
Abstract
Background:
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease (Mpro) is a key target for drug development in the fight against COVID-19 and computer-aided drug design (CADD) offers a promising way to discover potential drugs.
Aim:
Building on our team’s previous efforts against COVID-19, we have carefully curated a set of 80 semi-synthetic compounds. These compounds were selected matching the key pharmacophoric features of SARS-CoV-2 Mpro inhibitors.
Objective:
The aim of this study is to identify the most effective inhibitors of SARS-CoV-2 Mpro using a CAAD approach.
Method:
We employed a comprehensive approach, subjecting the selected compounds to molecular docking against Mpro (PDB ID: 6LU7). We then conducted thorough absorption, distribution, metabolism, excretion, and toxicity (ADMET) evaluations and toxicity assessments to determine the drug-likeness of the selected compounds. Further substantiation was provided by meticulous density functional theory (DFT) investigations and molecular dynamics (MD) simulations at 150 nanoseconds to study the electronic properties and stability of the complexes formed by the most promising candidates.
Result:
Molecular docking analysis revealed favorable binding modes and robust free energy profiles for compounds
Conclusion:
Our study designates compounds
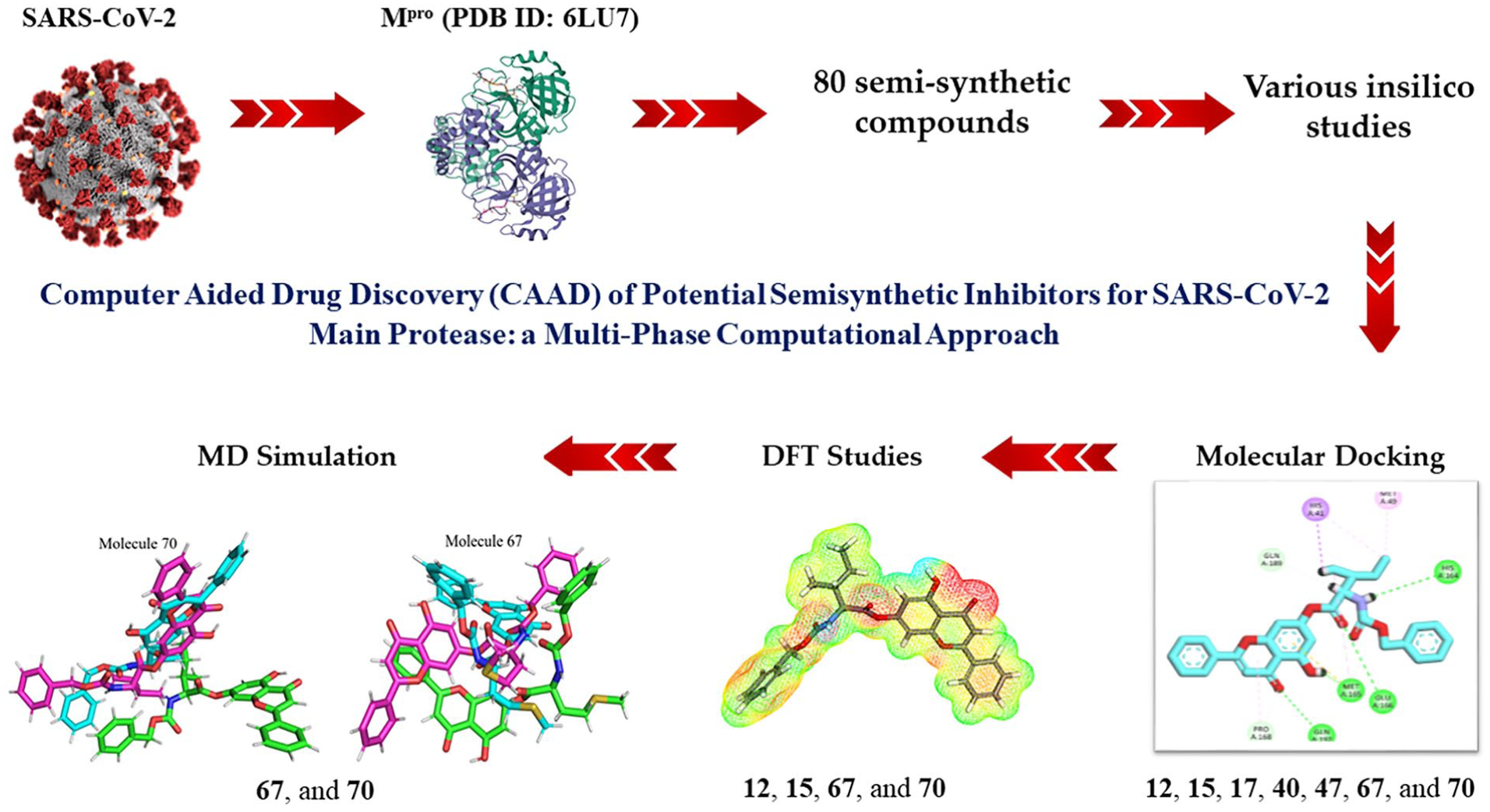
Introduction
In the past 20 years, computational (in silico) chemistry has become an essential instrument in the design and evaluation of pharmacological agents. 1 This increasing significance arises from several critical developments. The accurate determination of three-dimensional structures for various protein targets in the human body has been accomplished. 2 Second, notable advancements have occurred in computational software and hardware technologies. 3 Third, there has been an enhanced comprehension of structure-activity relationships (SAR) and quantitative structure-activity relationships (QSAR). 4 Consequently, in silico methods have been essential in investigating pharmacokinetic and pharmacodynamic properties, facilitating the identification of correlations between chemical structures and their biological activities. The approaches encompass QSAR modeling, 5 pharmacophore mapping, 6 homology modeling, 7 molecular docking,8,9 molecular dynamics (MD) simulations, 10 absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiling,11,12 and density functional theory (DFT) calculations. 13
Almost one-third of the Food and Drug Administration (FDA)-approved drugs are either based on or derived from natural sources in the time range of (1981–2014). 14 Utilizing semi-synthesis on natural products with the goal of producing diverse analogs offers the advantage of uncovering more potent drugs and enabling repurposing effort. 15 Furthermore, such cases facilitate easier and available SAR investigations, presenting a significant opportunity to discover novel bioactive compounds, improve drug-likeness, and enhance pharmacokinetic and pharmacodynamic properties. 16
X-ray structures of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease (Mpro) complexed with an α-ketoamide-type inhibitor were reported by Zhang et al.
17
In addition, four α-ketoamide inhibitors (

The active site of M.pro
Chemical structures of α-ketoamide inhibitors
To identify potential SARS-CoV-2 inhibitors, we employed a multi-stage computational screening approach against key viral enzymes. Among 310 natural antiviral compounds, the most potential inhibitors against SARS-CoV-2 papain-like protease, 23 Mpro, 23 and nsp10 24 were anticipated. Furthermore, between 3009 FDA-approved drugs, several inhibitors against SARS-CoV-2 RNA-Dependent RNA Polymerase, 25 papain-like protease,12,26 Mpro, 27 and nsp16-nsp10 2′-o-Methyltransferase Com-plex 28 were predicted. Furthermore, the SARS-CoV-2 helicase inhibitors were expected from 5956 traditional Chinese medicine candidates. 29 Finally, the most potent semisynthetic and natural inhibitors were selected amid 69 compounds 30 and 4924 African natural metabolites,31,32 respectively.
This study investigates a unique set of 80 semi-synthetic compounds that have not been previously studied as SARS-CoV-2 Mpro inhibitors. While they share key pharmacophoric features with known inhibitors, they represent unexplored chemical space. Our integrative computational approach—combining docking, ADMET analysis, DFT calculations, and MD simulations—adds novelty and depth, enhancing the reliability of identifying promising Mpro-targeting candidates.
Results and discussion
Pharmacophoric features study
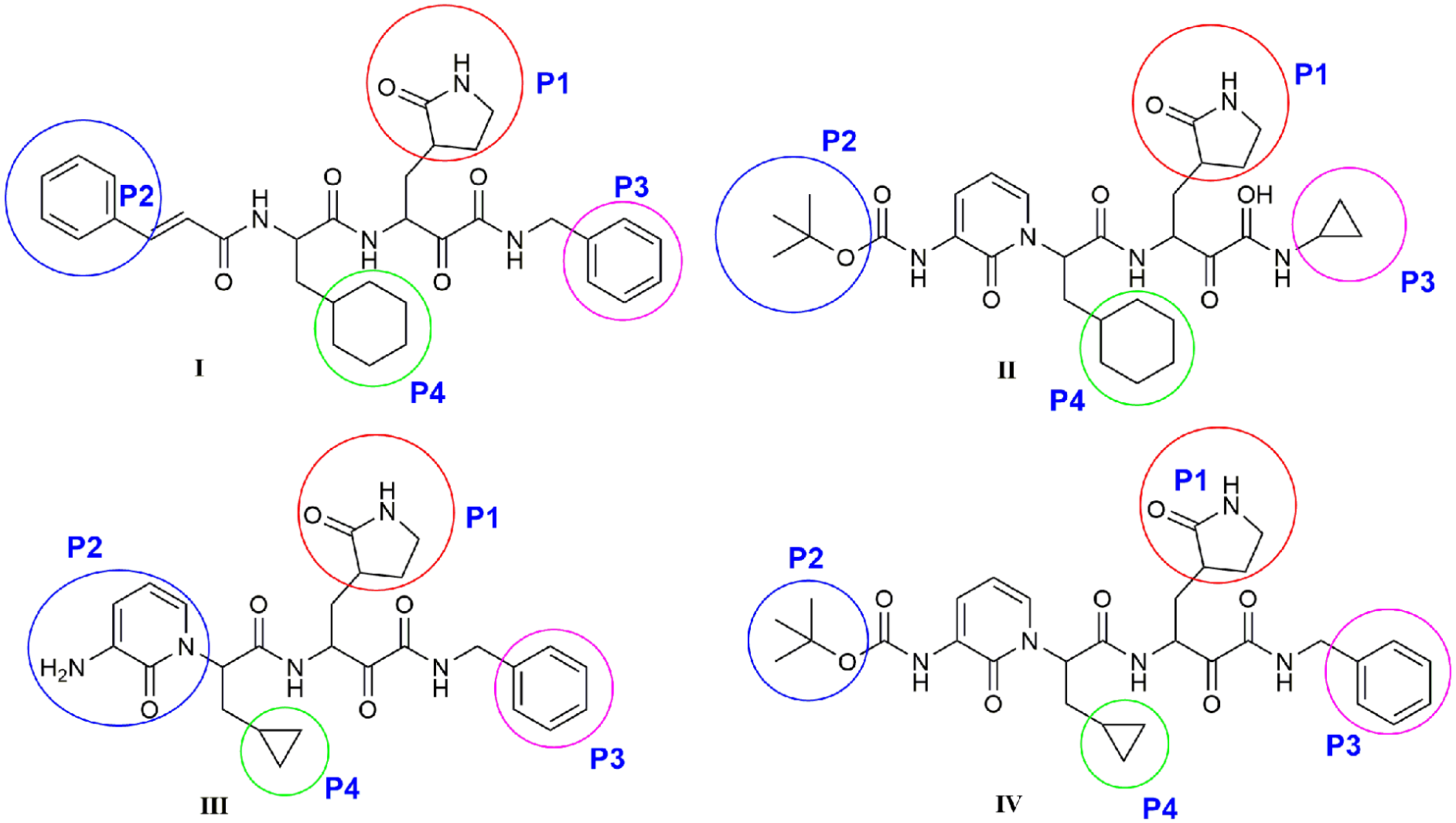
A curated set of 80 semi-synthetic compounds (Figure 3), each designed to match the essential pharmacophoric features of known Mpro inhibitors, was sourced from the Eximed Laboratory database 33 for computational screening in this study. The selection was guided by structural similarity to known Mpro inhibitors, particularly the presence of moieties capable of occupying the well-characterized S1, S1’, S2, and S4 binding pockets of the Mpro active site. An initial virtual screening was conducted on these compounds to evaluate their binding affinities toward Mpro using molecular docking techniques. As shown in Figure 4, representative compounds display the essential pharmacophoric elements—corresponding to P1, P2, P3, and P4—that are required for optimal interaction within the enzyme’s active site. This strategic alignment with known binding features provided a rationale for prioritizing these compounds for further computational evaluation.

The semi-synthetic compound’s chemical structures.

Samples of the tested compounds have the same essential pharmacophoric features as Mpro inhibitors.
Molecular docking studies
The SARS-CoV-2 Mpro, represented by PDB ID: 6LU7, is a well-established and critical therapeutic target due to its essential role in processing viral polyproteins necessary for viral replication and transcription. Its absence in human proteases makes it highly specific and a safe candidate for antiviral intervention. 34 As a result, 6LU7 has been widely employed in numerous structure-based drug design studies and virtual screening efforts aimed at identifying effective Mpro inhibitors.35–39
Molecular docking studies of 80 semisynthetic compounds were carried against SARS-CoV-2 Mpro (PDB ID: 6LU7) with the co-crystallized ligand (PRD_002214) as a reference (Table 1) to take a look at the binding free energies in addition to the binding modes of the endorsed metabolites.
The calculated ∆G (in Kcal/mol) of the tested semisynthetic compounds and (

The binding mode of (
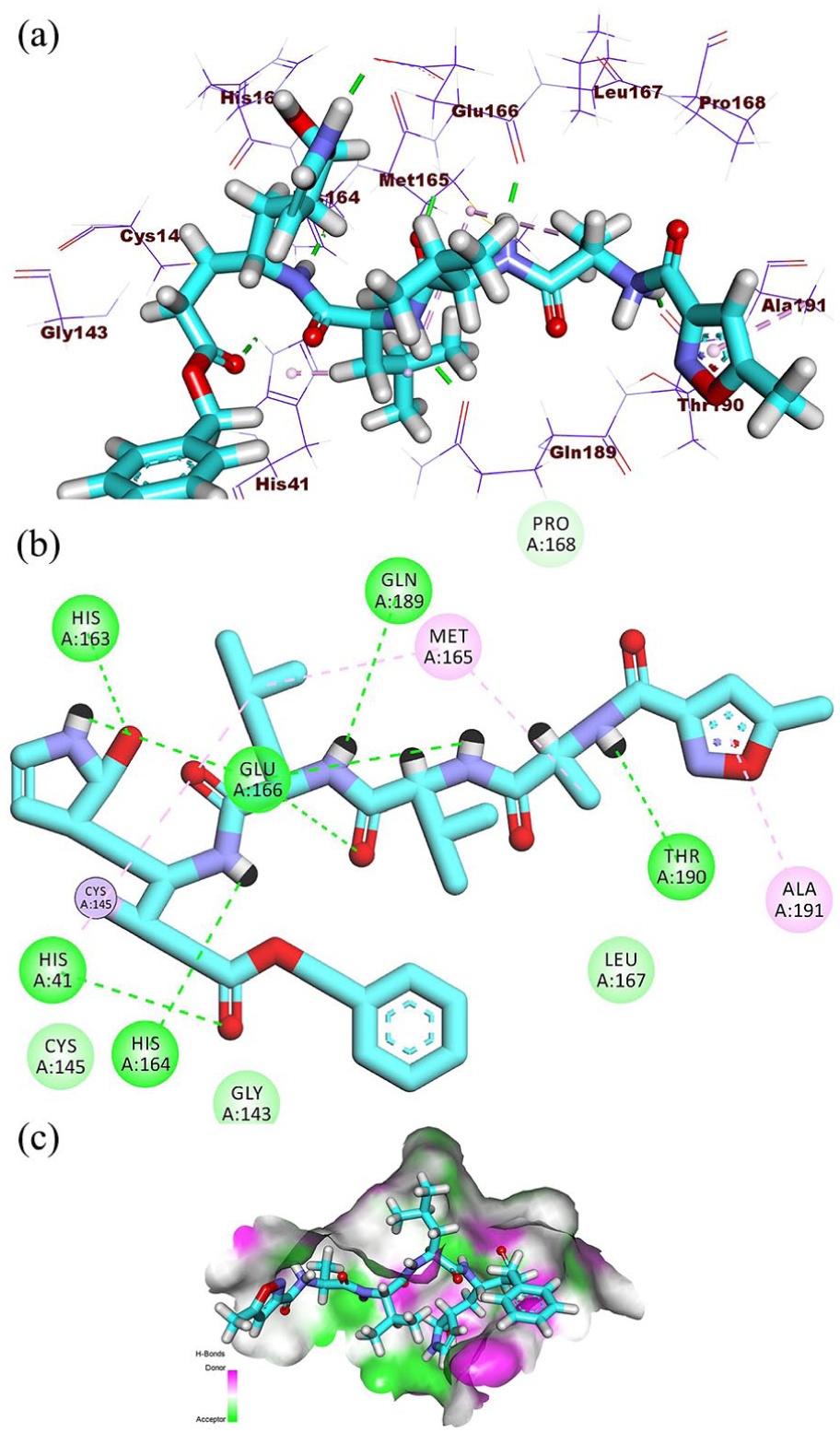
(a) Three-dimensional visualization, (b) 2D interaction diagram, and (c) surface mapping of (PRD_002214) in the Mpro active site.
Compound

(a) Three-dimensional visualization, (b) 2D interaction diagram, and (c) surface mapping of
Concerning compound

(a) Three-dimensional visualization, (b) 2D interaction diagram, and (c) surface mapping of
Respecting the docking results of
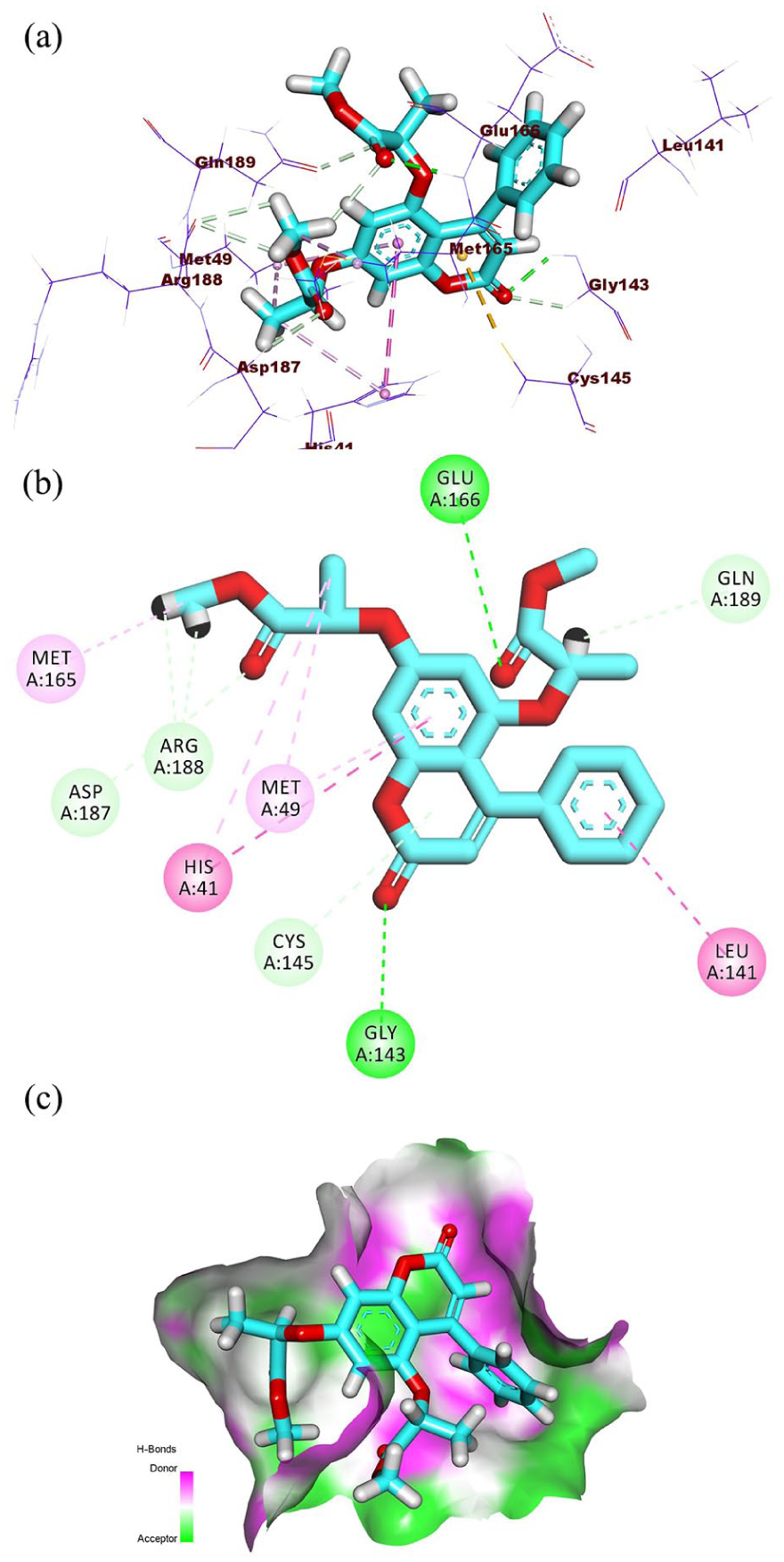
(a) Three-dimensional visualization, (b) 2D interaction diagram, and (c) surface mapping of
Compound

(a) Three-dimensional visualization, (b) 2D interaction diagram, and (c) surface mapping of
The predicted binding orientation of

(a) Three-dimensional visualization, (b) 2D interaction diagram, and (c) surface mapping of
Compound

(a) Three-dimensional visualization, (b) 2D interaction diagram, and (c) surface mapping of
Compound
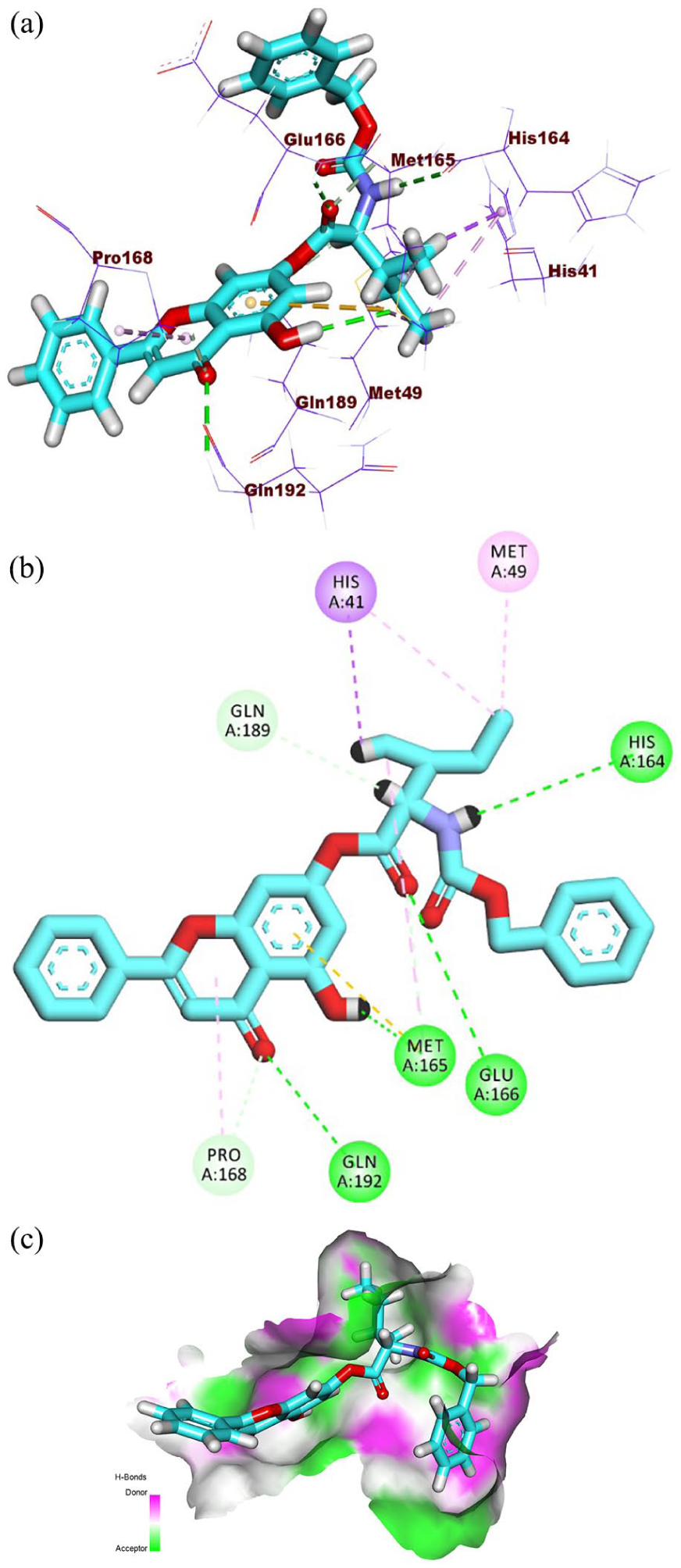
(a) Three-dimensional visualization, (b) 2D interaction diagram, and (c) surface mapping of
Binding mode analysis of semi-synthetic inhibitors versus reference molecule (PRD_002214)
The reference molecule, PRD_002214, binds to the SARS-CoV-2 Mpro by occupying all four key binding pockets—S1, S2, S1, and S4—forming eight H.B with key residues such as His163, Glu166, Gln189, and His164, along with four notable hydrophobic interactions involving Met165, His41, and Ala191. The tested semi-synthetic compounds (
Hydrophobic interactions were also prominently observed in the tested compounds, particularly in compounds
In silico ADMET analysis
Following the initial docking analysis, seven semisynthetic compounds—
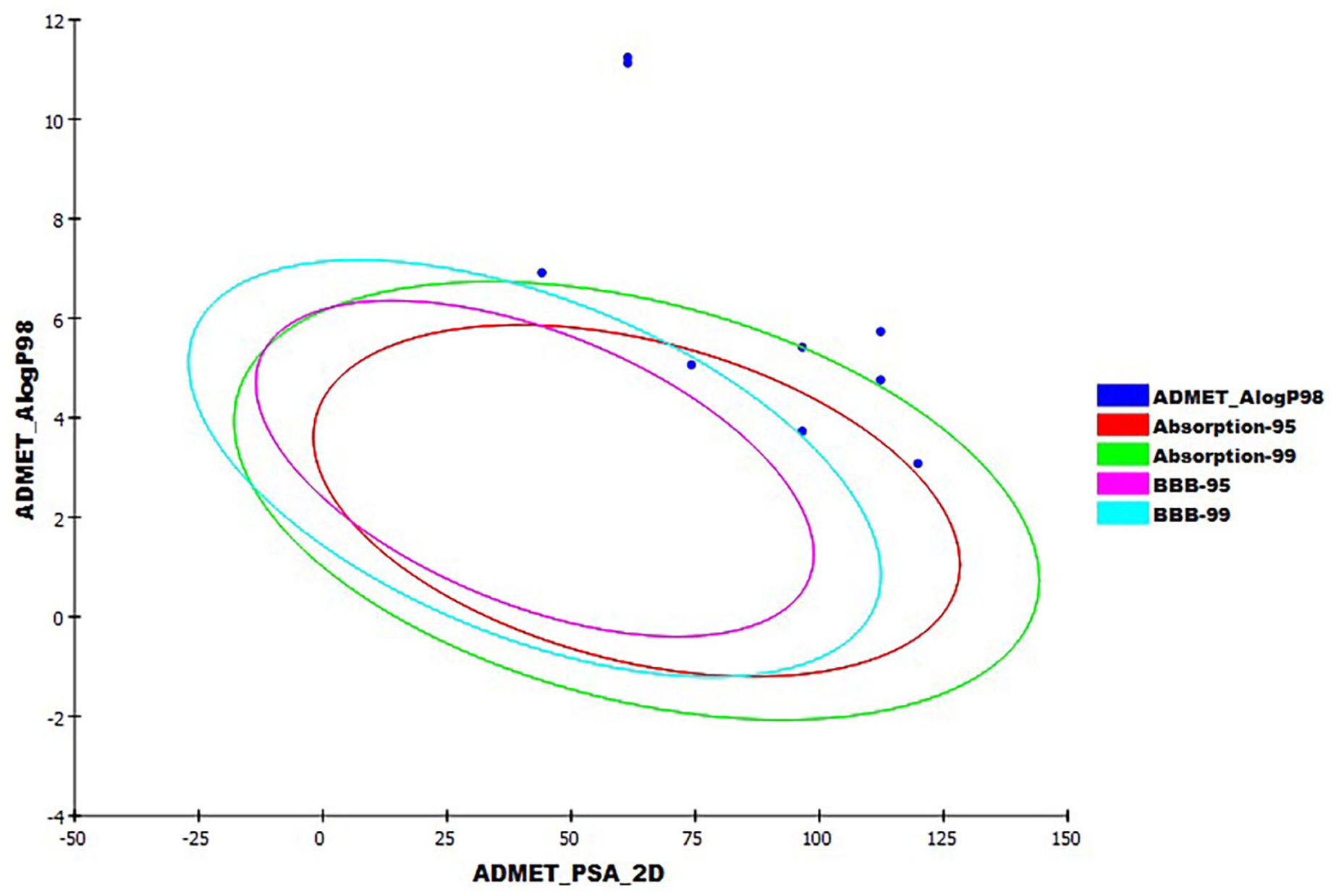
The anticipated ADMET analysis.
Predicted ADMET for compounds

BBB level, blood brain barrier level, h = high, v = very low.
Solubility level, v = very low, l = low, g = good.
Absorption level, g = good, m = moderate, p = poor, v = very poor.
CYP2D6, i = inhibitor, n = non inhibitor.
PBB, l means less than 90%, m means more than 90%.
The findings indicated that all semisynthetic compounds have very low levels of BBB penetration except compound
In silico toxicity studies
Subsequently, the toxicity profiles of the top seven semi-synthetic compounds
Toxicity properties of compounds

n = noncarcinogen, c = carcinogen.
Unit: g/kg.
In general, all semisynthetic compounds were predicted to be non-carcinogenic against FDA rodent carcinogenicity model except
DFT studies
To gain deeper insight into the electronic behavior and reactivity of the top candidates, DFT calculations were performed using Discovery studio software. Compounds
Molecular orbital analysis
The total energies of

Spatial distribution of of molecular orbitals for (a) PRD_002214, (b) 12, (c) 15, (d) 17, (e) 67, and (f) 70.
Thermodynamic parameters of compounds

Molecular electrostatic potential maps
The molecular electrostatic potential (MEP) map provides valuable insight into the electronic distribution of a compound, which in turn influences its potential interactions with the target receptor. 56 In MEP visualizations, color coding reflects electron density: red regions indicate electron-rich (electronegative) areas, blue regions correspond to electron-deficient (electropositive) areas, and green to yellow regions represent neutral zones. Typically, red regions act as H.B acceptors, blue regions serve as H.B donors, and green/yellow areas are involved in π-π stacking or other hydrophobic interactions. Based on these visual cues, potential interaction sites between ligands and their target receptors can be predicted. 57
The co-crystallized ligand PRD_002214 displayed eight red zones and four blue zones on its MEP map, suggesting the presence of eight H.B acceptor sites and four donor sites. Among the tested compounds, compound
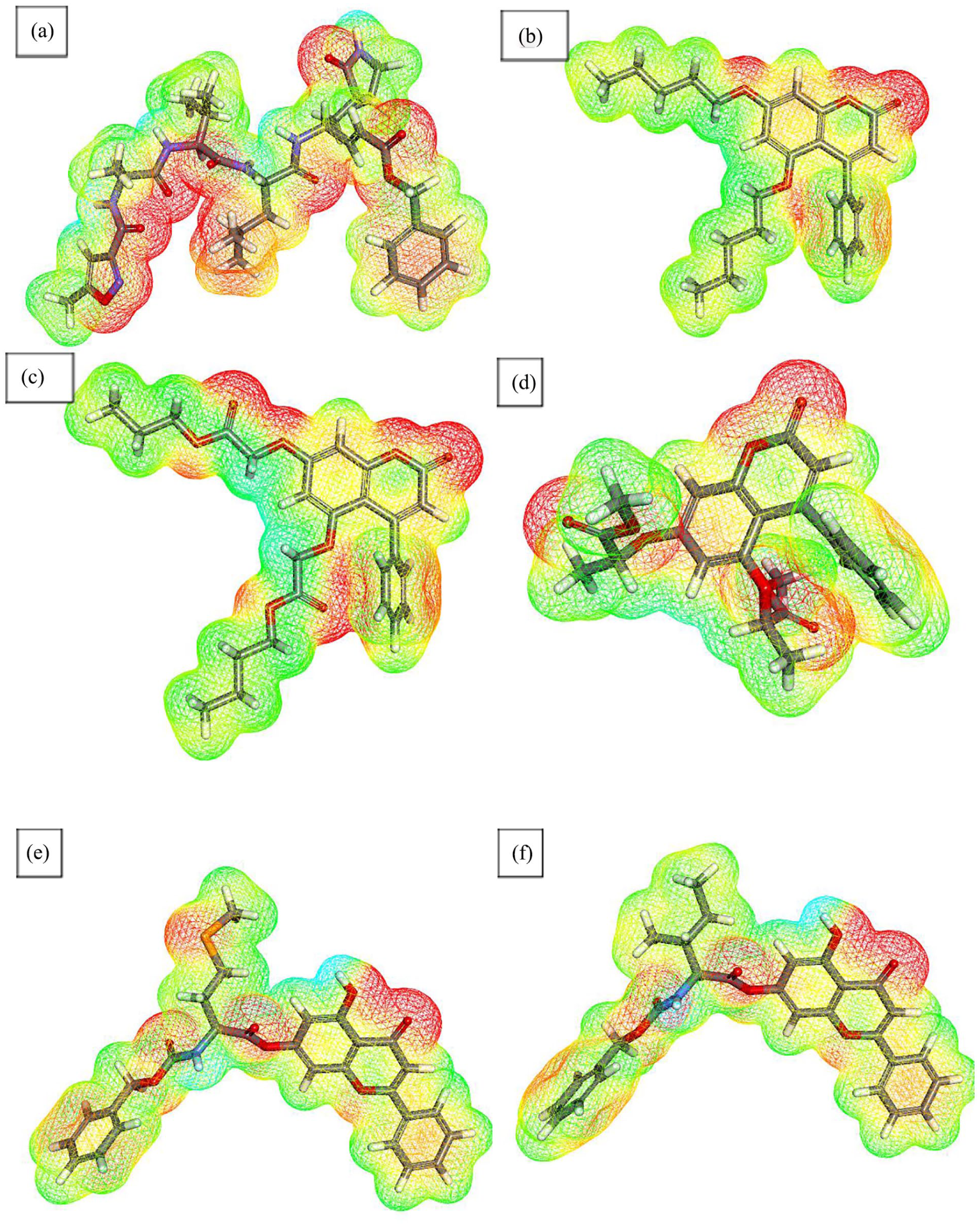
Molecular electrostatic potential map of (a) PRD_002214, (b) compound 12, (c) compound 15, (d) compound 17, (e) compound 67, and (f) compound 70.
SAR analysis
A detailed SAR evaluation of the top-performing semi-synthetic compounds (
Chromen or Coumarin Cores (e.g. compounds
Hydrophobic side chains (e.g. pentyloxy, butyloxy, phenyl, or steroidal moieties) effectively occupy S2 and S4 pockets, enhancing binding through van der Waals contacts. These features are visualized in Figure 7 (compound
Benzyloxycarbonyl-amino acid derivatives, such as those in compounds
Electron-rich regions supporting binding affinity, as evidenced by DFT and MEP analysis, align with the Mpro interaction maps of Figures 11 and 12, where compounds
These SAR trends correlate strongly with structural features required for optimal Mpro inhibition, supporting the rationale for prioritizing these compounds for further in vitro validation.
MD simulation
The stability and dynamic behavior of top candidate compounds
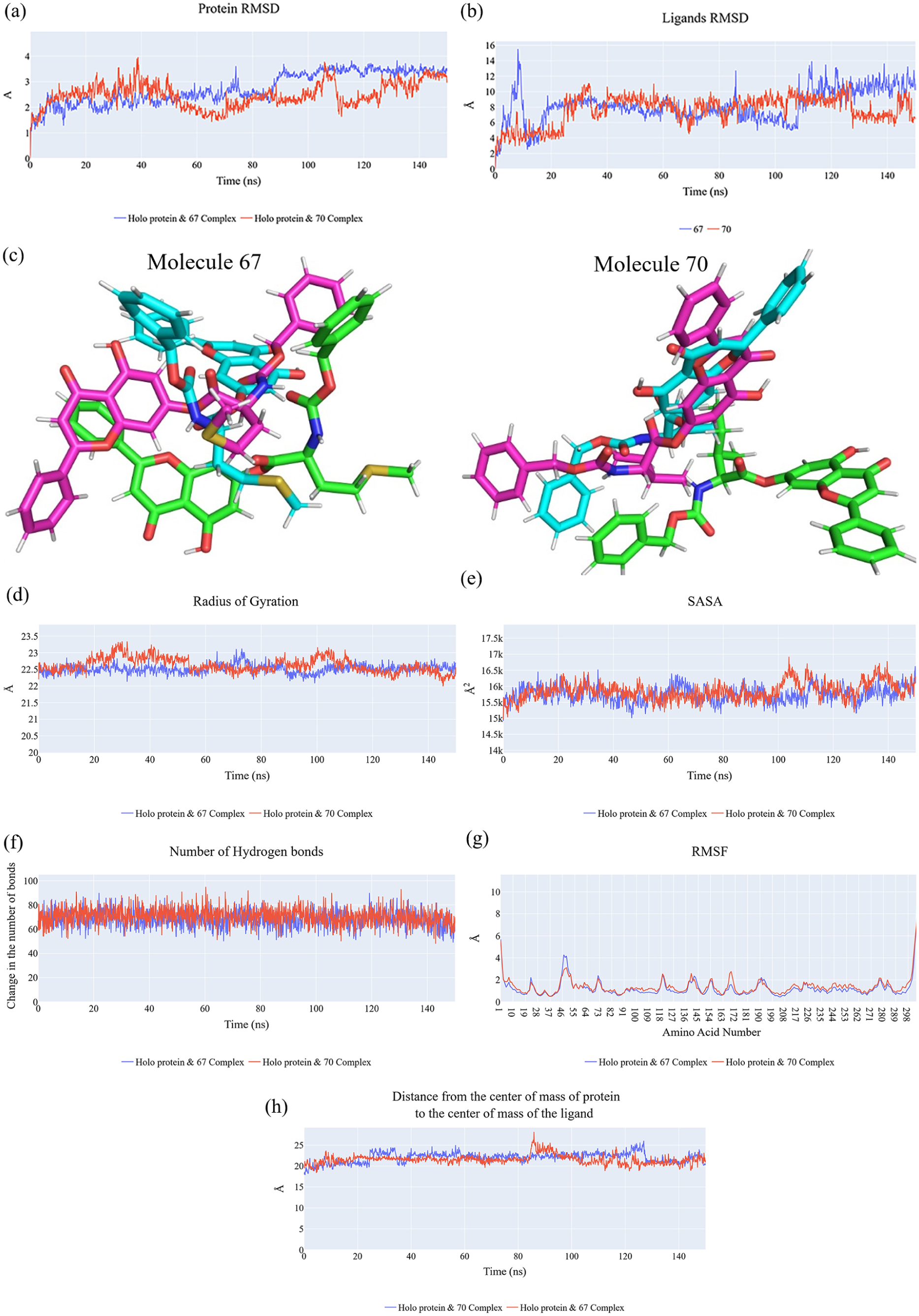
(a) RMSD values from the trajectory for the Mpro in Mpro_
MM-GBSA studies
Figure 17(a) and (b) shows the MM-GBSA components of the binding free energy for molecule

MM-GBSA energetic components and values for the Mpro_
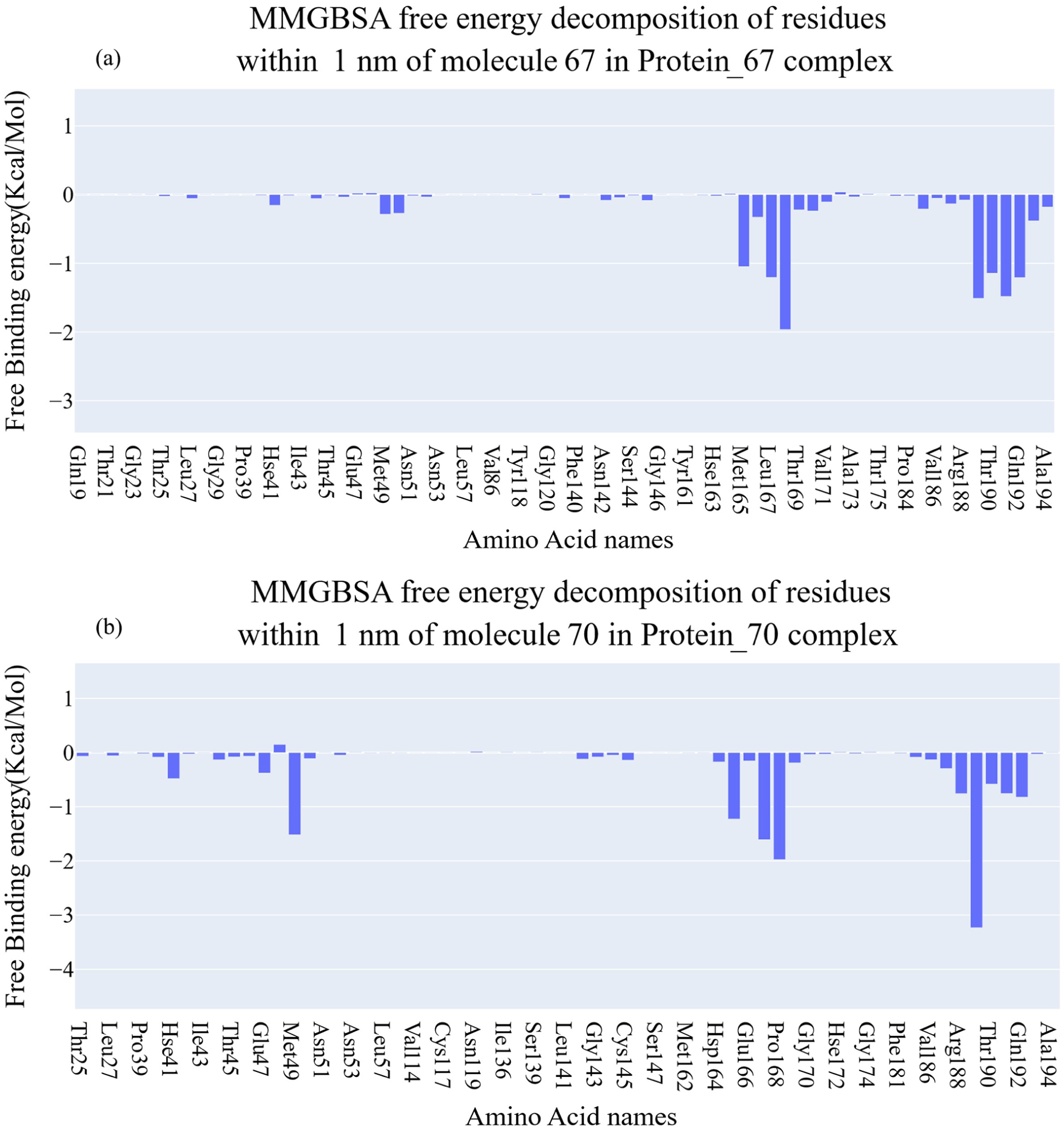
Decomposition of free binding energy of the Mpro_
Protein-ligand interaction fingerprint analysis
Protein-ligand interaction fingerprint (ProLIF) functions as a tool in computer-aided drug design and molecular docking investigations, focusing on the examination and characterization of connections between proteins and ligands. This approach comprises creating interaction fingerprints between a ligand and a protein, enabling the assessment of their binding interactions’ strength and characteristics.
58
These fingerprints allow for the quantification of diverse interaction types, encompassing H.B, hydrophobic forces, and other non-covalent associations. In MD simulations, ProLIF can be effectively employed to scrutinize the dynamic dynamics of protein-ligand complexes over time. This analysis provides valuable insights into the changing nature of interactions between the protein and ligand during the simulation, thereby aiding in comprehending the complex’s stability and binding affinity.
59
Very long-lasting hydrophobic interactions are detected from Met49 (85.2%), Met165 (98%), Leu167 (87.6%), Pro168 (90.1%), Asp187 (96%), Arg188 (79.8%), Gln189 (99.4%), and Thr190 (91.8%) and

ProLIF analysis of the Mpro_

ProLIF analysis of the Mpro_
Protein-ligand interactions profile studies
Protein-ligand interactions profiler (PLIP) emerges as a vital bioinformatics tool integral to the examination and visualization of interactions within molecular complexes between a protein and a ligand. This tool furnishes insightful data about the binding interactions and non-covalent connections formed between the protein and the ligand, a crucial aspect for grasping the MD of ligand-receptor associations.
33
Owing to its indispensable role in drug innovation, computational biology, and structure-focused bioinformatics, PLIP finds extensive application in the exploration of protein-ligand complexes and their interactions. By combining PLIP alongside other computational methods molecular docking, MD simulations, and calculations of free energy, a comprehensive comprehension of ligand-protein interactions is achieved, elevating its significance as a pivotal instrument in rational drug design initiatives.
60
The key frames identified via clustering of the compound

PLIP analysis of the Mpro_
Conclusion
This study utilized a comprehensive computer-aided drug design (CADD) approach to identify promising inhibitors of the SARS-CoV-2 Mpro from a curated set of 80 semi-synthetic compounds. Through molecular docking, ADMET profiling, DFT calculations, and 150-nanosecond MD simulations, compounds
Experimental
Docking analyses
Docking analyses were done for the pointed 80 semisynthetic candidates against SARS-CoV-2 Mpro (PDB ID: 6LU7) using Discovery studio software.61–63 (Please refer to the Method section available in the Supplemental Material for more details.)
ADMET analysis
Discovery Studio 4.0 was utilized for the prediction of the ADMET parameters.64,65 (Please refer to the Method section available in the Supplemental Material for more details.)
Toxicity studies
The software Discovery Studio 4.0 was employed for forecasting TD50, MTD, oral LD50, FDA rat carcinogenicity, LOAEL, as well as irritancy assessments for both ocular and dermal exposures.66,67 (Please refer to the Method section available in the Supplemental Material for more details.)
Density functional theory
Discovery Studio software was used to compute the DFT parameters, including total energy, binding energy, HOMO, LUMO, gap energy, dipole moment, and electrostatic potential. (Please refer to the Method section available in the Supplemental Material for more details.)
MD simulations studies
A 150-ns classical MD simulation was performed in GROMACS 2021 to examine the stability of the compound
Binding free energy calculation using MM-GBSA
The gmx_MMPBSA program was used to evaluate the binding strength of compound
PLIP studies
The elbow method was utilized to group the trajectory data obtained from the MD simulation of the compound
Supplemental Material
sj-pdf-1-chl-10.1177_17475198251367070 – Supplemental material for Computer-aided drug discovery of potential semisynthetic inhibitors for SARS-CoV-2 main protease: A multi-phase computational approach
Supplemental material, sj-pdf-1-chl-10.1177_17475198251367070 for Computer-aided drug discovery of potential semisynthetic inhibitors for SARS-CoV-2 main protease: A multi-phase computational approach by Ahmed M Metwaly, Hazem Elkady, Eslam B Elkaeed, Abdelrahman M Saleh, Aisha A Alsfouk, Ibrahim M Ibrahim and Ibrahim H Eissa in Journal of Chemical Research
Footnotes
Acknowledgements
Eslam Elkaeed would like to thank AlMaarefa University for supporting this research under project number MHIRSP2025-031.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2025R116), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data that support the findings of this study are enclosed in the manuscript and the Supplemental Materials.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.