
Abstract
Consent is one necessary foundation for ethical research and it’s one of the research ethics committee’s major roles to ensure that the consent process meets acceptable standards. Although on Oxford ‘A’ REC (an NHS Research Ethics Committee based in the UK) we’ve been impressed by the thought and work put into this aspect of research ethics, we’ve continued to have concerns about the suitability and effectiveness of consent processes in supporting decision making, particularly for clinical trials. There’s poor understanding of what people want to help them decide; current processes don’t provide the best grounding for informed consent and there’s inadequate public involvement. We’ve also found a lack of proportionality with researchers failing to adapt consent procedures in proportion to the burdens and consequences of the study. As a result, people are often not best helped to make an informed choice when asked to join a research study. To address these concerns, we considered how we might improve this aspect of research ethics review. Recognising the central importance of the dialogue between the volunteer and researcher, we’ve drawn up a model or flowchart of what we deem good consent practice, proposing consent should be built around four simple steps:
Step 1: Introducing the study and the choices: helping the potential participants get an overview of the proposal and introducing the key issues.
Step 2: Explaining all the details of the study using the detailed Participant Information Sheet.
Step 3: After a gap, if necessary, reviewing and checking understanding.
Step 4: Reaching agreement and recording consent.
These steps, we believe, could help all involved and this article lays out ways we might improve participant choice while complying with accepted principles and current regulations.
Oxford A research ethics committee’s concerns
Consent is one necessary foundation for ethical research and it’s one of the research ethics committee’s (REC’s) major roles to ensure the consent process meets acceptable standards to ensure fair and informed choice. Oxford ‘A’ REC is one of the three National Health Service (NHS) RECs based in Oxford that are overseen by the UK Health Research Authority. This REC includes both expert and lay volunteer members (15 in total) who review research proposals involving NHS staff and patients.
Several issues about the consent process raise concerns for Oxford A REC. Each of these concerns has been identified by others; they are not unique to our REC. However, we summarise them here to help contextualise our efforts to develop a model that might enhance the consent procedures. They include:
i. There’s a poor understanding of what people want to help them decide (Kirkby et al., 2012).
ii. Current processes and paperwork don’t provide the best grounding for consent (Brehaut et al., 2012; Innes et al., 2018; Wade et al., 2018). Both design and review place too much emphasis on the participant information sheet (PIS) when evidence is indicating that discussion is key (Flory and Emanuel, 2004) and people often don’t read what’s provided (Antoniou et al., 2011; Gillies et al., 2013; Reinert et al., 2014).
iii. There’s often a lack of proportionality, of failing to adapt consent procedures in line with the potential burdens and consequences of the study. This is particularly noticeable with simple and low risk studies where we often find the descriptions are out of proportion to reality.
iv. Much of the current material tends to focus on the interests of the researchers and sponsors rather than the participants (Xu et al., 2020). Consent is used to ensure the researchers and sponsors have ‘legal cover’, rather than as an aid to decision making. (Armstrong et al., 2012), and
v. There’s inadequate public involvement (Parris et al., 2015, Bhutta, 2004).
A possible model to shape consent
To address these concerns, we developed a model or flowchart that we use in our review. It proposes consent should follow four simple steps (Figure 1).
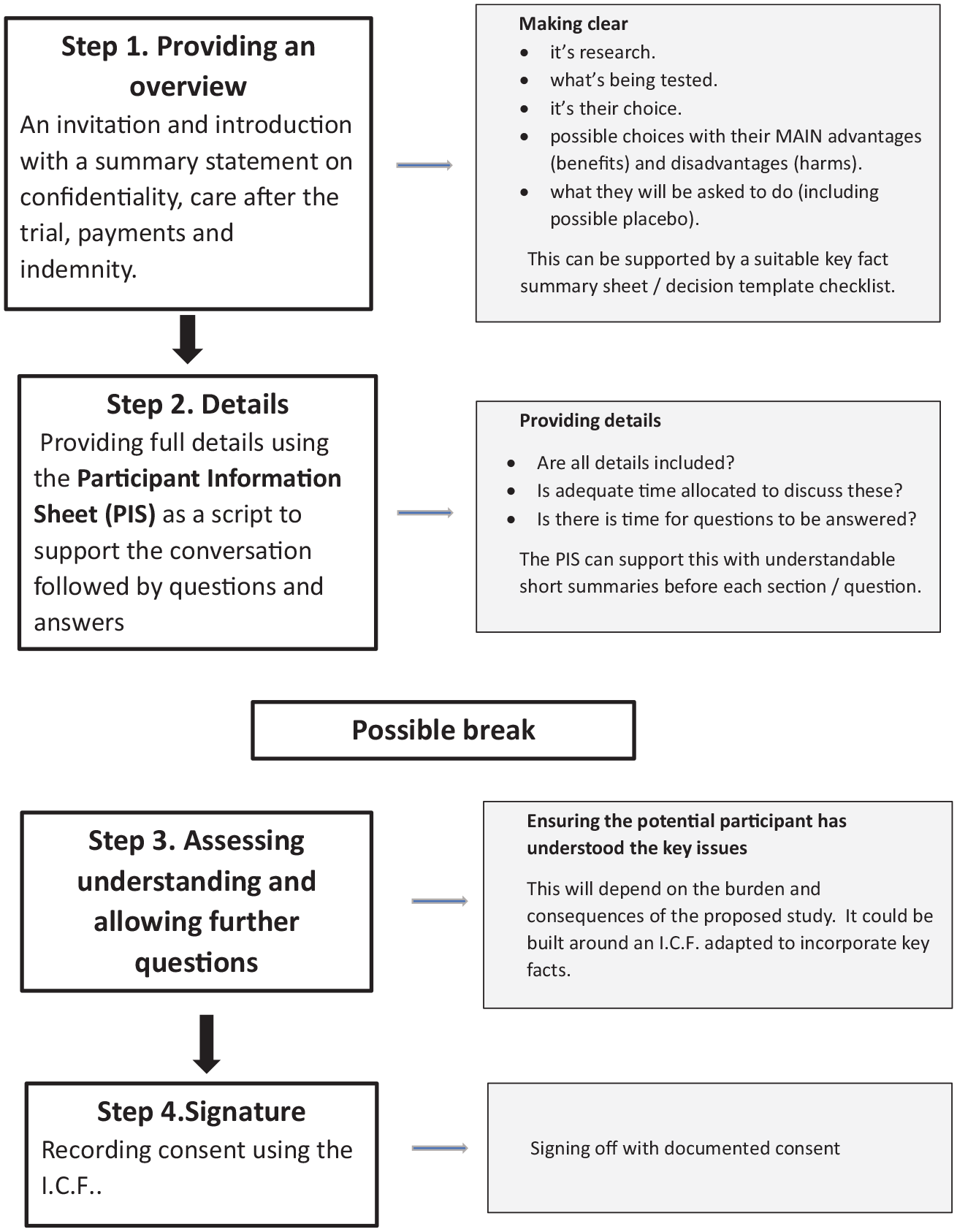
A four step model of consent.
Step 1: Introducing the study and the choices
This outlines the proposal introducing key, important issues to help the potential participant get an ‘overview’ of what he or she would be agreeing to.
Using evidence from published work on decision making (Innes et al., 2018; Wade et al., 2009; Elwyn et al., 2009) we designed a template ‘Key Facts Summary Sheet’ or ‘Decision making tool’ (Figure 2) that could underpin this introduction.

A template for a Key Fact Summary Sheet.
This is what we suggest for content:
• A straightforward and comprehensible title expanding the well-known acronym ‘I.P.O.C.’ to ‘I.P.P.O.C.’ – Intervention, Purpose, Population, Outcomes and (if relevant) Comparators.
• An invitation to join the research, including the fact they may withdraw without disadvantage.
• The study and its purpose in summary to include how it will provide benefit (and to whom).
• The choices for the potential participant and their significant, major consequences.
• How treatments will be allocated and the possibility of receiving a placebo.
• A short summary of confidentiality arrangements.
• Expenses and indemnity provided.
• End of study arrangements (if relevant).
Step 2: Explaining all details of the study
We accept that all details of a proposed study must be put before the potential participant. That’s the purpose of this step. It wouldn’t differ hugely from current practice, but we suggest placing greater emphasis on the PIS as a script for a conversation starting by explaining this and how long consent might take. Each question or section could start with a short summary to help the conversation. This then also provides material for any further consideration.
Steps 1 and 2 might need to be separated from Steps 3 and 4 by a suitable gap.
Step 3: Review and checking understanding
The purpose of this step is to ensure that the potential participants understand both the key facts and the details of the study after the preceding first two steps. To help, we took a standard informed consent form (ICF) and reshaped it using the key facts from Step 1 to lay out what the volunteer really needs to understand and is agreeing to (Figure 3).

A template for possible Informed Consent Forms.
Step 4: Reaching agreement
Confirming consent, either face to face or electronically, by initialling all items (to which they agree) on the ICF, then providing a full, dated, signature or mark, most likely in writing. This is no different to current practice.
Does our model address these concerns?
i. There’s a poor understanding of what people want to help them decide and current processes and paperwork don’t provide the best grounding for consent.
We’ve tried to use the evidence that is available to us.
• Step 1 is shaped around available decision-making evidence. In their work, Innes et al. (2018) tried to find out what a sample of participants and research nurses felt were necessary for fair decision-making. Of the items they identified, only eight were not in our Key Fact Summary Sheet (but would be in the PIS). All fell outside their ‘Top Ten’. Brehaut et al. (2012) modified a clinical evidence-based decision-making tool and identified 32 issues of relevance to consent for research. Only seven, minor ones, were not included in our Key Fact Summary Sheet.
• It’s generally accepted that discussion is the most important part of the consent process (Flory and Emanuel, 2004) so we’ve tried to shape our model around this dialogue. However, more is needed. Nusbaum indicated that it may not be adequate to simply ask participants if they have any questions; they may need help to frame these questions (Nusbaum et al., 2017) and the structure of the discussion is important. Techniques such as Teach Back/Talk Back in which important material is highlighted, repeated and discussed specifically to ensure understanding might help (Flory and Emanuel, 2004; Yen and Leasure, 2019).
• Timing and processes matter. Joffe et al. (2001) demonstrated that improved participant understanding was associated with time-separating the describing of the proposal and the seeking of consent. Hence, we suggest a possible gap between steps 2 and 3 where necessary, and reshaping the ICF in Step 3 to recap and ensure understanding.
• We haven’t abandoned the key role of a PIS. This, we thought, would be a step too far and unacceptable to many stakeholders. Instead, we suggest a Key Fact Summary Sheet to shape Step 1 and modifications to the PIS (Step 2) to put them both at the centre of a conversation, while retaining the PIS’ function as a resource and record for the potential participant to use whenever they wish.
ii. There’s often a lack of proportionality, failing to adapt consent procedures in line with the potential burdens and consequences of the study.
Recognising the wide range of research proposals that we review, these steps can be adjusted to a particular study’s potential consequences and burdens. Adequate for most questionnaires would be Step 1 alone. For minor studies (data collection, interview or studies with no new experimental treatment interventions), Steps 1 and 4 might suffice. While more challenging, complex studies would require all four steps. In these studies there may be things that the potential participants really must be informed about and understand. For these cases, we suggest researchers might:
• Ensure key issues are itemised on the consent form.
• Use a ‘Report back question and answer’ technique for which the potential participants describe what they understand. (Flory and Emanuel, 2004; Yen and Leasure, 2019).
• Involve the patient’s clinician or other clinical advocate, to ensure key issues are explained and understood.
• Ensure the standard Informed Consent Form (Figure 3) includes key issues raised during the consent processes.
• Set a short multiple-choice questionnaire before seeking final consent to assess understanding.
• Record the consent discussion (video or audio) for the participant and provide this for later reference and as a permanent record.
iii. Much of the current material tends to focus on the interests of the researchers and sponsors rather than the participants.
We believe that consent is often seen as a means to ensure the researchers and sponsors provide themselves with adequate ‘legal cover’, rather than as an aid to decision making. As a result, materials or processes are written to be comprehensive rather than comprehensible with focus on the researchers’, not the participants’ interests. Again, we recognise the need for a detailed PIS but feel the balance between ‘exculpation’ and a ‘respect for the potential participant’s autonomy’ needs redressing. We suggest three ways to address this:
• Providing a comprehensible introduction
• Situating the PIS into the context of a discussion, recognising that this is the critical part of consent.
• Modifying the ICF to cover issues that will be of concern to the potential participant.
iv. There’s inadequate public involvement.
We don’t address Public and Patient Involvement (PPI) directly in this model but we recognise its importance. In our routine review process we evaluate any PPI undertaken as, in our experience, contributions from patient groups and other relevant partners tend to improve the research design and the clarity of the consent process. This framework was drawn up by the full committee and the lay members have made significant contributions. We are actively seeking feedback from PPI groups.
Conclusions
Research ethics committees can be seen to have two primary spheres of influence on today’s research through:
(i) The modifications that are required for a favourable opinion and
(ii) Contributing to the broader conversation about ethical research and best practice beyond our committees.
We feel our approach can contribute to both; it can help REC members conduct their ethical review and it can help REC chairs and managers to correspond with applicants (much of which is around consent) more effectively. If these ideas are accepted and shared more widely, they might be incorporated into the broader debate with patients, public, professionals, researchers, regulators and reviewers.
However, we’re wary of suggesting further guidance. The library shelves groan and the overloaded i-clouds above are heavy with such documents. We also recognise the current reality that researchers and those designing consent processes may be straightjacketed by the requirements of their sponsors and institutions. However, we would argue that our proposals are neither radical nor revolutionary, requiring few modifications to current practice as they build upon current practice and guidance. Critics may say there’s little new in our approach (and this wouldn’t upset us). For most research proposals these suggestions would only require modifications of existing material. As an example, our Key Fact Summary Sheet matches 20 of the 22 consent recommendations in International Council for Harmonization Good Clinical Practice (ICH GCP) and also meets USA Food and Drug Administration Common Rule requirements (U.S. Common Rule 45 CFR 46.116(a)(5)(i)). Generic documents could still be developed which allow for modifications to accommodate local circumstances, culture and needs.
Our model, however, doesn’t cover all aspects of consent; we focussed upon processes and information. We didn’t include the necessary voluntariness of consent nor the need to ensure potential participants have the capacity to make a decision. We confined our attention to the necessary information for informed consent and how it might be presented. Even in just this respect, we recognise that determining the right level of detail requires judgement and that remains in the hands of researchers and RECs.
We believe our suggested approach, with its limited demands, is warranted and hope our ideas might help RECs and researchers improve consent procedures; we hope that it is not regarded as simply additional paperwork to be dutifully and unimaginatively filled in. We’ve been impressed by the thought and effort that so many applicants have put into the studies that this needn’t be the case. Finally, we recognise the need for our suggestions to be debated and tested. If care and research must be evidence based, then so should REC review.
Footnotes
Funding
All articles in Research Ethics are published as open access. There are no submission charges and no Article Processing Charges as these are fully funded by institutions through Knowledge Unlatched, resulting in no direct charge to authors. For more information about Knowledge Unlatched please see here: ![]() .
.