
Abstract
Papain-like protease (PLpro) is a crucial enzyme for SARS-CoV-2 replication and immune evasion. Inhibiting PLpro could be a promising strategy to fight against COVID-19. This study aimed to identify potent inhibitors of PLpro among FDA-approved drugs using an in silico approach. The study also aimed to examine and confirm the binding of the selected compounds to the active pocket of PLpro using a multi-phased in silico approach, involving the screening of 3009 FDA-approved drugs to pinpoint the most similar compounds to,
Keywords
A multi-phased in silico approach selected the most favorable FDA-approved drugs against SARS-CoV-2 PLpro. The study also examined and confirmed the binding of the selected compounds to the PLpro’s active pocket.

Introduction
The drugs that have been granted FDA approval have undergone rigorous evaluation by the Center for Drug Evaluation and Research (CDER) to assess their therapeutic benefits as well as potential adverse effects. 1 The FDA’s approval of a drug serves as an indication of its efficacy and overall safety profile. 2 Therefore, if a new use or indication for these drugs is discovered, they can be safely repurposed. 3 The discovery of new bioactive molecules is a costly and lengthy process, typically taking around 12 years and costing about US$2.6 billion per drug, 4 However, drug repositioning offers an efficient alternative for identifying and developing drugs with new pharmacological indications. 5 Drug repositioning controls and decreases the timeline of drug development, as the used drug exhibited good ADMT properties in humans. 6 Consequently, no need to conduct Phase I clinical trials. 7 The repurposing process was frequently used in the revelation of several drugs with promising activities against cancer,8,9 SARS-CoV-2, 10 inflammations 11 bacterial, 12 parasitic, 13 and viral 14 infections.
Computer-based (in silico) chemistry approaches have been extensively utilized in pharmacology development and testing over the past 20 years.15,16 This vast utilization was a result for various reasons. First, the identification of the exact 3D structures of various protein targets in the body of human.17,18 Second, the recent tremendous progress in the scope of computers software as well as hardware. 19 Third, the advancements in the understanding of the Structure Activity Relationship (SAR) and QSAR principles. 20 Computational chemistry encompasses two main approaches: ligand-based and structure-based. Ligand-based approaches, rely on the chemical structure of molecules that bind to the target of interest to predict the activity of new compounds. These methods allow to identify and optimize potential drug candidates based on their structural similarity to known active compounds such as structural similarity, 21 molecular fingerprinting, 22 Q-SAR, 23 pharmacophore, 24 ADMET properties, 25 and homology modeling. 26 However, structure-based approaches utilize the 3D structure of the target protein to identify and optimize binding interactions with potential ligands. These methods enable the visualization of how compounds interact with specific protein targets and aiding in the development of highly specific and potent drugs such as molecular docking, 27 and MD simulations. 28
To tackle the COVID-19 pandemic, our team employed various in silico techniques to identify natural inhibitors. From a collection of 310 natural antiviral metabolites, we pinpointed potential inhibitors for key SARS-CoV-2 proteins, including the main protease in silico(Mpro),29,30 nsp10, 31 helicase, 32 and papain-like protease (PLpro). 33 In addition, from 3009 FDA-approved drugs, the most potent inhibitors for the SARS-CoV-2 RdRp, 34 Mpro, 35 and the SARS-CoV-2 nsp16-nsp10 2′-O-Methyltransferase Complex 36 were selected. Also, the potential inhibitors of SARS-CoV-2 RdRp, 37 helicase, 38 and PLpro 39 were selected from 4924 natural African metabolites. Between 5956 of traditional Chinese medicine metabolites, the potential natural SARS-CoV-2 Helicase inhibitors were determined. 40
The PLpro is a key enzyme that helps the virus replicate and evade the immune system. It cuts the viral polyprotein to produce necessary non-structural proteins and removes ubiquitin from host proteins to weaken the immune response. 41 Because of its crucial role in the virus’s life cycle and immune suppression, PLpro is an important target for developing antiviral drugs. Inhibiting PLpro can stop the virus from replicating and boost the immune response, making these inhibitors potential treatments for COVID-19. 42
We herein report the preference of the most effective of SARS-CoV-2 PLpro (PDB ID: 3E9S) within 3009 clinical FDA-approved drugs. 43 The explored compounds’ chemical structures were retrieved from https://www.selleckchem.com/screening/fda-approved-drug-library.html.
Results and discussion
Filter using molecular fingerprints
A co-crystallized ligand refers to a compound (ligand) that demonstrates a remarkable binding affinity with the associated protein, leading to the formation of a crystallizable ligand-protein complex.
44
This complex provides valuable insights into the interactions between the ligand and protein, shedding light on the key structural and chemical features that contribute to the optimum binding affinity. Consequently, the co-crystallized ligand’s chemical structure serves as a valuable model for designing and developing inhibitors that can effectively bind to the target protein.
45
By studying the structural characteristics and functional groups present in the co-crystallized ligand, we can gain a deeper understanding of the key factors responsible for its strong binding affinity. We used this knowledge to guide the selection of compounds with structural similarities to
Molecular fingerprints study is a kind of ligand-based computer-based in silico methods. The utilization of molecular fingerprints involves establishing the degree of similarity between the chemical structures of the compounds under investigation.
47
The molecular fingerprints experiment outlined the presence and or absence of the below features in atoms and fragments of the investigated compounds and
Shared and absent structural fingerprints between the tested FDA-approved compounds and

Molecular Similarity
One key difference between molecular similarity and fingerprint studies is the level of molecular detail they capture. Molecular similarity studies typically consider a broader array of molecular descriptors and properties, allowing for a more thorough evaluation of structural and chemical similarities. These studies can account for factors such as shape, electrostatics, and pharmacophoric features. In contrast, fingerprint studies focus on capturing and comparing specific structural patterns encoded in binary fingerprints, offering a more concise representation of molecular structures.
57
Molecular similarity studies describe and compare the entire structures of the reference compound and the test set, using descriptors that are steric, electronic, topological, or physical.
58
To select the most similar 30 compounds to

Molecular similarity analysis of the FDA-approved drugs and
Molecular structural properties of the FDA-approved drugs and

The results demonstrated a high degree of similarity between

The chemical structures of the most like 30 FDA-approved compounds and
Docking studies
The most similar drugs with the co-crystallized ligand (
In these studies, it was depended on the binding mode of the co-crystallized ligand as a guide and reference. Table 3 summaries the binding energy of the tested compounds against PLpro (PDB ID: 3E9S).
The calculated ∆G of the FDA-approved drugs and

To confirm the accuracy of our docking processes, we began with a validation step. This involved docking

Superimposition of the docked form of
The co-crystallized ligand (

3D interaction of
The molecular docking studies revealed that seven drugs (Vismodegib
Summary of the binding modes of the tested compounds and
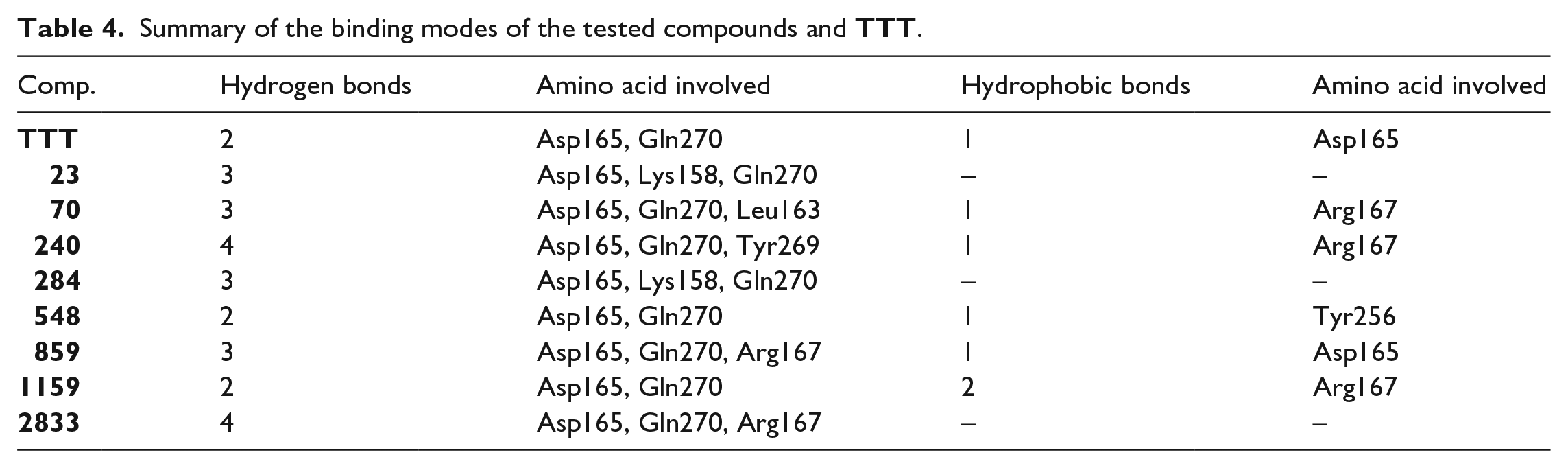
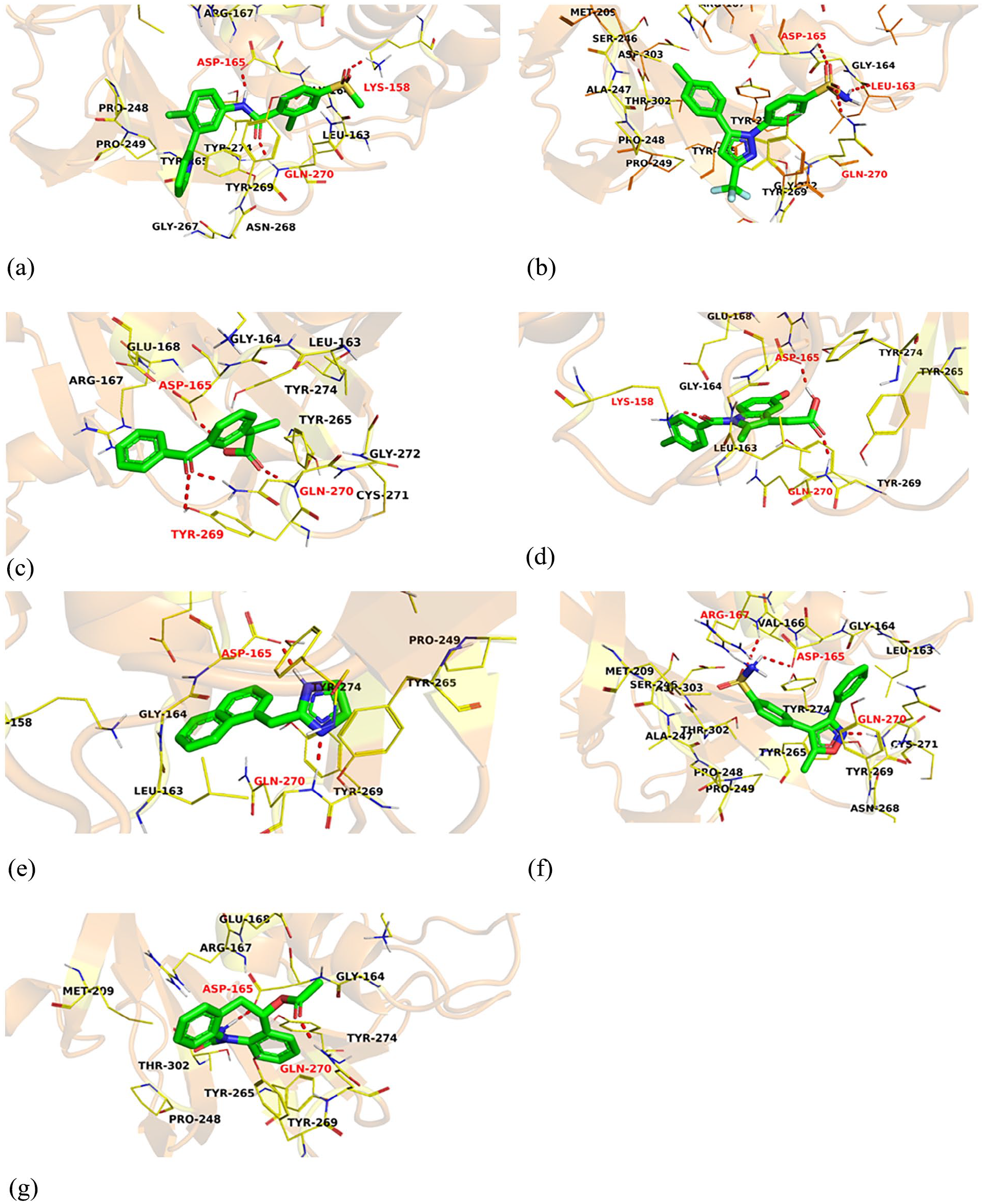
3D interactions of 23 (a), 70 (b), 240 (c), 284 (d), 548 (e), 859 (f), and 1159 (g) in the active site of PLpro.
MD simulations studies
Molecular docking is an in silico technique that reveals the binding site of a ligand inside of a protein structure as a result of in silico experiments. A docking study has the disadvantage of describing proteins' interactions as rigid entities. The conformational changes of a protein's structure after binding with an active compound are therefore not considered during docking experiments. 55 However, molecular dynamics (MD) simulations serve as a robust computational tool for investigating and comprehending protein dynamics and structural modifications at the atomic resolution. 68 By accurately representing the physical interactions of individual atoms within the targeted protein, MD simulations enable the exploration of conformational changes that occur upon ligand binding. Consequently, it provides valuable insights into the dynamic and energetic behaviors of that protein, shedding light on the mechanisms of ligand recognition and binding-induced structural alterations. 56
Upon investigation of the binding modes and binding energy of the tested compounds against SARS papain-like protease (PDB ID: 3E9S), it was found that celecoxib,

Different calculated measurements from the MD trajectories. (a) RMSD: PLpro (blue), celecoxib (red), and the complex (green), (b) RoG, (c) SASA, (d) Number of H. bonds over the 100 ns, (e) RMSF, and (f) Distance between the centers of mass of celecoxib and PLpro.
MM-GBSA calculations
The analysis of binding free energy was carried out by MM-GBSA (Figure 7) and showed the various components of energy that contributed to the binding of celecoxib B to the PLpro. Celecoxib showed a total exact binding energy with the average value of -19.04 Kcal/Mol. In addition, it was found that the van der Waals energy was the largest, and only, favorable contribution with an average value of -34.49 Kcal/Mol.
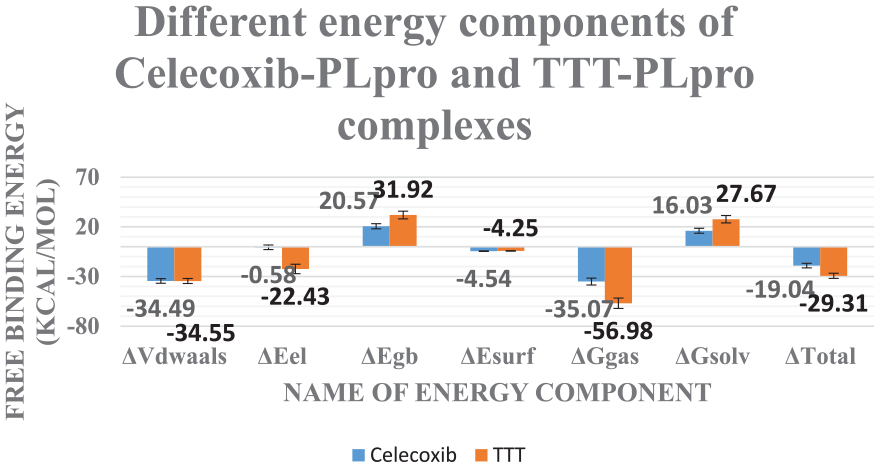
Different energetic components of MM-GBSA. Bars represent the standard deviations.
Moreover, a decomposition analysis (Figure 8) was preceded to identify which amino acid residues within 10 Å of PLpro that had the highest contribution to the interaction with both celecoxib and

Binding free energy decomposition of (a) the PLpro-
PLIP investigation
To have a deeper insight, protein-ligand interaction profiler (PLIP) studies were preceded. The resulted trajectory from the MD was clustered and separated into representative frames for every produced cluster. The number of the resulted clusters was automatically selected by the elbow method producing five clusters. The PLIP webserver was utilized for every cluster to explore the types and number of interactions between celecoxib and PLpro. Table 5 represents the obtained types and number of those interactions based on the PLIP webserver. The major interaction that was detected is the hydrophobic interaction with 32 interactions in all cluster representatives. This aligns with the significant difference in van der Waals and electrostatic energy values derived from the MM-GBSA analysis. The most common amino acid is Tyr269 (eight hydrophobic interaction) followed by Asp165 (four hydrophobic interaction), and Arg167 (four hydrophobic interaction). Besides the production of interaction numbers and types from PLIP, it also generated a .pse file to see celecoxib’s 3D conformations as well as celecoxib’s interaction with PLpro (Figure 9).
Number and types of interactions identified for celecoxib using the PLIP webserver. The amino acids in bold are the most frequently occurring ones in all cluster representatives.


PLIP interactions for each cluster representative for celecoxib. Hydrophobic interaction (dashed gray), amino acids (blue sticks), and celecoxib (orange sticks).
The PLIP webserver was utilized for every cluster to explore the types and number of interactions between
Number and types of interactions identified for


PLIP interactions for each cluster representative for
Conclusion
In conclusion, our study utilized a comprehensive in silico approach to identify potent inhibitors of the SARS-CoV-2 papain-like protease (PLpro) among 3009 FDA-approved drugs. Through rigorous screening and computational experiments, we identified seven promising candidates (Vismodegib, Celecoxib, Ketoprofen, Indomethacin, Naphazoline, Valdecoxib, and Eslicarbazepine) that demonstrated high structural similarity to the co-crystallized ligand (
Method
Fingerprint studies
Discovery Studio 4.0 software was employed to perform a molecular fingerprint analysis on 3009 FDA-approved drugs, ultimately selecting the 150 most similar candidates to
Molecular Similarity detection
Discovery Studio 4.0 software was employed to perform a molecular similarity analysis on the selected 150 FDA-approved drugs, ultimately selecting the 30 most similar candidates to
Docking studies
Discovery studio software was employed to run the docking studies against the PLpro (PDB ID: 3E9S) for the most like 30 FDA-approved drugs (
MD Simulations
PLpro-Celecoxib complex’s stability and structural changes were examined for 100 ns utilizing unbiased MD simulation in GROMACS 2021.70–72 Input files were prepared using the CHARMM-GUI server.73,74 Binding energy of the PLpro-Celecoxib and PLpro-
Supplemental Material
sj-docx-1-chl-10.1177_17475198241298547 – Supplemental material for Discovery of potential FDA-approved SARS-CoV-2 Papain-like protease inhibitors: A multi-phase in silico approach
Supplemental material, sj-docx-1-chl-10.1177_17475198241298547 for Discovery of potential FDA-approved SARS-CoV-2 Papain-like protease inhibitors: A multi-phase in silico approach by Ahmed M Metwaly, Eslam B Elkaeed, Mohamed M Khalifa, Aisha A Alsfouk, Fatma G Amin, Ibrahim M Ibrahim and Ibrahim H Eissa in Journal of Chemical Research
Footnotes
Acknowledgements
The authors thank AlMaarefa University, Riyadh, Saudi Arabia, for supporting this research
Author contributions
A.M.M. and I.H.E.: Supervision, conceptualization, and writing of the original draft. E.B.E.: Fingerprints and similarity analysis, writing, and manuscript revision. M.M.K.: Molecular docking studies. A.A.A.: Software development and support, writing, and manuscript revision. F.G.A.: MM-GBSA and PLIP calculations. I.M.I.: Molecular dynamics (MD) simulations. All authors revised and approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R116), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.