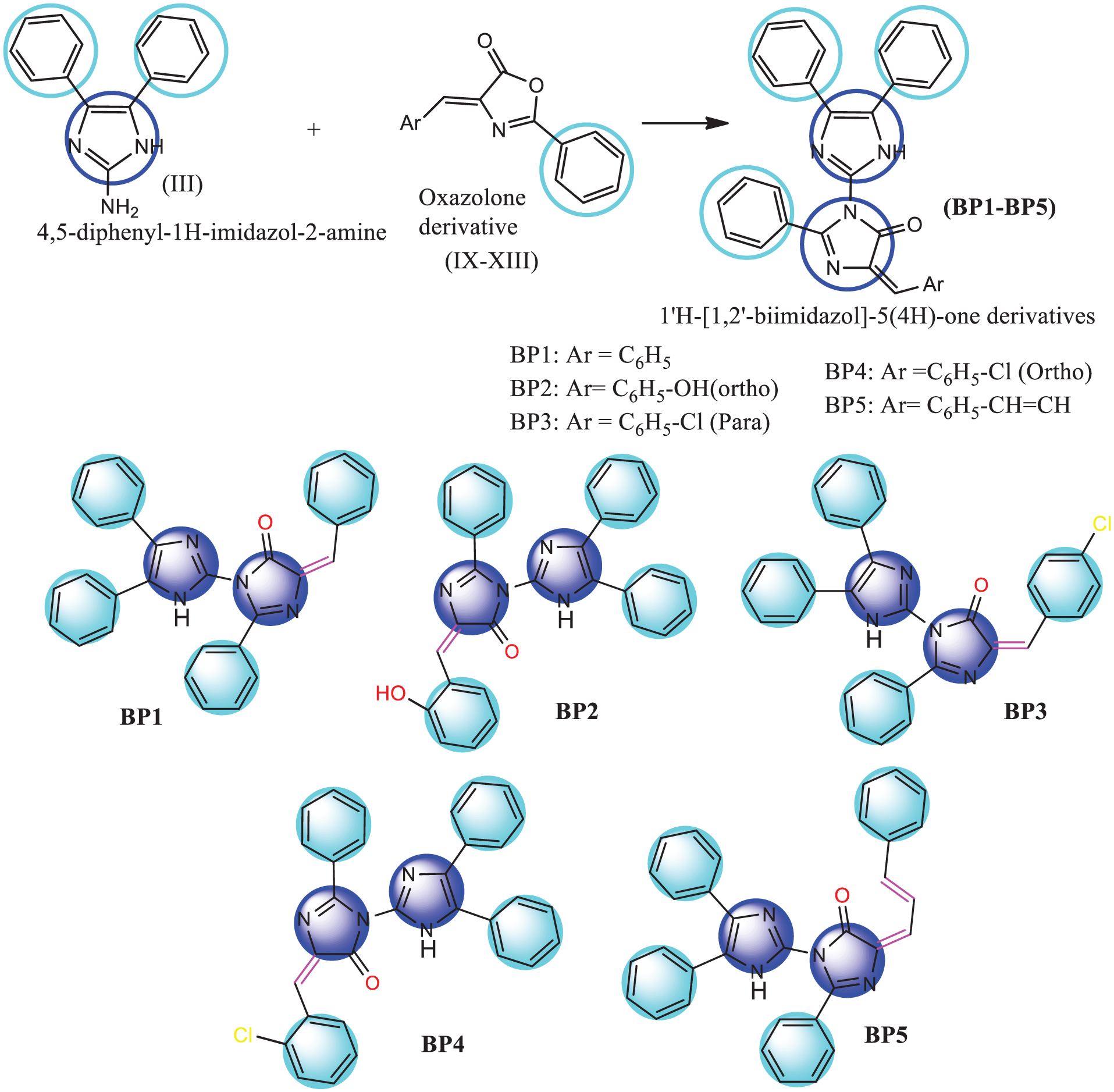
Synthesis of 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (
Molecular docking interaction
Molecular docking analysis of synthesized molecules (BP1-BP5) against both antibacterial and antifungal targets showed that BP1 and BP5 were the most interactive molecules. BP1 and BP5 expressed dock scores (kcal/mol) of -9.2, -8.7; -9.6, -9.5; -9.9, -9.8; -10.4, -10.1 with antibacterial targets 1KZN, 5TW8 and antifungal targets 1EAG, 3DJE respectively (Supplemental Table S1).21,22 In case of 1KZN BP1 interact to ASN46 with hydrogen bond and formed hydrophobic interactions with amino acids (ILE90, ILE78, PRO79, VAL 120). In the case of 1KZN, BP5 interacts with ASN46 with a hydrogen bond and forms hydrophobic interactions with amino acids (ARG76, ALA47, VAL167, ASP73, GLY117, ILE78). In the case of 5TW8, BP1 interacts with SER262 with a hydrogen bond and forms hydrophobic interactions with amino acids (PHE241, SER139, SER116, SER75, LYS78, ASN141, GLY181, GLU114, and ASP264). In the case of 5TW8 BP5, SER262 forms a hydrogen bond and forms hydrophobic interactions with amino acids (PHE241, SER139, SER116, SER75, LYS78, ASN141, GLY181, GLU114, ASP264, LEU115) (Figure 2). 23 In case of 1EAG BP1 showed hydrogen bond interactions with THR221, THR222 and hydrophobic interactions with amino acids (VAL12, ASP32, ASP218, GLY34, SER88, TYR84, GLY85, ILE305, ASP86, GLY220). In case of 1EAG BP5 showed hydrogen bond interaction with ASP86 and hydrophobic interactions with amino acids (THR221, TYR225, ASN301, ILE305, ALA303, GLY85, ASP2018, TYR84, ILE30, ILE123, ASP32). In case of 3DJE BP1 showed hydrogen bond interactions with GLY58, ARG112 and hydrophobic interactions with amino acids (VAL113, PHE258, LEU96, SER99, VAL54, LYS53, LYS368, GLY364, GLY366). In case of 1EAG BP5 showed three hydrogen bond interactions with LYS53, PHE367, GLY364 and hydrophobic interactions with amino acids (CYS337, ALA47, TRP236, GLU280, PHE264, LEU238, ARG365, LYS368, GLY366, ALA47, PHE258). Molecular docking interaction studies confirmed the well-docked features between ligand and receptors (Figure 3).24,25

Molecular docking interaction of BP1 with active sites on target proteins.

Molecular docking interaction of BP5 with active sites on target proteins.
Molecular dynamics simulation
RMSD analysis
Root Mean Square Deviation (RMSD) was used to measure the overall structural deviation of protein-ligand complexes by comparing atomic positions across different frames relative to the initial structure. This analysis provided insights into global stability, allowing the identification of significant conformational shifts over time. The RMSD plot revealed structural stability throughout the 200 ns simulation period, with the most stable complex exhibiting RMSD values ranging from 0.9 to 2.4 Å, indicating a well-equilibrated system with minimal deviations. The RMSD plot provides a detailed assessment of the structural stability and conformational behavior of 1KZN in both its unbound (apo) state and when complexed with BP1 and BP5 over a 200 ns molecular dynamics simulation. During the initial 10–20 ns, all three systems 1KZN (apo), 1KZN-BP1, and 1KZN-BP5exhibit noticeable fluctuations, which correspond to the equilibration phase where the structures adjust to the simulation environment. This phase is common in MD simulations as the system stabilizes after initial energy minimization and solvation effects. After equilibration, the unbound 1KZN (blue) stabilizes with RMSD values ranging from 1.2 to 1.8 Å, suggesting that the native conformation remains largely intact and serves as a reference for evaluating ligand-induced structural changes. When BP1 is introduced (red), the 1KZN-BP1 complex exhibits slightly higher RMSD fluctuations between 1.3 and 2.0 Å, indicating an increase in structural flexibility. This suggests that BP1 binding induces moderate adjustments in the protein structure, possibly due to the formation of new interactions that influence local conformational dynamics. Similarly, the 1KZN-BP5 complex (green) shows fluctuations ranging between 1.2 and 1.9 Å, following an intermediate trend between the unbound protein and the BP1-bound complex. This implies that BP5 binding does not introduce significant destabilization but may still cause minor conformational rearrangements. Throughout the remainder of the simulation (20–200 ns), all systems display stable RMSD values with minor oscillations, confirming that ligand binding does not cause major structural disruptions. The fact that RMSD remains below 2.5 Å in all cases is a strong indicator that 1KZN retains its global structural integrity, with only localized adjustments occurring upon ligand binding. These minor fluctuations could be attributed to side-chain flexibility, minor domain movements, or subtle alterations in protein-ligand interaction networks. The relatively stable RMSD trends suggest that BP1 and BP5 binding do not lead to large-scale unfolding or destabilization but rather induce targeted conformational modulations, which could be functionally relevant for ligand recognition or protein activity. The RMSD plot provides insights into the structural stability and conformational dynamics of the 5TW8 protein in its unbound (apo) form and when complexed with BP1 and BP5 over a 200 ns simulation. During the first 10–20 ns, all three systems, 5TW8 (blue), 5TW8-BP1 (orange), and 5TW8-BP5 (green), undergo an equilibration phase, as evidenced by initial fluctuations before reaching a stable trajectory. The RMSD values gradually increase and stabilize, indicating that the structures have equilibrated within the simulation environment. After 20 ns, the RMSD values for all systems remain within a low range of approximately 1.0–1.6 Å, suggesting that the 5TW8 protein maintains its structural stability throughout the simulation. The unbound 5TW8 (blue) stabilizes at around 1.3–1.6 Å, indicating that its native conformation is largely preserved. The 5TW8-BP1 complex (orange) exhibited slightly higher RMSD values, fluctuating between 1.2 and 1.6 Å, suggesting that BP1 binding introduces a minor increase in flexibility while maintaining overall stability. The 5TW8-BP5 complex (green) shows the lowest RMSD fluctuations, remaining between 1.0 and 1.3 Å, indicating that BP5 binding leads to a more rigid complex with reduced structural variations. Throughout the remainder of the simulation (20–200 ns), all three systems maintain stable RMSD values with minimal oscillations, confirming that ligand binding does not induce major conformational changes or destabilization. The fact that RMSD remains below 2.0 Å in all cases further reinforces that 5TW8 retains its overall structural integrity, with ligand-induced variations being localized rather than leading to large-scale unfolding or destabilization. Overall, these results indicate that both BP1 and BP5 binding have a stabilizing effect on 5TW8, with BP5 leading to a slightly more rigid complex, while BP1 introduces mild flexibility but does not compromise stability. The RMSD plot provides insights into the structural stability and conformational dynamics of the 1EAG protein in its unbound (apo) form and when complexed with BP1 and BP5 over a 200 ns simulation. During the initial 10–20 ns, all three systems (1EAG (blue), 1EAG-BP1 (orange), and 1EAG-BP5 (green) show fluctuations, reflecting the equilibration phase as the system adapts to the simulation conditions. The unbound 1EAG displays significant initial variations, reaching RMSD values above 3.0 Å before stabilizing, suggesting a higher degree of structural rearrangement during early equilibration. After 20 ns, the RMSD values for all systems stabilize with some minor fluctuations. The unbound 1EAG (blue) fluctuates between 2.0 and 3.5 Å, indicating moderate structural flexibility. The 1EAG-BP1 complex (orange) maintains lower RMSD values, ranging between 1.5 and 2.5 Å, suggesting that BP1 binding stabilizes the protein structure, potentially restricting its flexibility by forming stabilizing interactions. The 1EAG-BP5 complex (green) shows an intermediate RMSD trend, fluctuating between 2.0 and 3.0 Å, implying that BP5 binding does not significantly destabilize the protein but introduces some degree of structural adaptation. Overall, the lower RMSD values observed for the 1EAG-BP1 complex suggest that BP1 provides a stabilizing effect on the protein, potentially through stronger interactions that limit large conformational changes. Meanwhile, 1EAG-BP5 exhibits a moderate effect, indicating that its binding induces localized flexibility rather than global stabilization. The 1EAG (apo) structure remains the most flexible, with larger RMSD variations, reflecting its ability to explore more conformational space without ligand constraints. These findings indicate that BP1 and BP5 binding do not cause significant destabilization of 1EAG, with BP1 likely contributing to greater stabilization of the protein structure. The RMSD plot provides insights into the structural stability and conformational dynamics of the 3DJE protein in its unbound (apo) form and when complexed with BP1 and BP5 over a 200 ns simulation. During the first 10–20 ns, all three systems, 3DJE (blue), 3DJE-BP1 (orange), and 3DJE-BP5 (green), undergo an initial phase of fluctuations, corresponding to the equilibration period as the systems adjust to the simulation conditions. The RMSD values increase gradually before stabilizing, indicating that the structures reach equilibrium. After 20 ns, the RMSD values for all systems stabilize within a narrow range of approximately 1.5–2.5 Å, suggesting that the 3DJE protein remains structurally stable regardless of ligand binding.26,27 The unbound 3DJE (blue) shows relatively lower fluctuations, maintaining a steady RMSD around 1.5–2.0 Å, indicating that the protein retains its native conformation with minimal deviations. The 3DJE-BP1 complex (orange) exhibited slightly higher RMSD values, fluctuating between 1.7 and 2.5 Å, suggesting that BP1 binding induces some structural flexibility but does not destabilize the protein. The 3DJE-BP5 complex (green) follows a similar pattern, with RMSD values ranging between 1.6 and 2.3 Å, indicating that BP5 binding leads to minor conformational adjustments while maintaining overall structural stability. Throughout the remainder of the simulation (20–200 ns), all three systems maintain stable RMSD values with minimal oscillations, reinforcing that ligand binding does not induce significant conformational changes or destabilization. The fact that RMSD remains below 2.5 Å in all cases confirms that 3DJE retains its overall structural integrity, with ligand-induced variations likely reflecting localized binding site adjustments rather than large-scale unfolding or destabilization. Overall, these results suggest that both BP1 and BP5 binding have a modest impact on 3DJE’s structural flexibility, with no significant destabilization observed. BP1 appears to introduce slightly more flexibility compared to BP5, but both compounds remain within a stable conformational range. Based on the RMSD analysis across all three proteins, BP1 generally provides more consistent structural stabilization, with lower RMSD fluctuations compared to BP5, making it the more effective ligand in maintaining protein stability. The RMSD ligand plot reveals several key observations regarding ligand stability. BP1 exhibits more stable binding compared to BP5. During the initial 0–20 ns phase, BP1, like BP1_1KZN and BP1_3DJE, shows a steady rise in RMSD, stabilizing at 2–4 Å, suggesting strong and stable binding. In contrast, BP5, such as BP5_1KZN and BP5_1EAG, experience higher fluctuations (4–10 Å), indicating greater flexibility or weaker binding interactions. Over the 20–60 ns equilibration phase, BP1 maintains stable RMSD values, while BP5 continues to fluctuate, especially BP5_1KZN and BP5_1EAG, which exhibit persistent instability. The final 60–200 ns phase shows BP1 remaining consistently stable (~2–4 Å), reinforcing its strong binding, whereas BP5, especially BP5_5W18, shows significant RMSD increases, suggesting potential dissociation or major repositioning. The sharp rise in RMSD for BP5_1KZN around 95 ns hints at a final destabilization, possibly indicating partial unbinding or a conformational shift. Overall, BP1 is more stable throughout, while BP5 displays higher flexibility and potential instability, particularly in the later stages of the simulation (Figure 4). 28
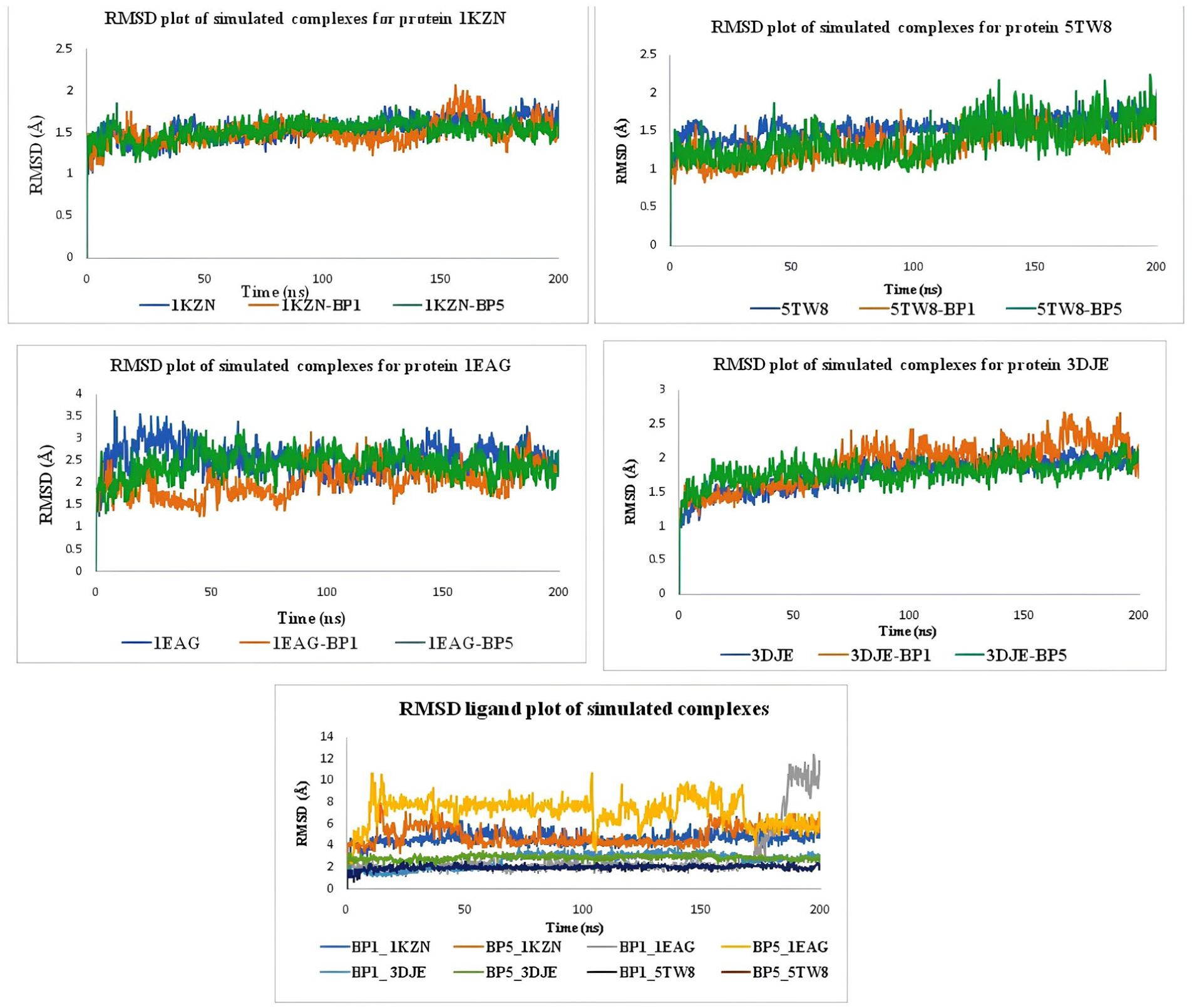
RMSD plot of BP1 and BP5 with 1KZN, 5TW8, 1EAG, and 3DJE.
RMSF analysis
Root Mean Square Fluctuation (RMSF) was employed to evaluate the flexibility of individual residues within the protein-ligand complexes. This analysis highlighted regions exhibiting significant conformational fluctuations, helping to differentiate between flexible and rigid segments of the complexes. RMSF analysis identified critical flexible and stable regions within the structures, providing a deeper understanding of dynamic conformational changes and interaction profiles. The RMSF plot of 1KZN provides a detailed view of residue-level flexibility across the simulated protein-ligand complexes (BP1 and BP5) compared to the native structure. The reference curve reflects the native dynamics of the protein, with most residues fluctuating between 0.5 and 1.5 Å, indicating overall stability. Two prominent peaks appear: one around residue 50, with RMSF values reaching 5 Å, likely corresponding to a flexible loop or unstructured region, and another peak around residue 100, with RMSF values around 4 Å, indicating additional flexibility in a potential binding or functional site. The BP1 complex follows the reference curve closely, with similar peaks around residues 50 and 100 but with slightly reduced fluctuations, suggesting that BP1 stabilizes these flexible regions without compromising overall protein dynamics. The BP5 complex shows slightly higher fluctuations, particularly around residues 50 and 100, where RMSF values exceed 4 Å, indicating that BP5 binding may introduce localized structural flexibility. Overall, both BP1 and BP5 influence flexibility at key regions, with BP1 stabilizing the protein, while BP5 induces more dynamic behavior in these flexible regions. Further analyses, such as interaction energy calculations, could provide deeper insights into the functional implications of these ligand-induced dynamics. 29 The RMSF plot of 1EAG provides a detailed view of residue-level flexibility across the simulated protein-ligand complexes (BP1 and BP5) compared to the native structure. The reference curve reflects the native dynamics of the protein, with most residues fluctuating between 0.5 and 2 Å, indicating general stability. Two distinct peaks appear: one around residue 50, with RMSF values reaching approximately 5 Å, likely corresponding to a flexible loop or unstructured region, and another increase around residues 250 and 300, with RMSF values around 4–5 Å, suggesting terminal flexibility or regions of functional importance. The BP1 complex follows the reference curve closely, with similar peaks around residues 50, 250, and 300 but slightly lower fluctuations, indicating minimal perturbation and partial stabilization of flexible regions. On the other hand, the BP5 complex shows slightly higher fluctuations, especially at residue 50, where RMSF exceeds 5 Å, along with increased fluctuations between residues 200–300 (around 3 Å), suggesting that BP5 may induce localized structural flexibility. Overall, BP1 appears to stabilize the protein structure with minor localized flexibility, while BP5 introduces more dynamic behavior, particularly in specific regions, which could indicate less stable binding or more flexible interactions. Further analyses, such as interaction energy calculations, could provide more detailed insights into the functional implications of these ligand-induced dynamics. The RMSF plot of 3DJE provides a detailed view of residue-level flexibility across the simulated protein-ligand complexes (BP1 and BP5) compared to the native structure. The reference curve reflects the native dynamics of the protein, with most residues fluctuating between 0.5 and 2 Å, indicating overall stability. Two distinct peaks appear: one around residue 100, with RMSF values reaching approximately 4 Å, likely corresponding to a flexible loop or binding site, and another around residues 290 and 300, with RMSF values around 4–5 Å, suggesting terminal flexibility or structural regions of functional importance. The BP1 complex shows similar fluctuation patterns to the native structure, with peaks around residues 100, 290, and 300, but with slightly lower RMSF values, indicating some stabilization in these flexible regions. In contrast, the BP5 complex exhibited higher fluctuations, especially at residues 100 and 290–300, where RMSF values exceed 4 Å, suggesting that BP5 induces more localized structural flexibility compared to BP1. Overall, both BP1 and BP5 interact with key flexible regions, with BP1 promoting more stability, while BP5 introduces more dynamic behavior, especially at the 100 and 290–300 regions, which could have implications for the protein’s functional state. Further analyses, such as interaction energy calculations, would help elucidate the effects of these dynamic changes. The RMSF plot of 5TW8 provides a detailed view of residue-level flexibility across the simulated protein-ligand complexes (BP1 and BP5) compared to the native structure. The reference curve reflects the native dynamics of the protein, with most residues fluctuating between 0.5 and 1.5 Å, indicating overall stability. Several prominent peaks appear: one around residue 50, with RMSF values reaching approximately 4–5 Å, suggesting flexibility in a loop or unstructured region; a second peak around residue 100, with RMSF values around 3.5–4 Å, likely corresponding to a flexible region or functional site; another peak around residue 200, with RMSF values around 3–3.5 Å, indicating additional flexibility; and a final peak around residues 290 and 300, with RMSF values reaching 4 Å, reflecting terminal flexibility or regions of functional importance (Figure 5).

RMSF plot of BP1 and BP5 with 1KZN, 5TW8, 1EAG, and 3DJE.
Analysis of interaction fraction histogram and residue-ligand contact numbers
The analysis of interaction fractions and residue-ligand contact numbers for BP1 and the various protein complexes (1KZN, 1EAG, 3DJE, and 5TW8) over 200 ns of molecular dynamics simulations reveals dynamic and complex binding patterns that contribute to the stability of the ligand-protein interactions. 30 Across all simulations, various stabilizing forces were observed, including hydrogen bonds, water-bridge networks, hydrophobic interactions, and occasional ionic contacts, which all play a crucial role in maintaining the conformational stability of the complexes. For BP1 in complex with 1KZN, stable hydrogen bonds with key residue ASN 46 and hydrophobic interactions are formed with residues like ARG 76, PRO 79, ILE 90, and ILE 78. In addition, water bridges interactions with the key residues ASN 46 and ASP 49 stabilize the complex during the simulation. These interactions maintain the structural integrity throughout the simulation, demonstrating stability during the simulation, with consistent contact over time, indicating a well-balanced and stable binding profile. Similarly, in the 1EAG complex, BP1 exhibits strong hydrogen bonding with significant residues such as THR 221, maintaining the consistency of the interaction, this residue shows stability during the simulation, with intense contact over time while also maintaining stable hydrophobic interactions with key residues TYR 84 and ILE 30, these residues maintain stable interactions throughout the simulation, ensuring sustained contact over time, supported by water-mediated interactions with residues GLY 85, this interaction profile suggests that BP1’s binding mode is stable and effective, as it minimizes structural deviations and maintains flexibility within key regions. For 3DJE, BP1 shows consistent hydrophobic bonding with residues like TRP 17, TRP 236, ALA 362, and PHE 237, as well as stable water bridges with the significant residue TRP 236. These interactions help maintain the structural stability of the protein, with BP1 providing a steady effect on the protein’s flexibility, preventing significant deviations during the simulation. In the 5TW8 complex, BP1 stabilizes the protein through consistent hydrogen bonds with residue ASP 28 and GLN 37, supported by hydrophobic contacts with residue TYR 121, resulting in a more rigid binding conformation. The lower RMSD values observed suggest that BP1 induces minimal structural perturbation and maintains the stability of the protein throughout the simulation. Overall, BP1 consistently induces stable binding in all four complexes, with hydrogen bonds, water bridges, and hydrophobic interactions contributing to the structural stability and conformational integrity of the protein-ligand complexes. BP1’s stabilizing effects make it effective at maintaining protein structure across a variety of protein environments. The interaction analysis of BP5 with the protein complexes 1KZN, 1EAG, 3DJE, and 5TW8 over a 200 ns molecular dynamics simulation highlights its dynamic binding patterns and stabilizing effects. Throughout the simulations, BP5 engages in key stabilizing interactions, including hydrogen bonds, water-bridge networks, hydrophobic interactions, and occasional ionic contacts, all of which contribute significantly to the structural integrity of the protein-ligand complexes. In the 1KZN complex, BP5 forms stable hydrophobic interactions with VAL 167 and PRO 79, and water bridges with ASN 46 and ASP 49, maintaining stability and consistent contact throughout the simulation. Similarly, in the 1EAG complex, BP5 interacts strongly through TYR 84 hydrogen bonding, while also forming hydrophobic interactions with TYR 84 and VAL 12, supported by water-mediated interactions with GLY 220 and VAL 12, which together reinforce the stability of the complex and maintain flexibility within key regions. In the 3DJE complex, BP5 establishes stable hydrophobic interactions with TRP 17, TRP 236, ALA 362, and PHE 237, supported by water bridges involving TRP 235 and ionic bonds with residue CYS 235, which help minimize conformational deviations and maintain overall protein stability. 31 Finally, in the 5TW8 complex, BP5 forms a hydrophobic interaction with PHE 225, GLN 229, and ALA 130, resulting in a binding conformation that is not maintained over time. Overall, BP5 consistently demonstrates a stabilizing effect across all four complexes, with hydrogen bonds, hydrophobic interactions, and water bridges playing a crucial role in maintaining structural stability and conformational integrity, reinforcing BP5’s potential as a stabilizing ligand across different protein environments (Figure 6).

Interaction histogram plot of BP1 and BP5 with 1KZN, 5TW8, 1EAG, and 3DJE.
DFT analyses
Frontier molecular orbital (FMO) analysis
HOMO and LUMO orbital energies (eV) of all the synthesized molecules (BP1-BP5) were -5.52, -5.38, -5.63, -5.46, -5.49, and -2.55, -2.50, -2.82, -2.63, -2.74, respectively. The HOMO and LUMO orbitals’ energy gap is associated with chemical strength and reactivity.31 HOMO and LUMO energy (eV) gaps of BP1, BP2, BP3, BP4, and BP5 were 2.97, 2.88, 2.81, 2.83, and 2.75, respectively. Table 1 includes the values for the energy gap (ΔE) between the HOMO and LUMO, softness, electronegativity, chemical hardness, and electrophilicity index. Among the synthesized molecules, BP5 is referred to as a soft molecule due to its minimal energy gap. A higher electrophilicity index is linked with higher chemical reactivity. BP3 showed maximum electrophilicity (Supplemental Figure S1). HOMO and LUMO orbitals of BP1, BP2, BP3, BP4, and BP5 focused on 4-methylidene-2,5′-diphenyl-1′H-[1,2′-biimidazol]-5(4H)-one, (5E)-5-benzylidene-2-phenyl-3,5-dihydro-4H-imidazol-4-one; 4′,5′-diphenyl-1′H-[1,2′-biimidazol]-5(4H)-one, (5E)-5-[(2-hydroxyphenyl)methylidene]-2-phenyl-3,5-dihydro-4H-imidazol-4-one; 4′,5′-diphenyl-1′H-[1,2′-biimidazol]-5(4H)-one, (5E)-5-[(4-chlorophenyl)methylidene]-2-phenyl-3,5-dihydro-4H-imidazol-4-one; 4′,5′-diphenyl-1′H-[1,2′-biimidazol]-5(4H)-one, (5E)-5-[(2-chlorophenyl)methylidene]-2-phenyl-3,5-dihydro-4H-imidazol-4-one; and (E)-4′,5′-diphenyl-4-((E)-3-phenylallylidene)-1′H-[1,2′-biimidazol]-5(4H)-one, (5E)-2-phenyl-5-[(2E)-3-phenylprop-2-en-1-ylidene]-3,5-dihydro-4H-imidazol-4-one, respectively. 32
FMO analysis of the synthesized molecules (BP1-BP5).

Molecular electrostatic potential (MEP) analysis
Different colors in the electrostatic map represent different zones in the structure, such as red color linked with electrophilic behavior, blue color linked with nucleophilic, and green color associated with the neutral zone. In BP1, BP2, and BP5: 1′H-[1,2′-biimidazol]-5(4H)-one, and some parts of the phenyl ring system are responsible for nucleophilic and electrophilic attack, respectively. In BP3: 1’H-[1,2′-biimidazol], 4-chlorophenyl, and some parts of the phenyl ring system are responsible for nucleophilic and electrophilic attack, respectively. In BP4: imidazole, 2-chlorophenyl, and some parts of the phenyl ring system are responsible for nucleophilic and electrophilic attack, respectively (Supplemental Figure S2). 33
Antibacterial activity
Antibacterial activity of 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (BP1-BP5) and standard ciprofloxacin against Bacillus subtilis (gram +ve), Staphylococcus aureus (gram +ve), Escherichia coli (gram -ve), and Salmonella typhi (gram -ve) was shown in Supplemental Table S2. BP1 showed inhibitions against all four bacterial strains with zone of inhibition values of 22 mm, 19 mm, 24 mm, and 26 mm, respectively. BP2 showed inhibitions against three bacterial strains (Bacillus subtilis, Staphylococcus aureus, and Salmonella typhi) with zone of inhibition values of 5 mm, 8 mm, and 12 mm, respectively. BP3 showed inhibitions against only two bacterial strains (Staphylococcus aureus and Escherichia coli) with zone of inhibition values of 3 mm and 9 mm, respectively. 34 BP4 showed inhibitions against three bacterial strains (Bacillus subtilis, Staphylococcus aureus, and Salmonella typhi) with zone of inhibition values of 5 mm, 8 mm, and 12 mm, respectively. BP5 also showed inhibitions against all four bacterial strains with zone of inhibition values of 24 mm, 25 mm, 23 mm, and 25 mm, respectively. Standard ciprofloxacin showed good bacterial growth inhibition with zone of inhibition values of 29 mm, 27 mm, 28 mm, and 29 mm, respectively. In addition to antibacterial screening, MIC and MBC values of the two most effective molecules (BP1 and BP5) were assessed. MIC values of BP1 and BP5 against Bacillus subtilis, Staphylococcus aureus, Escherichia coli, and Salmonella typhi were 2.5µg/mL, 3.0 µg/mL, 4.5 µg/mL, 3.5 µg/mL; and 3.0 µg/mL, 2.0 µg/mL, 4.0 µg/mL, 3.0 µg/mL, respectively. MBC values of BP1 and BP5 against Bacillus subtilis, Staphylococcus aureus, Escherichia coli, and Salmonella typhi were 10.0 µg/mL, 10.0 µg/mL, 10.0 µg/mL, 12.0 µg/mL; and 12.0 µg/mL, 8.0 µg/mL, 8.0 µg/mL, 10.0 µg/mL, respectively. MIC and MBC values of standard ciprofloxacin were 0.25 µg/mL, 0.5 µg/mL, 0.5 µg/mL, 0.5 µg/mL; and 1.5 µg/mL, 2.0 µg/mL, 2.0 µg/mL, 2.0 µg/mL, respectively. Antibacterial activity confirmed that synthesized molecules showed moderate activity against Bacillus subtilis and Staphylococcus aureus bacterial strains, as compared to standard ciprofloxacin (Figure 7 and Supplemental Table S3).35,36

MIC and MBC of BP1, BP5 and ciprofloxacin against different bacterial strains.
Antifungal activity
Antifungal activity of 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (BP1-BP5) and standard nystatin against Aspergillus niger and Aspergillus flavus were shown in Supplemental Table S4. Fixed concentrations of BP1, BP2, BP3, BP4, and BP5 showed percent inhibitions of fungal mycelia growth with 65.62, 38.44, 62.95, 28.22, and 68.25, respectively, against Aspergillus niger. In the same manner, BP1, BP2, BP3, BP4, and BP5 showed percent inhibitions of fungal mycelia growth with 63.30, 39.58, 65.65, 32.85, and 69.45, respectively, against Aspergillus flavus. Reference molecule nystatin showed percent inhibitions of 88.42 and 85.10 against Aspergillus niger and Aspergillus flavus, respectively. All synthesized molecules showed good antifungal activity in terms of mycelia growth inhibition and among them BP5 (E)-2,4′,5′-triphenyl-4-((E)-3-phenylallylidene)-1′H-[1,2′-biimidazol]-5(4H)-one), BP1 ((E)-4-benzylidene-2,4′,5′-triphenyl-1′H-[1,2′-biimidazol]-5(4H)-one), and BP3 ((E)-4-(4-chlorobenzylidene)-2,4′,5′-triphenyl-1′H-[1,2′-biimidazol]-5(4H)-one) showed good antifungal activities as compare to standard nystatin (Figure 8). Presence of ethenylbenzene, benzene, and 4-chlorobenzene group along with 4-methylidene-2,4′,5′-triphenyl-1′H-[1,2′-biimidazol]-5(4H)-one creates a positive impact on fungal growth. 37

Percent inhibition of fungal mycelial growth of synthesized molecules and nystatin.
Based on the structural features of miconazole, econazole, isoconazole, sulconazole, ketoconazole, clotrimazole, metronidazole CHEMBL5276104, CHEMBL5424433, and HY-144255, newer generation 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (BP1-BP5) were designed. Imidazole-linked imidazolone is the core of the synthesized compounds. Designed compounds were synthesized using basic organic chemistry reactions targeting antimicrobial efficacies. Then all the compounds were docked with two antibacterial (1KZN, 5TW8) and two antifungal (1EAG, 3DJE) receptors, respectively. BP1 and BP5 showed good docking interactions with both antibacterial and antifungal receptors. Phenyl and ethenylbenzene of BP1 and BP5 create a positive impact on bioactivity. 2-hydroxyphenyl, 4-chlorophenyl, and 2-chlorophenyl of BP2, BP3, and BP4 create a moderate effect on antimicrobial activity (Figure 9). Antimicrobial activity is well backed up by molecular docking, MD simulation, and DFT analysis.

SAR of synthesized of 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (BP1-BP5).
Conclusion
Novel 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (BP1-BP5) were synthesized by reacting 4,5-diphenyl-1H-imidazol-2-amine with various 1,3-oxazol-5(4H)-one derivatives and evaluated for their antibacterial and antifungal activities. BP1 and BP5 showed significant antibacterial effects against Bacillus subtilis, Staphylococcus aureus, Escherichia coli, and Salmonella typhi, with MIC values ranging from 2.0 to 4.5 µg/mL. In contrast, ciprofloxacin had MIC values of 0.25 to 0.5 µg/mL. The MBC values for BP1 and BP5 were higher than those for ciprofloxacin, indicating effective bactericidal properties. For antifungal activity, BP1, BP2, BP3, BP4, and BP5 inhibited the growth of Aspergillus niger and Aspergillus flavus, with BP5 achieving the highest inhibition rates (68.25% and 69.45%). In comparison to the standard nystatin, BP5, BP1, and BP3 demonstrated commendable antifungal efficacy. Molecular docking studies indicated that BP1 and BP5 were the most effective compounds, and MD simulations confirmed their structural integrity. DFT analysis revealed that BP5 is a soft molecule with a minimal energy gap, while BP3 showed maximum electrophilicity. Overall, these findings support the potential of the synthesized derivatives as effective antibacterial and antifungal agents.
Experimental
Synthesis
A solvent system consisting of n-hexane and ethyl acetate in a 7:3 ratio was employed to monitor the progression of the reaction. The melting points were measured using an EI digital melting point apparatus. For spectral analysis, a PerkinElmer FTIR spectrophotometer was utilized for FTIR measurements, while a Bruker Avance DRX300 300MHz FT-NMR spectrometer was used for NMR analysis. In addition, mass spectrometric analysis of the synthesized molecules was conducted using a Waters Alliance e2695/HPLC-TQD Mass spectrometer. Elemental analysis was performed with a Eurovector E 3000 instrument.
Synthesis of 2-hydroxy-1,2-diphenylethan-1-one (benzoin) (II) from benzaldehyde (I)
Benzaldehyde (0.047 M, 5.0 g), thiamine hydrochloride (0.003 M, 1.0 g), 4.0 g of water, and 6.0 g of ethanol were mixed well in a beaker and kept in the refrigerator for 30 minutes. Sodium hydroxide (0.047 M, 1.88 g) was mixed with 8.0 ml of water, and the mixture was refrigerated for 30 minutes. Dropwise sodium hydroxide solution was added to the benzaldehyde solution on a magnetic stirrer until the pH of the solution was near 9.0. The final solution was kept at 65°C for 3 h. Finally, the solution was kept in a refrigerator for 24 h to achieve crystallization.
Synthesis of 4,5-diphenyl-1H-imidazol-2-amine (III) from 2-hydroxy-1,2-diphenylethan-1-one (Benzoin) (II)
In a round-bottom flask, benzoin (II) (0.01 M, 2.12 g), guanidine hydrochloride (0.01 M, 0.95 g), and 15 mL of dimethyl formamide were mixed well and refluxed under a water bath for 3 h. The final product was collected by pouring the reaction mixture into ice-cold water
Synthesis of oxazolone derivatives (IX-XIII) from aromatic aldehyde derivatives (IV-VIII)
In a series of round bottom flasks, different substituted aromatic aldehydes (benzaldehyde/salisaldehyde/4-chlorobenzaldehyde/2-chlorobenzaldehyde/cinnamaldehyde) (IV-VIII) (0.02M), benzoyl glycine/hippuric acid (0.02 M), and fused sodium acetate (0.02 M) were added and mixed with glacial acetic acid (0.06 M) and refluxed under water bath for 3 h, followed by addition of ethanol (10 ml). Then the solution was stored overnight in the refrigerator. Furthermore, the product was collected by filtration and kept for further reaction (Scheme 2). 38
Synthesis of (E)-4-benzylidene-2,4′,5′-triphenyl-1′H-[1,2′-biimidazol]-5(4H)-one (BP1)
4,5-diphenyl-1H-imidazol-2-amine (III, 0.01 M), (4E)-4-benzylidene-2-phenyl-1,3-oxazol-5(4H)-one (IX, 0.01 M) dissolved in glacial acetic acid and fused sodium acetate. Then the solution was refluxed for 6–7 h, followed by cooling the solution at room temperature. The final product was collected by adding the solution to crushed ice, resulting in a white precipitate, and then recrystallized from ethanol to give final BP1.
Yield= 43.24%, m.p. (melting point): 112±0.4°C. Off white crystalline powder. Rf: 0.38. FT-IR (KBr) cm−1: 3149.82 (CH str), 1594.92 (C=C str), 755.84 (CH out of plane bending), 1672.77 (C=O str), 3370.28 (NH str). 1HNMR DMSO (300 MHz) δ ppm: δ 8.042 (s, 1H, NH), δ (6.1.00–7.993) (m, Ar-H). 13CNMR DMSO (300 MHz) δ ppm: 166.509 (C=O group), (127.263–139.755) (Aromatic ring). EIMS (%): 466 m/z (100%), and 167 m/z (50%). Molecular formula: C31H22N4O (M. Wt: 466.53 g/mol). Calculated C: 79.73% H: 4.71% N: 12.00% Analyzed C: 78.92% H: 4.25% N: 11.84%.
Synthesis of (E)-4-(2-hydroxybenzylidene)-2,4′,5′-triphenyl-1′H-[1,2′-biimidazol]-5(4H)-one (BP2)
4,5-diphenyl-1H-imidazol-2-amine (III, 0.01 M), (4E)-4-[(2-hydroxyphenyl)methylidene]-2-phenyl-1,3-oxazol-5(4H)-one (X, 0.01 M) dissolved in glacial acetic acid and fused sodium acetate. Then the solution was refluxed for 6–7 h, followed by cooling the solution at room temperature. The final product was collected by adding the solution to crushed ice, resulting in a white precipitate, and then recrystallized from ethanol to give final BP2.
Yield= 45.83%, m.p. (melting point): 95±0.4°C. Rf: 0.45. FT-IR (KBr) cm−1: 2929.53 (CH str), 1594.07 (C=C str), 759.19 (CH out of plane bending), 1675.73 (C=O str), 3381.14 (NH str), 1385.50 (OH def). 1HNMR DMSO (300 MHz) δ ppm: δ 8.857 (s, 1H, NH), δ 6.085 (s, 1H, OH), δ (7.226–8.029) (m, Ar-H). 13CNMR DMSO (300 MHz) δ ppm: 166.556 (C=O group), (127.278–139.763) (Aromatic ring). EIMS: 483 m/z (100%) and 414 m/z (50%). Molecular formula: C31H22N4O2 (M. Wt: 482.53 g/mol). Calculated C: 77.09% H: 4.55% N: 11.60% Analyzed C: 76.82% H: 4.12% N: 11.12%.
Synthesis of (E)-4-(4-chlorobenzylidene)-2,4′,5′-triphenyl-1′H-[1,2′-biimidazol]-5(4H)-one (BP3)
4,5-diphenyl-1H-imidazol-2-amine (III, 0.01 M), (4E)-4-[(4-chlorophenyl)methylidene]-2-phenyl-1,3-oxazol-5(4H)-one (XI, 0.01 M) dissolved in glacial acetic acid and fused sodium acetate. Then the solution was refluxed for 6–7 h. The solution was kept at room temperature. The final product was collected by adding the solution to crushed ice, resulting in a white precipitate, and then recrystallized from ethanol to give final BP3.
Yield= 39.25%, m.p. (melting point): 125±0.4°C. Rf: 0.38 FT-IR (KBr) cm−1: 3195 (CH str), 757.6 (CH out of plane bending), 1695.2 (C=O str), 694.4 (NH wagging), 833.4 (C-Cl def). 1HNMR DMSO (300 MHz) δ ppm: δ 8.866 (s, 1H, NH), δ 7.335 (-CH group), δ (7.404–8.005) (m, Ar-H). 13CNMR DMSO (300 MHz) δ ppm: 171.414 (C=O group), (127.275–139.762) (Aromatic ring). EIMS: 500 m/z (100%), and 414 m/z (50%). Molecular formula: C31H21ClN4O (M. Wt: 500.97 g/mol). Calculated C: 74.25% H: 4.19% N: 11.17% Analyzed C: 73.77% H: 4.02% N: 10.67%.
Synthesis of (E)-4-(2-chlorobenzylidene)-2,4′,5′-triphenyl-1′H-[1,2′-biimidazol]-5(4H)-one (BP4)
4,5-diphenyl-1H-imidazol-2-amine (III, 0.01 M), ((4E)-4-[(2-chlorophenyl)methylidene]-2-phenyl-1,3-oxazol-5(4H)-one (XII, 0.01 M) dissolved in glacial acetic acid and fused sodium acetate. After 6–7 hours of reflux, the solution was kept at room temperature. The final product was collected by adding the solution to crushed ice, which resulted in a white precipitate. This precipitate was then recrystallized from ethanol to yield the final product, BP4.
Yield= 40.84%, m.p. (melting point): 130±0.4°C. Rf: 0.42. FT-IR (KBr) cm−1: 1594.45 (C=C str), 765.23 (CH out of plane bending), 1674.73 (C=O str), 3375.65(NH str), 833.85 (C-Cl def). 1HNMR DMSO (300 MHz) δ ppm: δ 8.870 (s, 1H, NH), δ 7.332 (-CH group), δ (7.096–7.982) (m, Ar-H). 13CNMR DMSO (300 MHz) δ ppm: 171.405 (C=O group), (127.272–139.758) (Aromatic ring). EIMS: 500 m/z (100%), and 414 m/z (50%). C31H21ClN4O (M. Wt: 500.97 g/mol). Calculated C: 74.25% H: 4.19% N: 11.17% Analyzed C: 73.21% H: 3.85% N: 10.21%.
Synthesis of (E)-2,4′,5′-triphenyl-4-((E)-3-phenylallylidene)-1′H-[1,2′-biimidazol]-5(4H)-one (BP5)
4,5-diphenyl-1H-imidazol-2-amine (III, 0.01 M), (4Z)-2-phenyl-4-[(2Z)-3-phenylprop-2-en-1-ylidene]-1,3-oxazol-5(4H)-one (XIII, 0.01 M) dissolved in glacial acetic acid and fused sodium acetate. After refluxing for 6–7 hours, the solution was allowed to cool to room temperature. The final product was obtained by adding the solution to crushed ice, which resulted in a white precipitate. This precipitate was then recrystallized from ethanol to yield the final product, designated as BP5 (Scheme 3). 39
Yield= 48.23%, m.p. (melting point): 125±0.4°C. Rf: 0.32. FT-IR (KBr) cm−1: 3153.33 (CH str), 1592.13 (C=C str), 755.36 (CH out of plane bending), 1672.67 (C=O str), 3375.28 (NH str). 1HNMR DMSO (300 MHz) δ ppm: δ 8.865 (s, 1H, NH), δ (6.086–7.226) (=CH-CH=CH group), δ (7.250–7.979) (m, Ar-H). 13CNMR DMSO (300 MHz) δ ppm: 171.430 (C=O group), (127.289–139.771) (Aromatic ring). EIMS: 493 m/z (100%), and 414 m/z (50%). C33H24N4O (M. Wt: 492.56 g/mol). Calculated C: 80.39% H: 4.87% N: 11.36% Analyzed C: 79.29% H: 4.14% N: 10.61%. 40
Molecular docking studies
In the present study, molecular docking simulations were conducted using AutoDock Vina software to investigate the binding interactions and modes of the selected compounds with target proteins associated with antibacterial and antifungal activities. 41 The focus was on understanding the mechanisms of interaction between the compounds and the selected proteins to assess their potential as therapeutic agents for bacterial and fungal infections. The objective was to identify promising candidates for targeting antibacterial and antifungal pathways. 42
Selection of protein
In this manuscript, 1KZN and 5TW8 receptors belong to antibacterial targets. 1KZN and 5TW8 receptors were associated with crystal structures of E. coli complexed with clorobiocin and wild-type S. aureus penicillin binding protein 4 (PBP4) in complex with ceftaroline, respectively. 1EAG and 3DJE receptors belonged to antifungal targets. 1EAG and 3DJE receptors were associated with aspartic proteinase (SAP2) from Candida albicans complexed with A70450 (N-ethyl-N-[(4-methylpiperazin-1-yl)carbonyl]-D-phenylalanyl-N-[(1S,2S,4R)-4-(butylcarbamoyl)-1-(cyclohexylmethyl)-2-hyd roxy-5-methylhexyl]-L-norleucinamide) and fructosamine oxidase from Aspergillus fumigatus (Amadoriase II) in complex with 1-S-(carboxymethyl)-1-thio-beta-D-fructopyranose, respectively. The target proteins involved in antibacterial and antifungal activities were retrieved in PDB format from the RCSB Protein Data Bank (PDB) and visualized using BIOVIA Discovery Studio (Supplemental Table S5). 43
Protein preparation
The proteins were then processed with the addition of polar hydrogen atoms, Kollman, and gasteiger charges. Finally, the structure of the protein was saved in PDBQT format. During the process, the deviation in coordinates was rectified by energy minimization using Swiss PDB viewer (SPDBV 4.1.0, Swiss Institute of Bioinformatics). The energy-minimized protein in pdb format was then subjected to the Python Molecular Viewer. Afterward, bond orders were assigned, polar and missing hydrogens were merged with the inclusion of partial Gasteiger atomic charges. The missing hydrogens and protonation states were determined by the H++ server. Finally, to make the docking software compatible, all the atoms in the protein were made to Autodock4 type (t), and the pdb file of the protein was converted to pdbqt, where q defines the charge and t for Autodock4 type. 44
Ligand preparation
The selected compounds (BP1-BP5) were prepared using Chem3D and optimized with Avogadro software, employing its auto-optimization tool under the MMFF94 force field and the steepest descent algorithm. The optimized ligand and protein structures were then converted into the PDBQT format for molecular docking simulations. AutoDock Vina was used to explore the conformational space of ligand-protein binding. The docking scores obtained for the compounds were compared with reference molecules known for their antibacterial and antifungal activity. 45
Validation of molecular docking
Validation is a crucial step in ensuring the reliability and reproducibility of the docking process. This is achieved by re-docking the ligand that occupies the active site. To do this, the active site of the receptor was cleared by removing the complexed ligand from its structure, and then the ligand was re-docked into the active site of the protein. The root mean square deviation (RMSD) between the re-docked conformation and the original crystallographic conformation of the compound was found to be less than 2.0 Å. This result confirms the reliability of the docking method in accurately reproducing the experimentally observed binding mode for the receptor. 46
Molecular dynamics simulation
The top 2 docked molecules (BP1 and BP5) were subjected to molecular dynamics (MD) simulations to evaluate their stability and dynamic behavior within their respective target complexes. The Desmond module (Schrödinger) was employed for 200 ns MD simulations, allowing for an in-depth analysis of the structural evolution over time. 47 The systems were solvated using the TIP3P water model within an orthorhombic water box to ensure proper hydration and structural accuracy. Na+/Cl− ions were introduced to maintain electrostatic neutrality, and the OPLS3e force field was applied for accurate molecular interaction modeling. Temperature and pressure were controlled at 300°K and 1 ATM using the Nose–Hoover chain thermostat and isotropic pressure scaling barostat to maintain stable thermodynamic conditions. 48 Throughout the simulations, interaction histograms, RMSD, and RMSF analyses were conducted to assess complex stability and flexibility. 49 These histograms provided insights into the persistence of non-covalent interactions such as hydrogen bonds, hydrophobic interactions, and salt bridges. The analysis helped identify key residues contributing to binding affinity, confirming the dynamic stability and potential efficacy of the investigated compounds. 50
Density functional theory analyses
Frontier molecular orbital (FMO) analysis
Electronic behavior of all the synthesized molecules (BP1-BP5) was determined by Beck’s (B) three-parameter hybrid model and Lee, Yang, and Parr’s (LYP) correlation functional under the B3LYP/6-31G (d, p) basis set. Distance between the highest occupied and lowest unoccupied molecular orbital energies associated with softness, electronegativity, hardness, and electrophilicity properties of the structures. GAMESS software and WxMacMolPlt (version 7.7.3) were used for FMO analysis. 51
Chemical hardness:
Molecular electrostatic potential (MEP) analysis
MEP map analysis all the synthesized molecules (BP1-BP5) was also performed by Beck’s (B) three-parameter hybrid model and Lee, Yang, and Parr’s (LYP) correlation functional B3LYP/6-31G (d, p) basis set to identify electrophilic and nucleophilic attack zone using GAMESS software (version R2 released on June 30, 2024). Different colors in the electrostatic map represent different zones in the structure, such as red color linked with electrophilic behavior, blue color linked with nucleophilic, and green color associated with the neutral zone. 52
Antimicrobial screening of synthesized molecules (BP1-BP5)
All synthesized molecules (BP1-BP5) were screened for antimicrobial properties against four bacterial strains (Bacillus subtilis (gram +ve), Staphylococcus aureus (gram +ve), Escherichia coli (gram -ve), and Salmonella typhi (gram -ve) and two fungal strains (Aspergillus niger and Aspergillus flavus).
In vitro antibacterial activity of synthesized molecules (BP1-BP5)
The disk diffusion method was used to evaluate the synthesized compounds (BP1-BP5) for in vitro antibacterial activities, using ciprofloxacin as a reference. The bactericidal tests were conducted on standard nutritional agar medium. Bacterial stock cultures were purchased from IMTECH, Chandigarh, and kept at 4°C in nutrient agar slants. To create active cultures for testing, cells from the stock cultures were transferred using a sterile loop into test tubes filled with nutritional agar media. To encourage bacterial development, the injected test tubes were subsequently incubated for 24 hours at 37°C under static conditions, that is, without agitation. To reach a consistent optical density of roughly 2.0 x 10⁶ colony-forming units (CFU) per milliliter, the bacterial cultures were diluted with nutrient broth after incubation. 53
Assessment of MIC and MBC of selected synthesized molecules
Different dilutions of two synthesized molecules (BP1 and BP5) and standard ciprofloxacin were evaluated for minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) using the micro broth dilution method against the selected bacterial microorganism. The Clinical and Laboratory Standards Institute (CLSI) method was used to determine MIC and MBC.
Screening of mycelium growth of synthesized molecules (BP1-BP5)
In vitro antifungal assessment of 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (BP1-BP5) and standard nystatin were evaluated against two fungal strains. 54 The investigation was based on the poisoned food technique. In vitro antifungal assessment of 1′H-[1,2′-biimidazol]-5(4H)-one derivatives (BP1-BP5) and standard nystatin were evaluated against two fungal strains. 54 The investigation was based on the poisoned food technique. 55
Supplemental Material
sj-docx-1-chl-10.1177_17475198251378174 – Supplemental material for Efficient synthesis of new 1′H-[1,2′-biimidazol]-5(4H)-one derivatives as antibacterial and antifungal agents with characterization, molecular docking, MD simulation, DFT analysis and in vitro bioactivity
Supplemental material, sj-docx-1-chl-10.1177_17475198251378174 for Efficient synthesis of new 1′H-[1,2′-biimidazol]-5(4H)-one derivatives as antibacterial and antifungal agents with characterization, molecular docking, MD simulation, DFT analysis and in vitro bioactivity by Supriyo Saha, Farheen Parveen, Mohit Bisht, Abul Hasan, Mazen Almehmadi, Mamdouh Allahyani, Mohammed A Alshamrani, Lamiae ElBouamri, Samir Chtita and Vikash Jakhmola in Journal of Chemical Research
Footnotes
Acknowledgements
The authors extend their appreciation to Taif Unviversity, Saudi Arabia, for supporting this work through project number (TU-DSPP-2024-31).
Consent to participate
All the authors are consent for participations.
Consent for publication
All the authors are given consent for publication.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Taif University, Saudi Arabia, Project No. (TU-DSPP-2024-31).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data are available in Supplementary file.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.