
Abstract
Inducible nitric oxide synthase is known as a potential biological target that plays a crucial role in regulating the release of nitric oxide and is responsible for the amount of nitric oxide released during the inflammation process. Searching for compounds from natural sources that inhibit inducible nitric oxide synthase may reduce excessive nitric oxide production and counteract metabolic diseases originating from prolonged inflammation. One of the valuable medicinal plants with significant anti-inflammatory activity evaluated in this study is Millettia dielsiana. The current work focuses on the molecular docking analysis of compounds derived from Millettia dielsiana to identify potential candidates against the inducible nitric oxide synthase enzyme. As a result, four compounds (D10 (Tupichinol C), D20 (Durmillone), D46 (Glycitin), and D50 (5,7,4′-trihydroxyisoflavone 7-O-β-

Introduction
Inflammation is a pathological condition that encompasses a wide range of diseases, including rheumatic and immune-mediated disorders, diabetes, cardiovascular disease, and so on. Large amounts of nitric oxide (NO) produced by inducible nitric oxide synthase (iNOS) are known to be responsible for the vasodilation and hypotension seen in septic shock and inflammation.
1
NO is produced by the oxidation of
Millettia dielsiana Harms ex Diels is a type of woody vine plant in the Fabaceae family, primarily found in Laos, China, and Vietnam, and grows in closed evergreen forests or semi-dry, semi-deciduous forests. 6 In folk medicine, this plant is used as a tonic and to effectively treat muscle aches and rheumatoid arthritis. 7 Numerous studies have demonstrated its anti-inflammatory activity. According to previous research, flavonoids, the major chemical components of this plant, are responsible for its anti-inflammatory activity. These compounds inhibit NO biosynthesis, which is regarded as a pro-inflammatory mediator that causes inflammation in abnormal situations due to overproduction.1,8 Their results suggest that M. dielsiana Harms ex Diels can be effective in anti-inflammatory activity and NO biosynthesis inhibition. The EtOAc extract of M. dielsiana stems significantly inhibited the production of NO and tumor necrosis factor (TNF)-α in RAW 264.7 cells. Furthermore, isoflavones were isolated from the bioactive fractions of EtOAc extract from M. dielsiana stems which indicated millesianin C exhibited excellent NO and TNF-α inhibitory activity. 9 On the other hand, among isolated compounds from the ethanolic (EtOH) extract, (3S)-vestitol had the strongest inhibitory effect on NO production, with an IC50 value of 16.0 ± 1.5 μM, while isoliquiritigenin and tupichinol C had moderate inhibitory effects, with IC50 values of 31.2 ± 2.5 and 8.4 ± 1.9 μM, respectively. 10
Therefore, our study focused on in silico screening for the iNOS inhibitory activities of selected compounds from M. dielsiana using the molecular docking method.
Materials and methods
Preparation of ligands and macromolecule
From previous reference materials, compounds isolated from the M. dielsiana plant were depicted using the Marvin JS software.9–13 All two-dimensional (2D) structures were converted into three-dimensional (3D) structures using the OpenBabel software. 14 Subsequently, the converted structure files were energy and geometry optimized using the Gaussian software with the M06-2X/6-311 g++ (d,p) basis set. 15 For protein preparation, the crystal structure of the Human iNOS Reductase and Calmodulin Complex, with the PDB ID 3HR4, was obtained from the RCSB Protein Data Bank to serve as the receptor for docking. 2 The Discovery Studio Visualizer software was used to remove water molecules and co-crystallized molecules from the protein model. Finally, AutoDockTools software was used to prepare input files for the molecular docking program.
Molecular docking simulation
The docking studies were conducted using the AutoDock Vina v1.2.3 program on the Ubuntu operating system. 16 The prepared compounds were docked into the active site of the target protein 3HR4. The grid box parameters were set with dimensions of XxYxZ as 22 × 22 × 22, and the center coordinates were specified as x = 1.0 Å, y = 9.2 Å, z = −64.7 Å. A grid spacing of 1 Å was chosen, and the exhaustiveness value was set to 400. Other parameters were kept at their default settings. The AutoDock Tools software and Discovery Studio Visualizer software were utilized for post-docking analysis. The docked poses with the strongest binding affinities were selected for analyzing the interactions between the target receptor and ligands using the Discovery Studio Visualizer software.
DFT calculations
The investigation into the bioactive potential of certain compounds through their interaction with biological active sites has been advanced using theoretical chemistry methodologies, particularly focusing on their molecular electronic structures. This study applied geometric optimization to the compounds of interest in the gas phase, utilizing the Gaussian 09 software suite. The optimization process involves determining the optimal structural parameters using density functional theory (DFT) calculations with the M062X method at the 6-311++g(d,p) level. 15 The M062X functional has been reported to optimize effectively and compute good energies for small molecules.17,18 Further analysis explored the compounds’ local reactivity by examining the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), and the molecular electrostatic potential (MEP) surface. Critical to this study, vital molecular parameters derived from DFT, including HOMO energy (εHOMO), LUMO energy (εLUMO), band gap energy (Δε), electron chemical potential (μ), softness (σ), hardness (η), electrophilicity (ω), and electronegativity (χ), were calculated using the following relationships. 19 This comprehensive approach aims to elucidate the compounds’ bioactivities by correlating their electronic configurations with potential biological interactions







Toxicity prediction
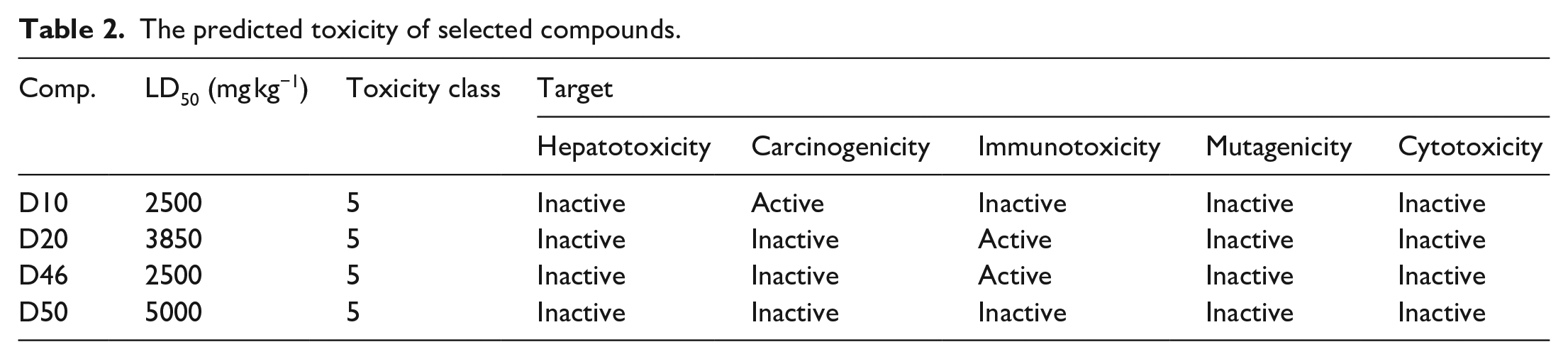
The online tool “Prediction of Toxicity of Chemicals (ProTox-II)” (https://tox-new.charite.de/protox_II/) is specifically designed for anticipating the toxicity of compounds before embarking on the drug discovery journey. 20 ProTox-II uses an advanced method that considers the proximity of unknown molecules to the average lethal dose (LD50) of known compounds, amalgamating identified hazardous components. To estimate toxicity, the structural files of potential compounds are converted into SMILES format and input into the ProTox web server. The output provides predicted LD50 values, toxicity levels, and additional factors such as hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, and cytotoxicity. Moreover, the ProTox-II web server assesses the toxicity class of a chemical value, categorizing it from I to VI based on the LD50 value according to the guidelines of the globally harmonized system of classification and labeling of chemicals. 21
Results and discussion
Molecular docking analysis
The molecular docking study of natural compounds derived from M. dielsiana reveals binding affinities in the active site of iNOS ranging from −5.542 to −9.261 kcal mol−1, with an average value of −7.967 kcal mol−1. Among the docked compounds, D20 exhibits the strongest binding affinity with an ΔG of −9.261 kcal mol−1. In addition, 30 compounds were found to have higher binding efficiency than the control compound diclofenac. The binding energies and interaction details of the studied compounds and diclofenac are presented in Table 1 and Supplemental Table S1. To investigate the inhibitory mechanism against iNOS, the interaction between potential compounds with ΔG values less than −9.0 kcal mol−1 and iNOS protein was further explored. Compounds with high-binding affinities, including D10, D20, D46, and D50, were analyzed for protein–ligand interactions as shown in Figure 1. In Figure 1, compound D10 forms hydrogen bonds with Thr547 and π-anion interactions with Glu661. In addition, two types of π-alkyl and π-π stacked interactions are observed in the D10-iNOS complex with residues Tyr631 and Phe593. Compound D20 forms two hydrogen bonds with residues Ser628 and Thr547 and also exhibits π-anion interactions similar to D10. Moreover, the aromatic ring system in this compound forms two π-π stacked interactions with amino acid residues Phe593 and Tyr631. Similar interactions are found in the D46-iNOS complex. Furthermore, a hydrogen bond with residue Arg633 is observed in the D46-iNOS complex. For compound D50 in the iNOS enzyme active site, three hydrogen bond interactions are observed with amino acid residues Glu661, Asp597, and Asn595. In addition, D50 forms π-π stacked interactions with Tyr631 and π-anion interactions with Asp597. Notably, π-anion interactions (with Glu661) and π-π stacked interactions (with Phe593, Tyr631) are also present in the diclofenac-iNOS inhibitory complex. Important hydrogen bonding interacting amino acid residues in the complexes D10, D20, D46, and D50 include Thr547, Ser628, Arg633, and Glu661, which are also found in the crystallographic complex (Calmodulin) as seen in Figure 1. As reported previously, tupichinol C (D10) exhibits anti-inflammatory activity with moderate inhibitory potential against NO production (IC50 = 38.4 ± 1.9 μM), while 5,7,4′-trihydroxy isoflavone 7-O-β-
Binding affinities and interactions of potential compounds within the active site of the iNOS enzyme.


The 2D interactions between potential compounds (ΔG < –9.0 kcal mol−1) within the active site of the iNOS enzyme.
Oral toxicity prediction
To assess the potential harm of specific compounds to the human body, supportive tools such as ProTox II, 20 ToxAlerts, 23 and Pred-hERG 24 have been developed to quickly predict the potentially toxic effects of candidate drug compounds. In this section, potential compounds with the ability to inhibit iNOS through molecular docking assessments continue to predict toxicity using the ProTox II web server. The detailed prediction results in Table 2 show that the predicted lethal dose (LD50) for the researched compounds ranges from 2500 to 5000 mg kg−1. According to the toxicity grading scale, where an LD50 value between 2000 and 5000 mg kg−1 classifies a compound as toxicity level 5, all four researched compounds fall within this category. This indicates low toxicity and high safety for these compounds at level 5. In addition, Table 2 shows that none of the compounds exhibit hepatotoxic or cytotoxic activities. However, compound D10 is predicted to exhibit carcinogenic activity, and compounds D20 and D46 are associated with immunotoxicity, warranting caution in their further research use. In contrast, compound D50 meets all safety criteria, suggesting its potential as a candidate for the development of new anti-inflammatory drugs.
The predicted toxicity of selected compounds.
Frontier molecular orbital analysis
MEP surface
DFT is a pivotal tool for investigating the biological attributes of a wide array of biomolecules. By employing DFT, the complexity of intermolecular interactions can be elucidated, mainly by analyzing the relationship between various metrics indicative of their inhibitory potential.19–25 In the current study, DFT analysis was used to predict the inhibitory effects by scrutinizing the electronic properties of the four principal components identified across all notable compounds. The documentation includes Figure 2, which illustrates the optimized molecular structures achieved using the M062X/6-311++g(d,p) level of theory, alongside the distributions of the lowest unoccupied and highest occupied molecular orbitals (LUMO-HOMO) and the MEP plots for these compounds, respectively. In addition, quantum chemical parameters are detailed in Table 3.
Quantum chemical parameters of compounds calculated at the level of theory M062X/6-311++g(d,p).

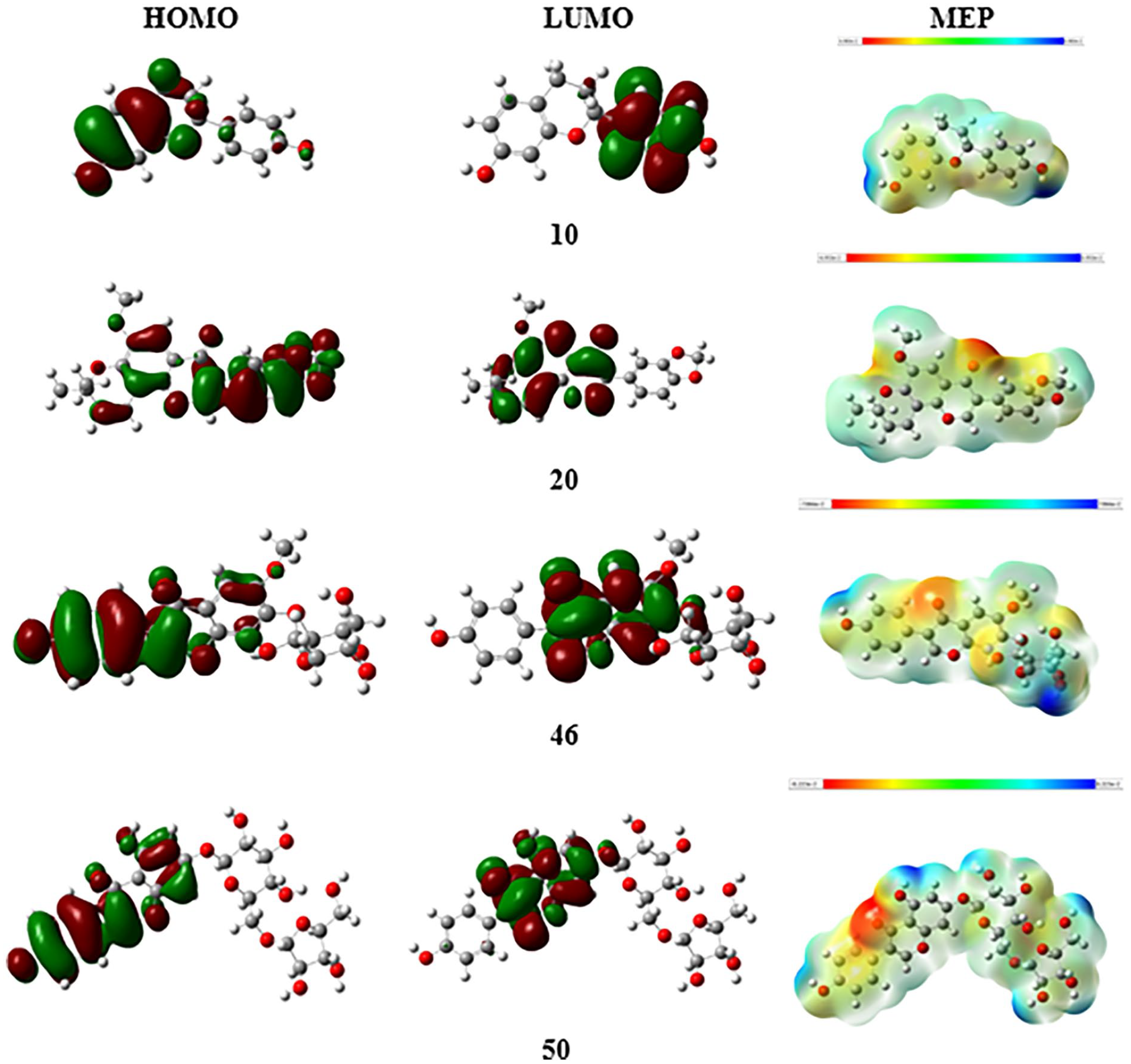
The HOMO and LUMO molecular orbitals and molecular electrostatic potential (MEP) surfaces of potential compounds.
The biological activity of compounds is assessed through the values of HOMO and LUMO according to the Frontier Molecular Orbital theory. This argument is founded upon the theoretical charge-transfer properties of HOMO (which signifies intermolecular electron-donation tendency) and LUMO (which represents electron-accepting capability).
26
All structures exhibit electronic stability in parameterization given their significantly low EHOMO with values ranging from −6.996 to −7.322 eV. Besides, the band gap energy (ΔEgap) varies between 6.257 and 7.814 eV. The energy gap (ΔEgap) plays a crucial role in determining the chemical stability of molecules and can provide insights into charge transfer interactions.25,27 This study observed an increasing trend in ΔEgap among the four compounds, specifically in
MEP surface analysis emerges as a crucial visualization method for identifying potential interaction sites within biomolecules, crucial for understanding drug-receptor and enzyme engagements. 28 The analysis in Figure 2 elucidates the compound’s electron density distribution, highlighting negatively charged areas (in red) predominantly around oxygen atoms. These regions, characterized by their susceptibility to electrophilic attacks, indicate reactive sites potentially involved in polar interactions with active site polar residues of biomolecules. In contrast, the light blue areas on the MEP map denote electron-deficient zones, suggesting sites that are particularly reactive toward nucleophilic attacks. The visualization reveals that positive MEP points are located around the carbon and hydrogen atoms of the molecules, meaning these areas are reactive sites.
The dipole moment values derived from the optimized structures range from 1.796 to 2.941 Debye, highlighting the electronic diversity among the studied molecules. In the evaluation of potential bond formation or complexation between the ligand and the target protein, a higher dipole moment value is correlated with a greater advantage.
29
Accordingly, while some compounds exhibited weaker interactions,
Data analysis in Table 3 reveals that compound D20 has a significantly greater likelihood of engaging in chemical reactions, as indicated by its notably low chemical hardness (η) of 3.129 eV. This is complemented by substantial negative values for the electron chemical potential (μ) at −3.868 eV and a high softness (σ) measure of 0.320 eV–1, collectively characterizing
Conclusion
In this study, molecular docking methods were used to screen natural compounds from M. dielsiana to identify potential compounds with the best binding affinities. Based on the docking simulation results, four compounds were identified as having strong binding affinities, with ΔG < –9.0 kcal mol−1: tupichinol C (–9.024 kcal mol−1), durmillone (–9.261 kcal mol−1), glycitin (–9.116 kcal mol−1), and 5,7,4′-trihydroxyisoflavone 7-O-β-
Supplemental Material
sj-docx-1-chl-10.1177_17475198241263837 – Supplemental material for In silico molecular docking, DFT, and toxicity studies of potential inhibitors derived from Millettia dielsiana against human inducible nitric oxide synthase
Supplemental material, sj-docx-1-chl-10.1177_17475198241263837 for In silico molecular docking, DFT, and toxicity studies of potential inhibitors derived from Millettia dielsiana against human inducible nitric oxide synthase by Hoang Thi Tue Trang, Nguyen Xuan Ha, Cao Hong Le, Truong Thi Thuy Nhung, Dinh Thi Truong Giang, Nguyen Thi Diem Hang and Phan Thi Thuy in Journal of Chemical Research
Footnotes
Author contributions
P.T.T. supervised the project. P.T.T. and H.T.T.T. designed the project. H.T.T.T., N.X.H., T.T.T.N., D.T.T.G., and N.T.D.H. calculated and analyzed the data. P.T.T., C.H.L., and N.X.H. wrote the paper. All authors discussed and analyzed the results of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.