
Abstract
The repercussions of the COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are catastrophic, and the world has yet to achieve full recovery. Several inhibitors targeting SARS-CoV-2 main protease are experiencing diminished efficacy owing to resistance-inducing mutations. The current situation implies that the quest to find potent and resilient SARS-CoV-2 main protease drugs to overcome resistance must be a continuous effort. Here, multiple receptor virtual screening and molecular dynamics (MD) simulation techniques were employed to identify novel binders from an integrated small-molecule database as leads for the discovery, design, and development of antivirals immune to resistance by SARS-CoV-2 main protease. The small-molecule database was initially screened separately against five SARS-CoV-2 main protease structures with different substrate-binding site conformations using the GOLD program, after which the fitness score of a control compound was used as the cutoff to create a shortlist of potential hits in each case. Then, 21 compounds at the intersection of all five shortlists were selected as virtual screening hits. The hits were subjected to MD simulations, identifying four novel compounds capable of remaining bound to SARS-CoV-2 main protease for up to 100 ns. Analysis of the mode of binding and interactions between each of the four compounds and SARS-CoV-2 main protease revealed that the compounds fit better into the conserved subpockets of the substrate-binding site than the control and interact with important amino acid residues. Conjointly, MD simulations, binding energy, and toxicity analysis results further demonstrated that the compounds are promising leads for the discovery, design, and development of potent drugs to augment the fight against SARS-CoV-2 main protease resistance.

Introduction
The repercussions of the COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a positive-sense single-stranded RNA virus, on health, education, economies, geopolitics, and sociocultural lifestyles are catastrophic, and the world has yet to achieve full recovery.1 –5 The World Health Organization reports that COVID-19 remains a concern because new SARS-CoV-2 variants, infections, and related deaths are being recorded, and the global death toll from COVID-19 stood over seven million as of September 2024. 6 Despite the identification of numerous SARS-CoV-2 drug targets7 –9 and potential therapeutic agents,10,11 the consistent emergence of new SARS-CoV-2 variants poses a threat to COVID-19 treatment owing to drug resistance.12 –20 To tackle these challenges, the discovery of a variety of SARS-CoV-2 inhibitors or leads for the development of effective drugs is required.
The SARS-CoV-2 main protease (SARS-CoV-2-Mpro), referred to as 3-chymotrypsin-like protease, is a cysteine protease that plays an important role in the viral life cycle because it is associated with the processing of polyproteins to produce functional proteins and enzymes essential for viral replication and assembly. SARS-CoV-2-Mpro is functional in the dimeric form,21,22 and its monomeric structure consists of three domains (I, II, and III) with the catalytic H41-C145 dyad situated within the cleft of domains I and II (see Figure 1). Surrounding the catalytic dyad is a binding pocket (defined by residues from all three domains) consisting of subpockets S1, S2, S3, S4, and S5 to contain the substrate (see Figure 1).23,24 Upon binding of the polyprotein substrate to Mpro, H41 abstracts a proton from the thiol group of C145 to give rise to a thiol nucleophile. The nucleophile attacks the scissile amide bond, resulting in bond breaking, the release of the first product, and deprotonation of H41 to restore its original state. Subsequently, the generated thioester is attacked by a nucleophilic hydroxide ion (OH−) formed by the deprotonation of a water molecule by H41, leading to product release and restoration of the catalytic dyad.25,26 Considering the critical role of Mpro and the fact that it is conserved among coronaviruses and lacks a human analog, it has been recognized as an attractive target for SARS-CoV-2 drug development.27,28 Compounds that strongly bind to the substrate-binding site of Mpro can act as inhibitors by competing with or dislodging the polyprotein substrate. Although many antiSARS-CoV-2-Mpro inhibitors have been identified,29 –31 the majority experience diminished efficacy owing to mutations in Mpro that confer drug resistance.32 –37 For instance, a decreased potency of nirmatrelvir, the first SARS-CoV-2-Mpro inhibitor approved for clinical use, has been observed following T21I, L50F, N142L, E166M/V, Q189E/I, Q192T, P252L, and T304I mutations in Mpro.35,38 The situation implies that the quest to find potent and resilient SARS-CoV-2-Mpro drugs to overcome resistance must be a continuous effort.

Structure of the main protease (Mpro) of SARS-CoV-2. The domains 1, 2, and 3 are colored cyan, gray, and green, respectively. The substrate-binding site and subpockets (S1, S2, S3, and S4) are shown.
In this study, multiple receptor virtual screening (MRVS) and molecular dynamics (MD) simulation techniques were employed to identify novel binders from a small-molecule database generated by merging peptidomimetic and Mpro-targeted libraries as leads for the discovery, design, and development of antiviral agents immune to resistance by SARS-CoV-2-Mpro. The small-molecule database was originally screened independently against five structures of Mpro with varying substrate-binding site conformations using the GOLD program,39 –41 after which the fitness score of a control compound was used as the cutoff to create a shortlist of potential hits in each instance. Subsequently, 21 compounds at the intersection of all five shortlists were selected as virtual screening hits. The hits were subjected to MD simulations, identifying four novel compounds capable of remaining bound to Mpro for up to 100 ns. Analysis of the mode of binding and interactions between each of the four compounds and Mpro revealed that the compounds fit better into the conserved subpockets of the substrate-binding site than the control and interact with important amino acid residues. Collectively, MD simulations, binding energy, and toxicity analysis results further demonstrated that the compounds are promising leads for the discovery, design, and development of potent drugs to augment the fight against SARS-CoV-2-Mpro resistance. This study covers a wider chemical space and identifies novel compounds as drug leads.
Methods
Selection and preparation of SARS-CoV-2-Mpro receptor models
The three-dimensional coordinates of SARS-CoV-2-Mpro in both apo and holo forms were obtained from the Protein Data Bank (PDB). Three apo crystal structures with PDB IDs 7BAJ, 42 7DAV, 43 and 7JP1 44 were downloaded. For the holo forms of Mpro, crystal structures with PDB IDs 6LU7 (Mpro complexed with covalent inhibitor N3), 45 6LZE (Mpro complexed with covalent inhibitor 11a), 46 6WTT (Mprocomplexed with covalent inhibitor GC376), 47 6Y2F (Mpro complexed with covalent inhibitor 13b), 48 7D3I (Mpro complexed with covalent inhibitor MI-23), 49 7CA8 (Mpro complexed with noncovalent inhibitor shikonin), 50 and 7P2G (Mpro complexed with noncovalent inhibitor Z222979552) 51 were retrieved. These holo structures were selected based on the presence of well-characterized inhibitors. 52 All retrieved structures were examined for suitability as receptors for structure-based virtual screening (SBVS). The final selection of receptor models for MRVS was based on the configuration, orientation of amino acid side chains, and uniqueness of the substrate-binding site. Receptors with substrate-binding sites blocked by the side chains of amino acid residues were rejected. Ultimately, five structures, including 7BAJ (apo Mpro), 6LU7 (N3-bound Mpro), 6Y2F (13b-bound Mpro), 7CA8 (shikonin-bound Mpro), and 7P2G (Z222979552-bound Mpro), were selected for MRVS exercises. All water molecules were deleted from each of the selected structures, followed by replacement of all missing atoms and amino acid residues using the SWISS-MODEL server.53,54 Each structure was fully protonated, energy-minimized, and saved as a mol2 file using the protein preparation tool incorporated in the BIOVIA Discovery Studio 2016.
Construction and preparation of small-molecule library
An integrated small-molecule library was constructed by merging a peptidomimetic library obtained from Life Chemicals and Mpro-targeted libraries sourced from Life Chemicals, Enamine, Otava, and TargetMol databases. A peptidomimetic library is a collection of compounds with special features that allow them to mimic the natural peptide substrate/ligand of a receptor. 55 A targeted library, on the contrary, comprises a selection of compounds possessing unique characteristics that may enhance their interaction with a particular target. 56 Four known inhibitors of Mpro, including N3, 45 13b, 48 shikonin, 50 and Z222979552, 51 were incorporated into the library as control compounds. These inhibitors are ligands for the holo-Mpro receptors selected for MRVS. The automatic ligand preparation tool in BIOVIA Discovery Studio 2016 was used to prepare the library, which entailed protonation, energy optimization, deduplication, and fixing the bad valences of the constituent compounds. A total of 15,427 unique compounds were prepared and saved as a mol2 file. Table 1 displays a breakdown of the number of compounds in the databases that constitute the integrated small-molecule library.
Composition of the integrated small-molecule library.

Mpro: main protease.
MRVS and hits selection
The GOLD docking program39 –41 was used for all screening exercises. The suitability of the GOLD program for this study was validated by its ability to reproduce the experimental binding mode of the control with a root mean square deviation (RMSD) of 1.99 Å (see Supplemental Figure S1). GOLD employs a genetic algorithm (GA) to search the conformational space and predict the possible modes of ligand binding to a receptor. The predicted binding modes can be ranked using one of the built-in scoring functions (CHEMPLP, GoldScore, ChemScore, and ASP) or a user-defined function. The GoldScore function used in this study comprises a GOLD fitness score, which is the sum of four terms: the protein–ligand hydrogen bond (H-bond) score (Shb_ext), protein–ligand van der Waals score (Svdw_ext), contribution due to ligand intramolecular H-bonds (Shb_int), which is switched off by default, and contribution of the ligand intramolecular strain (Svdw_int). The GOLD fitness score is given by equation (1)

The MRVS model adopted in this study requires sequential screening of the small-molecule library against the five Mpro receptor models. In each instance, the prepared receptor was uploaded to the GOLD wizard. The substrate-binding site of each receptor for the VS exercise was defined by the following amino acid residues: T24, T25, T26, L27, N28, H41, C44, T45, S46, E47, D48, M49, L50, P52, Y54, Y118, N119, F140, L141, N142, G143, S144, C145, H163, H164, M165, E66, L167, P168, T169, G170, V171, H172, F185, D187, R188, Q189, T190, A191, and Q192. After uploading the small-molecule library to the GOLD wizard, selecting the GoldScore scoring function, and specifying the default VS settings as the GA search option, the compounds were docked onto Mpro.
The protocol for selecting screening hits requires GOLD-generated best-ranking files, which contain the highest scoring poses for all compounds, along with a control to determine the selection cutoff. The control compounds N3 and 13b emerged as the best; however, compound 13b was chosen for hit selection because N3 obtained a very high score in its receptor (N3-bound Mpro) and would exclude most promising candidates. First, the best-ranking file corresponding to each of the five receptors was sorted in decreasing order of GOLD fitness score. The fitness score of the control compound in each sorted best-ranking file was used to set the selection cutoff, resulting in the shortlisting of all compounds with scores higher than the control as potential hits and the rejection of those with lower scores than the control. Thus, five sets of shortlisted compounds consisting of 532, 2488, 3006, 1353, and 546 entries for the apo, N3-bound, 13b-bound, shikonin-bound, and Z222979552-bound receptors, respectively, were generated. Eventually, the compounds at the intersection of all five shortlists were selected as VS hits, resulting in the identification of 21 promising leads. The 21 compounds were redocked onto the control-bound receptor (13b-bound Mpro) using the default settings of GOLD, and complexes involving the best ligand poses were saved as PDB files for further analyses. Although each of the selected 21 compounds is a potential Mpro binder, the complex structure of each compound with Mpro was subjected to MD simulations to identify compounds and scaffolds that could remain stably bound to Mpro. The simulation of each compound was categorized into phases 1, 2, and 3, involving 20 ns, 50 ns, and 100 ns simulation times, respectively. The RMSD and final pose of each compound were analyzed in each phase to ascertain stability, which served as a yardstick to determine progression to the next phase. The focus was then placed on the compounds that remained stably bound to Mpro for up to 100 ns. Two additional independent simulations were conducted for each stable compound.
MD simulations
Conventional MD simulations were conducted using the GROMACS 2020.4 simulation package.57,58 The topology of Mpro was constructed using the Amber 99SB force field 59 and the TIP3P water model. 60 In contrast, the topologies of all ligands (selected compounds) were generated and the corresponding atom types and restrained electrostatic potential (RESP) charges 61 were assigned using the antechamber tool included in the AMBER 21 suite.62,63 In each instance, the Mpro–compound complex was situated in the middle of a cubic box with a minimum separation distance of 10 Å between the protein surface and box edges. Subsequent to the solvation of the simulation box with an explicit TIP3P water model, 60 the system’s net charge was neutralized at an ionic concentration of 0.1 M by incorporating the requisite quantities of Na+ and Cl− ions. The Particle Mesh Ewald method 64 was employed to address long-range charge–charge interactions. The LINCS (linear constraint solver) approach 65 was used to restrain all bonds that involved hydrogen atoms at constant equilibrium lengths. The system was energy-optimized using the steepest descent algorithm. Position restraints were applied to the non-hydrogen atoms of Mpro and the compound, and initial velocities were arbitrarily apportioned to all atoms of the system. A 5 ns equilibration was conducted at 1 bar and 300 K in the NPT ensemble, utilizing the Berendsen barostat for pressure regulation and the V-rescale thermostat for temperature coupling. 66 A production run in the NPT ensemble was performed for the requisite simulation duration following the removal of position restraints, with data saved every 1 ps.
Binding free energy calculations
The binding free energies (BFEs) and per-residue contributions (PRCs) by Mpro to the BFEs of Mpro–ligand complexes were estimated using the g_mmpbsa tool. 67 In each case, the calculations were performed on 1000 frames sampled from the final 50 ns simulation trajectory using a dielectric constant (pdie) of 6 and the default settings of all other parameters of the tool. The selection of the pdie of 6 was based on the observation that the Mpro–control complex obtained unfavorable BFEs at pdie values < 6. The BFE was calculated as the summation of the van der Waals energy (Evdw), electrostatic energy (Eelec), electrostatic contribution to the solvation free energy (Gpolar), and nonelectrostatic contribution to the solvation free energy (Gnon-polar) terms, as summarized in equation (2).

Pose, interaction, and MD simulation analyses
Ligand poses and interactions were analyzed using PyMol 1.7.4, 68 BIOVIA Discovery Studio 2016, and LigPlot 2.2 software. 69 Simulation trajectories were analyzed using the GROMACS 2020.4 tools,57,58 including gmx rms for RMSD calculations, gmx gyrate for radius of gyration (Rg) calculations, gmx rmsf for root mean square fluctuation (RMSF) calculations, gmx hbond for H-bond analyses, and gmx covar and gmx anaeig tools for free energy landscape (FEL) analyses. For ligand RMSD and FEL analyses, the atoms of the receptor (Mpro) were least-squares fitted, and the calculations were performed on the ligand. For Mpro(receptor) backbone RMSD analysis, calculations were executed on the backbone atoms after the same backbone atoms were least-squares fitted. Regarding FEL analyses, the gmx covar utility was used to create eigenvectors, which were used as gmx anaeig input data to project principal components 1 (PC1) and 2 (PC2). The PC1–PC2 projections were used to generate FEL diagrams. For H-bond analyses, the default donor–acceptor angle of 30° and donor–acceptor separation distance of 3.5 Å of the gmx hbond tool were used.
Results
Selected compounds
Following the MRVS and ligand-selection criteria, 21 compounds emerged as hits. The chemical structures of the 21 compounds and the docking scores corresponding to each receptor model are shown in Figure 2 and Supplemental Table S1, respectively. Supplemental Table S1 shows that the compounds obtained better fitness scores than the control. For the 21 compounds, 18 entries (compounds with IDs starting with “F”) belong to the Life Chemical database, whereas two compounds (T2598 and T2727) and one compound (Z24395724) belong to the TargetMol and Enamine databases, respectively. The RMSD and pose analysis results of the 21 compounds obtained from the MD simulations and used to assess stability are presented in Supplemental Figures S2 and S3, respectively. The results show that four compounds, including T2727, F1044-0016, F0514-3956, and F0514-4563, as well as the control, remained stably bound to Mpro for up to 100 ns. Thus, the remainder of this study focused on compounds T2727, F1044-0016, F0514-3956, and F0514-4563 as representatives of the 21 VS hits.

Chemical structures of the 21 compounds identified as virtual screening hits. The control and compounds that remained stably bound to SARS-CoV-2-Mpro for up to 100ns during MD simulations are indicated by green labels.
Default docking scores, poses, and interactions of compounds T2727, F1044-0016, F0514-3956, and F0514-4563
The default GOLD fitness, H-bond, and van der Waals scores obtained by compounds T2727, F1044-0016, F0514-3956, F0514-4563, and the control are presented in Supplemental Table S2. Compounds T2727, F1044-0016, F0514-3956, and F0514-4563, as well as the control, obtained fitness scores of 83.70, 84.12, 71.94, 69.35, and 68.39, respectively. Various chemical interactions and bonds occur between Mpro and compounds T2727, F1044-0016, F0514-3956, and F0514-4563. As illustrated in Figure 3(a), compound T2727 occupies subpockets S2, S3, S4, and S5 of the substrate-binding site of Mpro and forms H-bonds with amino acid residues T25, H41, F140, H164, E166, and Q192. Compound T2727 is also engaged in pi–pi interactions with H41 and P168. There are many other nonbonding contacts and interactions between T2727 and Mpro residues: M49, L141, N142, H163, M165, L167, H172, V186, D187, R188, Q189, T190, and A191. Compound F1044-0016 occupies subpockets S1, S3, S4, and S4 and makes H-bonds with E166 and Q192, as shown in Figure 3(b). A pi–pi interaction is observed between H41 of Mpro and compound F1044-0016, which makes numerous nonbonded contacts with T25, T26, L27, M49, L141, N142, G143, C145, H163, H164, M165, P168, D187, R188, and T190 of Mpro (see Figure 3(b)). Figure 3(c) shows that compound F0514-3956 occupies subpockets S1, S3, S4, and S5 of the substrate-binding site of Mpro. H-bonds are formed between G143 and E189 of Mproand compound F0514-3956. Residues T24, T25, T26, H41, M49, L141, N142, C145, H163, M165, E166, P168, D187, R188, T190, A191, and Q192 of Mpro establish nonbonded interactions with compound F0514-3956. Compound F0514-4563, which occupies subpockets S1, S3, S4, and S5, forms an H-bond with Glu166 of Mpro along with many nonbonded interactions with H41, M49, T54, F140, N142, G143, S144, C145, H163, H164, M165, P168, D187, R188, Q189, T190, and Q192, as illustrated in Figure 3(d). As shown in Supplemental Figure S4(a), the control occupies only subpockets S1, S3, and S4 and forms H-bonds with H41, G143, C145, H163, H164, and E166. A covalent bond is formed between the control compound and the thiol group of C145. Nonbonded interactions exist between the control compound and residues T26, M49, F140, L141, N142, M165, L167, P168, H172, D187, and Q189 of Mpro. The ability of compounds T2727, F1044-0016, F0514-3956, and F0514-4563 to occupy the subpockets of the substrate-binding site of Mpro better than the control explains why they obtained better docking fitness scores.

Interactions between Mpro and the four representative compounds. (a) Interactions between Mpro and compound T2727. (b) Interactions between Mpro and compound F1044-0016. (c) Interactions between Mpro and compound F0514-3956. (d) Interactions between Mpro and compound F0514-4563. Carbon, nitrogen, oxygen, sulfur, fluorine atoms are depicted by balls colored in black, blue, red, yellow, and cyan, respectively. Bonds linking the atoms of T2727, F1044-0016, F0514-3956, and F0514-4563 are colored magenta. Bonds connecting the atoms of amino acid residues involved in H-bonding are colored gray. The red “eyelashes” depict nonbonded contacts. H-bonds are illustrated as green dashes.
Ligand RMSD and convergence analyses
Three independent 100 ns simulations were conducted for each of the final selected Mpro–T2727, Mpro–F1044-0016, Mpro–F0514-3956, and Mpro–F0514-4563 complexes. Three independent simulations of the Mpro–control complex were also carried out. All trajectories in each case were used to compute the RMSDs of the compound during the simulations to ascertain the repeatability of the data and ligand stability. The RMSD results for compounds T2727, F1044-0016, F0514-3956, and F0514-4563, along with the control, are presented in Figure 4(a)–(e), respectively. The similarity of the curves in each case (see Figure 4) suggests reproducibility of the results. Furthermore, the stable RMSD curves in each case are indicative of the stability of the compound and its interactions with Mpro. The RMSD trajectories in Figure 4 show that the simulations converged by 50 ns. Therefore, to compare the RMSDs of the four compounds, the average RMSD of each compound was calculated using the last 50 ns of the three corresponding simulations. Compounds T2727, F1044-0016, F0514-3956, and F0514-4563, along with the control, obtained average RMSDs of 3.87 ± 0.25 Å, 2.48 ± 0.04 Å, 4.13 ± 0.22 Å, 2.01 ± 0.04 Å, and 3.15 ± 0.11 Å, respectively. Overall, the average RMSDs of the four compounds are less than 4.20 Å, indicating stability, and they compare well with that of the control compound. Given that the standard error is narrow in each case, it is noteworthy that the most stable trajectory associated with each compound was used for the remaining analyses.

Ligand RMSD data. (a) RMSD of compound T2727. (b) RMSD of compound F1044-0016. (c) RMSD of compound F0514-3956. (d) RMSD of compound F0514-4563. (e) RMSD of the control. In each case, the red, green, and black curves show results for three independent simulations.
Receptor RMSD, RG, and RMSF analyses
RMSD, RG, and RMSF analyses were performed on the backbone of Mpro in the T2727-bound, F1044-0016-bound, F0514-3956-bound, F0514-4563-bound, and control-bound forms, as well as the apo form, to monitor receptor conformation and stability during the simulations. Receptor-backbone RMSD, RG, and RMSF analyses provide valuable insights into receptor stability, compactness, and local-atomic fluctuations, respectively, which can aid in understanding biological mechanisms and phenomena. However, large changes in RMSD, RG, and RMSF could indicate receptor instability. RMSD and RG are functions of simulation time and thus were calculated using the entire 100 ns of each trajectory. The RMSF, which requires sampling conformations from the stable region of a trajectory, was computed using the last 50 ns of each trajectory. The RMSD, RG, and RMSF results are presented in Figure 5(a)–(c), respectively. Compared to the apo form, no significant conformational and structural variations attributable to ligand binding can be observed in Mpro in the T2727-bound, F1044-0016-bound, F0514-3956-bound, F0514-4563-bound, and control-bound forms. The results suggest that Mpro was stable in all ligand-bound forms during the simulations.

RMSD, RG, RMSF, and hydrogen bond data. (a) Backbone RMSDs of Mpro in T2727-bound, F1044-0016-bound, F0514-3956-bound, F0514-4563-bound, control-bound, and apo forms shown as green, cyan, blue, yellow, red, and black curves, respectively. (b) Backbone RGs of Mpro in the T2727-bound (green), F1044-0016-bound (cyan), F0514-3956-bound (blue), F0514-4563-bound (yellow), control-bound (red), and apo (black) forms. (c) Backbone RMSFs of Mpro in T2727-bound (green), F1044-0016-bound (cyan), F0514-3956-bound (blue), F0514-4563-bound (yellow), control-bound (red), and apo (black) forms. (d) The number of H-bonds between Mpro and T2727, F1044-0016, F0514-3956, F0514-4563, and control shown as green, cyan, blue, yellow, and red curves, respectively.
Hydrogen bond analyses
The number of H-bonds between Mpro and each of the compounds T2727, F1044-0016, F0514-3956, and F0514-4563, as well as the control, was calculated along the corresponding simulation trajectory. The results are shown in Figure 5(d), where the consistent H-bond counts over the simulation time, as illustrated by the curves, suggest that the interactions between Mpro and the compounds are stable. For each compound, the results from the last 50 ns were used to calculate the average H-bond count. Compounds T2727, F1044-0016, F0514-3956, and F0514-4563, as well as the control, obtained average H-bond counts of approximately 4, 2, 2, 1, and 4, respectively. Compound T2727 had the same H-bond count of 4 as the control; however, the overall results suggest that the other forces described in Figure 3 play significant roles in stabilizing the interactions between Mpro and compounds F1044-0016, F0514-3956, and F0514-4563 in particular.
FEL and conformational analyses
FEL analyses were performed on compounds T2727, F1044-0016, F0514-3956, and F0514-4563, as well as the control, to determine the energy minima and most stable binding orientations explored during the simulations. The two-dimensional (2D) FEL diagrams for compounds T2727, F1044-0016, F0514-3956, and F0514-4563 are presented in Figure 6(a)–(d), respectively. The FEL diagram for the control is shown in Supplemental Figure S4(b). The energy minima in the FEL plots are colored deep red and indicate the stable binding modes of the compounds to Mpro. Thus, a single energy minimum indicates one main ligand-binding mode, whereas many energy minima with distinct energy barriers imply several potential ligand-binding modes. As illustrated in Figure 6, the FEL diagrams for compounds T2727, F1044-0016, and F0514-3956 each show one main energy minimum, whereas the FEL plot for compound F0514-4563 shows two distinct energy minima. The FEL diagram of the control compound shows two energy minima, as illustrated in Supplemental Figure S4(b).

FEL diagrams for the four representative compounds. (a–d) FEL diagrams for compounds T2727, F1044-0016, F0514-3956, and F0514-4563, respectively.
To examine the ligand poses and binding modes explored during the simulations, Mpro–compound complex structures were extracted from the main energy minimum/minima of each compound and compared with the corresponding initial docking complexes through all-ligand atom superimposition. The results of the superimposition are shown in Figure 7, where the carbon atoms of the compounds in the docking complexes are colored magenta, whereas the carbon atoms of T2727, F1044-0016, F0514-3956, and F0514-4563 in the corresponding simulation complexes are colored green, cyan, blue, and yellow, respectively. The results of the superimposition involving the control are shown in Supplemental Figure S4(c). The superimposition of the atoms of compounds T2727, F1044-0016, F0514-3956, and F0514-4563 in the simulation complexes on the same atoms in the corresponding docking complexes produced all-atom RMSDs of approximately 2.38 Å, 1.73 Å, 1.70 Å, and 0.88/0.11 Å, respectively. Figure 7(d) shows minor differences between the structures extracted from bins 1 and 2 of the FEL diagram for compound F0514-4563 and the docking structure. The superimposition of the simulation poses of the control extracted from bins 1 and 2 on the initial pose generated an all-atom RMSD of 2.00 Å in each case (see Supplemental Figure S4(c)), indicating that there is no major difference between the simulated poses. All compounds, including the control, maintained the ability to occupy the subpockets of the substrate-binding site of Mpro during the simulations. Generally, similar to the control, the pose and RMSD (< 2.5 Å) results of each compound indicate that the predicted binding mode and the corresponding interactions are stable.
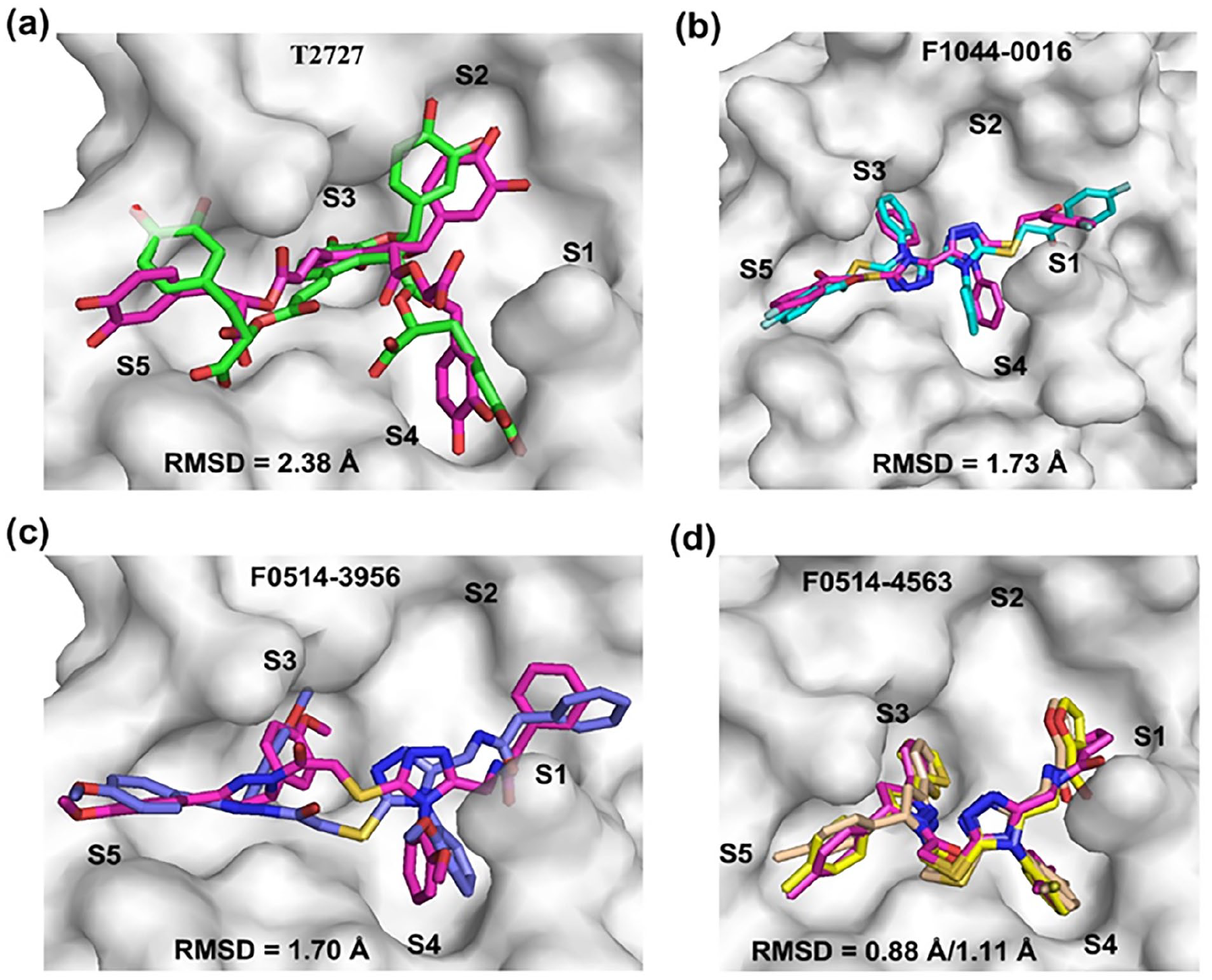
Comparison of simulated poses of the four representative compounds with the corresponding docking poses. (a–d) Superimposed simulated poses of T2727 (green sticks), F1044-0016 (cyan sticks), F0514-3956 (blue sticks), and F0514-4563 (yellow sticks) on the corresponding docking poses (magenta sticks), respectively.
BFE and PRC analyses
The BFE analyses of the Mpro–T2727, Mpro–F1044-0016, Mpro–F0514-3956, Mpro–F0514-4563, and Mpro–control interactions produced BFEs of −112.79 ± 26.79 kJ mol−1, −177.07 ± 16.70 kJ mol−1, −177.83 ± 22.81 kJ mol−1, −165.54 ± 12.95 kJ mol−1, and −94.50 ± 15.98 kJ mol−1, respectively. The PRC by Mpro to the BFEs of the Mpro–T2727, Mpro–F1044-0016, Mpro–F0514-3956, and Mpro–F0514-4563 complexes are illustrated in Figure 8(a)–(d), respectively, where only residues with favorable energy contributions ⩽ −2 kJ mol−1 and unfavorable contributions ⩾ +2 kJ mol−1 are labeled. As shown in Figure 8(a), the amino acid residues H41, N142, C145, M165, P168, Q189, T190, and A191 of Mpro made favorable contributions to the BFE of the Mpro–T2727 complex. Figure 8(b) shows that L27, H41, M49, C145, M165, C168, and Q189 of Mpro made favorable contributions to the BFE of the Mpro–F1044-0016 complex. The Mpro residues L27, H41, M49, N142, C145, M165, D187, and Q189 made favorable contributions to the BFE of the Mpro–F0514-3956 complex, as illustrated in Figure 8(c). For the Mpro–F0514-4563 complex, Figure 8(d) shows that L27, H41, M49, N142, C145, M165, D187, and Q189 made favorable contributions to the BFE of the Mpro–F0514-4563 complex. In each of the four complexes, E166 made the most unfavorable contribution to the BFE. For the Mpro–control complex, R4, H41, R60, R131, K137, L141, N142, C145, M165, and H172 are the main favorable energy contributors to the BFE, as shown in Supplemental Figure S4(d), where only residues with favorable energy contributions ⩽ −3 kJ mol−1 and unfavorable contributions ⩾ +3 kJ mol−1 are labeled. Supplemental Figure S4(d) also show that residues D48, E55, E166, D197, E240, and D289 are the main unfavorable contributors to the BFE of the Mpro–control complex.

Per-residue contributions (PRCs) from Mpro to the binding free energies (BFEs) of complexes involving Mpro and the four representative compounds. (a–d) PRCs from Mpro to the BFEs of the Mpro–T2727, Mpro–F1044-0016, Mpro–F0514-3956, and Mpro–F0514-4563 complexes, respectively.
Toxicity assessment
The ProTox 3.0 web server, 70 which integrates molecular similarity, fragment propensities, pharmacophores, and machine-learning models for the prediction of various organ toxicity and toxicity endpoints, was used to assess the toxicity potentials of compounds T2727, F1044-0016, F0514-3956, and F0514-4563, as well as the control. For organ toxicity, the hepatotoxic, neurotoxic, nephrotoxic, and cardiotoxic potentials of each compound were assessed. Regarding toxicity endpoints, the carcinogenic, mutagenic, immunotoxic, and cytotoxic potentials were evaluated. Table 2 shows the results obtained where the prediction as “+” (toxic) or “−” (nontoxic) is made with the corresponding probability of the prediction indicated. The deep green, light green, light red, and deep red colors in Table 2 represent inactive, moderately inactive, moderately active, and active predictions, respectively. Overall, the compounds predominantly exhibit nontoxic or moderately toxic potentials, with compound T2727 flagged as a potential immunotoxin with high certainty. It is possible to reduce the toxic potentials of the compounds during lead optimization processes.
Toxicity assessment of T2727, F1044-0016, F0514-3956, F0514-4563, and control.


The deep green, light green, light red, and deep red colors represent inactive, moderately inactive, moderately active, and active predictions, respectively.
Discussion and future directions
Notwithstanding the computational cost, incorporating receptor flexibility into SBVS is considered a useful step toward identifying true binders and potential inhibitors.71 –74 Although various strategies for integrating receptor flexibility into SBVS have been suggested, MRVS, which involves docking small molecules against different receptor conformations, enables extensive exploration of receptor flexibility.71,75 Typically, MRVS involves docking small molecules onto superimposed structures of the receptor (ensemble docking)72,76 or docking sequentially onto multiple conformations of the receptor. 77 Despite its speed, the ensemble docking approach only selects the best binding mode involving a compound (ligand) and one receptor conformation as the hit. Conversely, sequential MRVS offers a more robust approach to exploring the receptor conformational space by shortlisting receptor–ligand complexes involving all receptor conformations for further evaluation and selection of the best. The selection of compounds at the intersection of the top-ranked docking hits of all receptor models in sequential MRVS has been shown to be a reliable strategy for identifying true binders and reducing the associated challenge of false positives. 78
The sequential MRVS strategy and selection of intersection ligands, which is based on the proposition that a true binder can effectively bind to various binding site conformations, 78 identified 21 compounds (see Figure 2) as potential binders of SARS-CoV-2-Mpro. The selection of 21 compounds as hits underscores the usefulness of docking onto different receptor conformations and selecting the intersection ligands in the top-ranked solutions, considering that 532, 2488, 3006, 1353, and 546 compounds were shortlisted for the apo, N3-bound, 13b-bound, shikonin-bound, and Z222979552-bound receptor models, respectively. Inspection of the substrate-binding site in the five Mpro receptor models reveals backbone conformational variations in an unstructured loop (Supplemental Figure S5(a)) comprising residues T45, S46, E47, D48, and M49. Different side-chain orientations are also observed for some binding-site residues such as T24, H41, M49, N142, M165, and Q189, as illustrated in Supplemental Figure S5(b). These structural variations account for the differences in the number of compounds shortlisted for each receptor model, which is in agreement with the observation that each binding site conformation introduces a unique set of false-positive docking predictions.77,78 Thus, the ligands at the intersection of the top predictions for all receptor models are true binders, immune to the effects of variations in backbone structure and side-chain orientations, owing to their ability to target the conserved conformation of the binding site. The receptor-binding potencies of such ligands may be immune to perturbations by side-chain mutations responsible for resistance.
By comparing the docking fitness scores of the 21 selected compounds with the control (see Table S1), it is suggestive that each of these compounds can bind to SARS-CoV-2-Mpro and serve as lead for the development of potent inhibitors. However, four of the compounds, including T2727, F1044-0016, F0514-3956, and F0514-4563, demonstrated, through MD simulations, that they are capable of remaining stably bound to Mpro for up to100 ns. Close examination of the binding modes and interactions of compounds T2727, F1044-0016, F0514-3956, and F0514-4563 reveals that they interact with important Mpro residues, and their scaffolds fit into the subpockets of the substrate-binding site better than the control (see Figures 3 and 7, and Supplemental Figures S4(a) and S4(c)). The RMSD analysis results (see Figure 4) show that the four compounds are stable, and the average RMSD values compare well with the control compound, with compounds F0514-4563 (2.01 ± 0.04 Å) and F1044-0016 (2.48 ± 0.04 Å) projected as being relatively stable than the control (3.15 ± 0.11 Å). Furthermore, the estimated BFEs between the compounds and Mpro compare well with the control and confirm that the compounds have reasonable affinities for Mpro.
The results suggest that compounds T2727, F1044-0016, F0514-3956, and F0514-4563 strongly bind to SARS-CoV-2-Mpro, indicating the possibility of their optimization to produce novel inhibitors with improved pharmacological properties. By fitting perfectly into the conserved subpockets of the substrate-binding site of Mpro (see Figures 3 and 7), the scaffolds of these compounds could serve as starting materials for the design of potent noncovalent and covalent inhibitors with immunity to resistance conferred by mutations in SARS-CoV-2-Mpro. Given that structure is more conserved than sequence, 79 such inhibitors, like the substrate, could be immune to resistance induced by mutations in amino acid side chains. Structure–activity relationship (SAR) and chemical mutagenicity 80 studies on compounds T2727, F1044-0016, F0514-3956, and F0514-4563 could generate potent noncovalent inhibitors of SARS-CoV-2-Mpro. Covalent inhibitors of SARS-CoV-2-Mpro, such as the control, 48 form covalent bonds with the thiol group of C145. As illustrated in Figure 9, comparing the poses of T2727, F1044-0016, F0514-3956, and F0514-4563 to the control, along with the close proximity of some atoms to the thiol group of C145, indicates the possibility of covalent interactions. More importantly, Figure 9 also suggests the possibility of modifying the compounds by introducing the appropriate covalent warheads to achieve covalent interactions with C145. Many covalent warheads have been shown to readily form covalent interactions with cysteine amino acid residues. 81

Covalent bond-forming possibilities of the four representative compounds. (a–d) Poses of compounds T2727, F1044-0016, F0514-3956, and F0514-4563 compared with the control, respectively. Carbon atoms of compounds T2727, F1044-0016, F0514-3956, F0514-4563, and the control are colored green, cyan, blue, yellow, and magenta, respectively.
Conclusion
MRVS and MD simulation techniques were employed to identify novel compounds that could target the conserved subpockets of the substrate-binding site of SARS-CoV-2-Mpro and thus serve as starting materials for the discovery, design, and development of antiviral therapeutics to address the resistance challenge. An integrated database of small molecules was sequentially screened against five receptor models of SARS-CoV-2-Mpro, followed by the identification of compounds at the intersection of the top-ranked docking solutions from all five receptors as hits. A total of 21 VS hits were identified, out of which four compounds, including T2727, F1044-0016, F0514-3956, and F0514-4563, could remain stably bound to Mpro for up to 100 ns during MD simulations. The scaffolds of T2727, F1044-0016, F0514-3956, and F0514-4563 fit the conserved subpockets of the substrate-binding site of Mpro better than the control, which suggests that mutations and structural variations in amino acid side chains may not perturb their binding efficacies. Conjointly, the results suggest that the selected compounds are promising leads for the discovery, design, and development of potent drugs to buttress the fight against SARS-CoV-2-Mpro resistance. In vitro, in vivo, SAR, and mutagenicity studies are recommended to further evaluate the Mpro-binding and anti-SARS-CoV-2 characteristics of the compounds, as well as to optimize their general pharmacological features.
Supplemental Material
sj-docx-1-chl-10.1177_17475198251326083 – Supplemental material for Identification of lead small molecules for the design and development of potent severe acute respiratory syndrome coronavirus 2 main protease inhibitors
Supplemental material, sj-docx-1-chl-10.1177_17475198251326083 for Identification of lead small molecules for the design and development of potent severe acute respiratory syndrome coronavirus 2 main protease inhibitors by Elvis Awuni in Journal of Chemical Research
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.