
Abstract
In the chaotic market of fur goods, genetic distinction is increasingly important for identifying species. A vast diversity of species identification methods has been proposed, while little is developed, particularly those easy, fast, and cost-effective ones. In this study, a simple and reliable novel loop-mediated isothermal amplification method for identifying cytochrome c oxidase I of felis and vulpes was established. It saves laborious post–polymerase chain reaction procedures and shortens the time for high-fidelity gene amplification. The sensitivity of this method for felis and vulpes identification, which is well matched to quantitative polymerase chain reaction, could be 10 or 1.0 pg, respectively. Predominantly, the sensitivity of loop-mediated isothermal amplification is more tolerant to those polymerase chain reaction inhibitors such as pigments, dyes, or other fur ingredients, compared to quantitative polymerase chain reaction. Even without costly specialized equipment, a water bath is sufficient for genetic distinction. Our approach is a new technique with broad application perspective, such as on-site species identity tests, commercial fraud, and wildlife crimes.
Introduction
Fur has long served as an important source of clothing for humans for its aesthetic and insulating properties. Usual animal sources for fur garments and trimmed accessories include cat, fox, rabbit, otter, sable, beaver, sheep, and possum. Nowadays, there are various kinds of fur goods of varying quality on market including fake and shoddy products. For great fortune, sham manufacturers use dyed artificial or cheap animal fur to mimic exotic animal patterns and to imitate the feel of a soft velvet fabric. For customers, it is difficult to choose a genuine fur product just judging from its appearance. Therefore, species identification provides an alternative method for verification.
Coupled with two specific primers, conventional polymerase chain reaction (PCR) method uses DNA polymerase, dNTP (deoxy-nucleotide-triphosphates), Ck, and bivalent cations to amplify the targeted DNA region. Currently, it contains complex steps such as multiple thermal cycles and electrophoresis and has been widely used for gene amplification since the concept was put forward in the early of 1980s. 1 However, the extraction of pure high-quality DNA is the key for conventional PCR. Ironically, it is difficult to extract pure high-quality DNA from fur products. 2 The complicated processing techniques cause damages to the target’s fragile DNA by oxidation. On the other side, most dyes in fur samples suppress the gene amplification of PCR, even for natural fibers which contain pigments as PCR inhibitors. 3 All these factors restrict the conventional PCR method’s wide application in fur products. Moreover, conventional PCR method requires special techniques and costly sophisticated equipment which may be unavailable to laboratories in low resource areas. Thus, simplification of gene amplification with more tolerance to sample ingredients as awaited in many fields has yet to be realized.
An alternative DNA technique known as loop-mediated isothermal amplification (LAMP) which was put forward by Notomi and colleagues4,5 in 2000 sheds light on the simplification of genetic testing, making possible a more rapid and more affordable technique. Using four specific primers and DNA polymerase with strand displacement activity, LAMP forms the loop structure at the end. This stem-loop structure allows the 3′ end to initiate self-extension process and synthesize new DNA strand with strand displacement manner. Besides, it has been reported that LAMP’s sensitivity is less affected by various components of samples. 6 The LAMP assay is a novel nucleic acid amplification method with high sensitivity and specificity to discriminate single nucleotide differences. Coupled with a strand-displacing Bst DNA polymerase, two or three sets of specially designed inner primers (FIP (forward inner primer) and BIP (backward inner primer)), outer primers (F3 and B3), and loop primers guarantee a higher specificity. Loop primers are not requisites. However, the amplification time could be shortened to one-third as usual by the usage of loop primers. 7 Moreover, LAMP is conducted under isothermal conditions (60°C–65°C); Therefore, a simple water bath or block heater is sufficient to the highly specific gene amplification. It also has other advantages over conventional PCR in detection of DNA amplification. This assay allows visual detection by naked eyes through the addition of fluorescent dye. The usage of DNA-intercalating dyes enables real-time monitoring of LAMP reactions by measuring the fluorescence. Shorter reaction time, more cost-effective, higher specificity and sensitivity make it an attractive testing method for field application.1,8
Cytochrome c oxidase I (COI) has been used as a DNA barcode to identify animal species. On one side, mutation rate is fast enough to distinguish species; on the other side, COI sequence is conserved among conspecifics. It is universally acknowledged that COI is suitable for this role. Therefore, a highly sensitive and specific fluorescence-based real-time LAMP assay for conserved COI gene has been developed to identify felis and vulpes from fur goods. The aim of this study was to develop a novel method suitable for on-site inspection. It has dominant advantages over quantitative polymerase chain reaction (qPCR) in practical application.
Materials and methods
Chemicals and samples
Proteinase K solution (40 U/mg) was purchased from Sangon Biotech Co., Ltd. (Shanghai, China) and tris (hydroxymethyl) aminomethane (Tris) from Sigma-Aldrich Co. LLC (Germany). Phenol was purchased from Beijing Solarbio Technology Co., Ltd. (Beijing, China). Hexadecyltrimethylammonium bromide (CTAB), chloroform, and iso-amyl alcohol were provided by Sinopharm Group Co., Ltd. (Shanghai, China). The DNA amplification kit was purchased from Loopamp, Eiken Chemical Co., Ltd. (Tokyo, Japan).
Fur samples of eight commonly used species, namely, cat, rabbit, weasel, sheep, mink fox, muskrat, and otter were purchased from International Furs Trading Center, Daying, China. All samples were transported under ice-chilled condition (4°C) and stored at −20°C for future work. The authenticity of all samples was verified by PCR method.
Preparation of DNA
DNA was extracted by using the classical phenol-chloroform extraction (CTAB method). 9 A total of 3 g of the homogenized sample was mixed with 10 mL CTAB extraction solution and 10 µL proteinase K solution. The mixture was incubated at 50°C under shaking overnight. DNA was extracted in duplicate. After centrifugation at 8000 r/min for 5 min, 1 mL of supernatant was transferred into a new 2 mL tube and mixed with 500 µL phenol and 500 µL chloroform/iso-amyl alcohol (25:24:1, v/v/v). After 30-s vibration, the mixture was centrifuged at 13,000 r/min for 10 min. Aqueous phase of 800 µL was transferred into a new 2 mL tube and mixed with 400 µL chloroform /iso-amyl alcohol (24:1, v/v). After centrifugation at 13,000 r/min for 10 min, 600 µL of aqueous phase was transferred into a new 1.5 mL tube and mixed with 600 µL iso-propanol. The mixture was incubated at −20°C for at least 30 min before centrifugation at 13,000 r/min for 5 min. After that, the supernatant was carefully drawn off with a pipette and the pellet was dried. The dried pellet was dissolved in100 µL sterile water. The concentration and quality of the DNA was determined by measuring its absorbance at 260 nm (A260) and 280 nm (A280) with a Nano-spectrophotometer (WPA-Biowave II, England).
Primer design
Appropriate primer design is essential for amplification by loop-mediated isothermal amplification. Use of primer designing software exclusively for the LAMP method is recommended. Refer to LAMP designing software, PrimerExplorer, at the website: http://primerexplorer.jp/e/.4,5 PrimerExplorer is a primer designing software, which generates the primer sets based on the target sequence information and meets the primer designing requirements such as Tm calculation and primer end’s stability calculation. One primer set contains four primers, FIP, F3, BIP, and B3. Primers for LAMP are listed in Table 1.
Sequence of LAMP primers.

LAMP: loop-mediated isothermal amplification; FIP: forward inner primer; BIP: backward inner primer.
LAMP
The oligonucleotide primers used for the LAMP assay amplification were selected based on highly conserved region (COI) of the target species. A commercial DNA amplification kit (Loopamp; Eiken, China) was used in this study. This kit is a simple reagent kit for the amplification of DNA by the loop-mediated isothermal amplification method. It contains 0.6 mL 2× reaction mix, 60 µL Bst DNA polymerase, 1.0 mL distilled water, 25 µL primer mix DNA (primers for positive control DNA amplification), and 0.1 mL positive control DNA. Briefly, 11.5 µL of the reaction mix was dispensed into each reaction tube. Then 1 µL sample DNA was added to the reaction tube and the final volume of the solution should be 25 µL in total. For control reactions, 1.0 µL of distilled water was used as negative control and 1.0 µL positive control DNA as positive control. Mix the solution well by pipetting. It was run in an ABI 7500 PCR system (Applied Biosystems, Foster City, CA, USA). For visual detection, 1.0 µL of the Loopamp Fluorescent Detection Reagent was also added before reaction. The reaction procedure for target DNA was conducted by 80 cycles of 63°C, 60 s and 64°C, 60 s, and then 95°C, 5 min for denaturation. All LAMP reactions were carried out in duplicate. The fluorescent signals of the solutions were observed under ultraviolet (UV) lamp (240 nm). Furthermore, the LAMP products were also visualized by 2% agarose gel electrophoresis stained with ethidium bromide solution (1 µg/mL).
Analytical sensitivity and specificity of LAMP and qPCR
The LAMP specificity was tested using the DNA samples of other animals, including rabbit, weasel, sheep, mink, muskrat, and otter. The analytical sensitivity of LAMP and qPCR for COI gene was evaluated against 10-fold serial dilutions of DNA ranging from 10 ng to 1 pg. The dilution series were tested in duplicate with the LAMP to determine the minimum amount of template DNA per reaction that could be detected by each protocol.
Real-time PCR
COI gene was selected as the target for qPCR. The primer sequences and probes used in this study are shown in Table 2, which are provided and defined in our National Standard10,11 (SN/T 3730.6-2013 for felis and SN/T 3730.3-2013 for vulpes). The qPCR reaction mixture (25 µL) contained 2 µL DNA template, 5 pmol probe, 10 pmol of each forward and reverse primers, 12.5 µL Premix Ex Taq™ (2X, TAKARA), and sterile water. A negative control (distilled water instead of template DNA) was included in each run. The reactions were carried out at 30 s at 94°C, and then 80 cycles for 94°C, 5 s and 60°C, 34 s.
Sequence of qPCR primers.
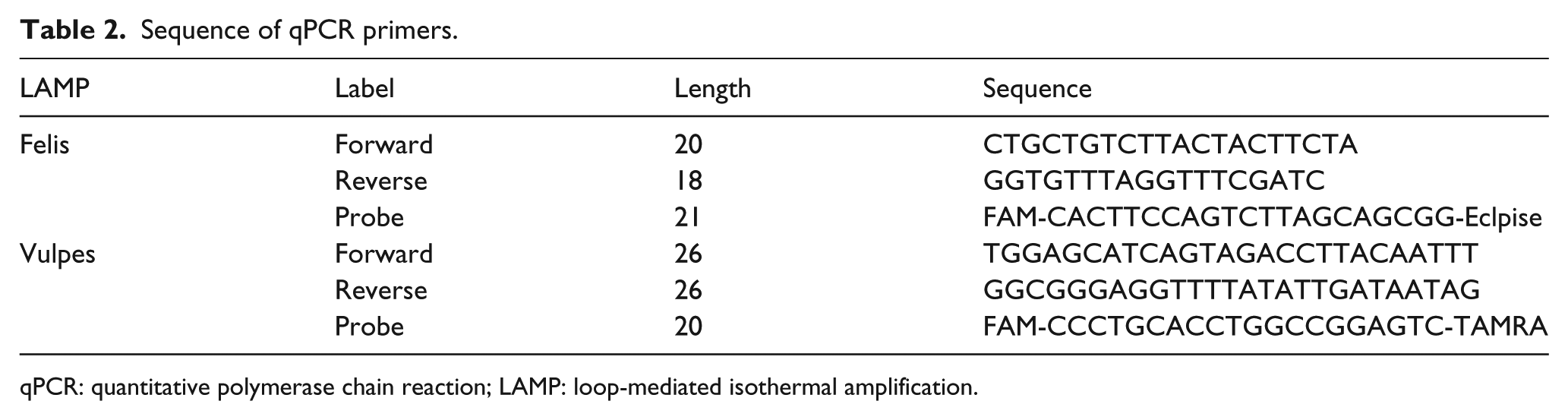
qPCR: quantitative polymerase chain reaction; LAMP: loop-mediated isothermal amplification.
Results
Specificity of LAMP and its product confirmation
Only cat DNA could be detected with naked eyes by observing the color changes (see Figure 1(a) and (c)). The positive reaction visually turned green (especially under UV lamp) (see Figure 1(d)) and the amplicons appeared as a characteristic ladder of multiple bands of different sizes on a 2.0% ethidium bromide–stained agarose gel (see Figure 1(g)), while the negative responses remained pale orange in normal light (see Figure 1(c)). Besides, the amplified target’s signal was real-time recorded in the positive reaction (see Figure 1(a)). No amplification was observed in other animals’ DNA samples, suggesting that the primers for felis’ COI were highly specific in this study. The specificity test for vulpes’ LAMP similarly showed that no amplification occurred in DNA samples other than that of fox (see Figure 1(b), (e), (f), and (h)).

The specificity of the LAMP method among eight kinds of commercial animals. (a, b) The real-time records of amplification reaction per tube run in ABI 7500 PCR system. (c–f) The detection of LAMP products by naked eyes (c, e) and under UV light (d, f). (g, h) The visualization of LAMP products on 2% agarose gel.
The specificity of the LAMP primers was further confirmed by PCR amplification and subsequent sequencing of the products using two outer primers B3 and F3 (see Table 1). Sequence analysis showed that the obtained amplified sequences were identical to the corresponding COI sequences recorded in GenBank (accession numbers KJ192803 for felis and JF443559 for vulpes). Besides, the primers in the LAMP reaction did not induce self-amplification in the absence of the DNA template.
Sensitivity of LAMP
The analytical sensitivity of LAMP was evaluated against 10-fold serial dilution of target DNA from 10 ng to 1 pg. The results are shown in Figure 2. Concentrations of 10 ng–10 pg of the felis DNA could be amplified, whereas concentrations below this range were not detectable (see Figure 2(a), (c) and (d)). For vulpes, the detection limit of LAMP could be as low as 1.0 pg (see Figure 2(b), (e) and (f)).

The sensitivity of the LAMP method evaluated from 10 ng to 1 pg. (a, b) The real-time records of DNA amplification reaction per tube in ABI 7500 PCR system (a: felis and b: vulpes). (c–f) The detection of LAMP products under UV light (c, e) and on 2 % agarose gel (d, f). Lanes 1–8: Maker; LAMP products for 10 ng, 1 ng, 0.1 ng, 10 pg, and 1 pg target’s DNA per reaction, respectively; NC: negative control; PC: positive control, a digested plasmid as template.
Comparison of LAMP and real-time PCR
For real-time PCR, the sensitivity of both species could reach 1 pg (see Figure 3(a) and (c)), and the standard curve generated by qPCR using 10-fold serial dilutions of DNA was linear over five orders of magnitude (see Figure 3(b) and (d)). R2, the goodness of fit, reached 0.9644 for felis and 0.9344 for vulpes. The LAMP sensitivity was well matched to that of qPCR.

Results of real-time PCR analysis. (a, c) The amplification curves with a table of Ct values of different DNA concentrations from 10 ng to 1 pg. (b, d) Relationships between the serial dilution of DNA and Ct values. The crossing point values are plotted against the log of the initial template concentration.
Application in examples
Three pieces of fox fur products (Samples 8, 9, and 10) and one piece of cat hair goods (Sample 11) were prepared for functional comparison between this LAMP method and qPCR (see Figure 4). Those extracted DNA were diluted to10, 5, 1, and 0.1 ng in per reaction and run in two different methods.

Appearances of four samples. (a) Sample 8, thick, soft green fox fur; (b) Sample 9, abundant black fox fur; (c) Sample 10, hairy brownish fox fur; and (d) Sample 11, pubescent white cat fur.
As shown in Figure 5, all these samples of four concentrations proved positive by LAMP method (see Figure 5(a) to (c) and (g)). However, by qPCR method, only Sample 9 of all four DNA concentrations was fully validated (see Figure 5(e)) and Sample 11 of 10 and 5 ng was confirmed positive (see Figure 5(h)). Through qPCR method, Samples 8 and 10 could not be validated (see Figure 5(d) and (f)), suggesting that the qPCR method applied in fur species distinction may drastically increase the possibility of false-negative. The validation results were also supported by the observation of color changes (see Figure 5(i) and (j)) and visualization of LAMP products on agarose gel (see Figure 5(k) and (l)). This may result from the PCR-inhibitory ingredients in sample itself. In contrast, LAMP is more tolerant to those PCR inhibitors such as pigments, dyes, or other fur ingredients.

Results of LAMP tests on these samples. (a–c) The real-time records of LAMP tests for 10, 5, 1, and 0.1 ng vulpes’ DNA per reaction. (d–f) The real-time records of qPCR tests for 10, 5, 1, and 0.1 ng vulpes’ DNA per reaction. (g) The real-time record of LAMP tests for 10, 5, 1, and 0.1 ng felis’ DNA per reaction. (h) The real-time record of qPCR tests for 10, 5, 1, and 0.1 ng felis’ DNA per reaction. (i, j) Detection of LAMP products for10 ng sample DNA under UV light (at 240 nm). (k, l) Visualization of LAMP products on 2% agarose gel.
Discussion
COI has been the barcode for genetic distinction. 12 This maker here allows us to identify felis and vulpes among other seven common livestock animals. As reviewed by Bottero and Dalmasso, 13 various PCR-based methods have been proposed for identifying farm animals. Notwithstanding, little is documented about methods for distinguishing world-widely distributed poultry and livestock animals, particularly those easy, fast, and cost-effective ones. 14 In this study, a highly sensitive, specific, and tolerant fluorescence-based real-time LAMP assay has been developed to identify felis and vulpes from fur goods. The ABI Gene-Amp PCR system is not requisite, which is replaceable of a water bath but loses the capability of real-time monitoring. The herein LAMP method has considerable improvement in efficiency, simplicity, and operability, compared with other molecular methods, such as qPCR, DNA sequencing, and terminal restriction fragment length polymorphism. 15
Those advantages render the LAMP technique a broad perspective of many usages, especially in the rapid genetic on-site identification. 16 It has the most potential to be an important tool at points of entry such as sea- and airports. It could be used to prevent introduction and spread of economically harmful pest species that are unintentionally transported by the global trade. It could be applicable to the wildlife forensics in the fight against poaching and rapid detection of epidemic disease due to parasites and viruses.17–19
Admittedly, it is difficult to acquire high-quality DNA from fur products, in terms of quantity and pureness. 20 This is due to the complicated processing technologies. Coloration may make DNA more fragile and fragmented, while bleaching with hyperoxide may cause DNA damages by oxidation. LAMP is a robust reaction and it can tolerate inhibitory substances in biological samples better than PCR. 6 Besides, those primers (two to three pairs) specially designed for LAMP recognize six distinct regions on target DNA, conferring a high degree of specificity to the reaction. 21 These features guarantee the positive chances in testing. Comparably, due to those inhibitory factors in sample itself, the qPCR method increases the false-negative possibility in fur products. Moreover, a hypothesis that further diluted sample DNA, more chances of false-negative is proposed.
In this study, this LAMP method was employed for qualitative analysis. However, more advances have been achieved in the LAMP technology. Dugan et.al. 22 demonstrated that those positive LAMP results were obtained without nucleic acid extraction such as boiling, lysis, and DNA purification. It greatly simplified and shortened the detection process. Moreover, simultaneous multiple target detection in one single real-time LAMP reaction tube has already been developed. 23 Nathan et.al. modified the standard LAMP primers to contain a quencher–fluorophore duplex region so that a series of fluorescent signals could be gained upon strand separation. This method permits one to four target sequences in one reaction, extending the utility of this technique. In the near future comes more improvement in detection method.
Conclusion
A novel loop-mediated isothermal amplification assay for identifying felis and vulpes in fur goods was developed. To the best of our knowledge, there is little literature about the application of LAMP into textiles and fibers. Although qPCR method has been used into this area, factors such as natural dyes and fur processing technique have a major influence on amplification. Moreover, our article provides strong evidence for the high tolerance of LAMP to dye and poor-quality DNA.
LAMP is an efficient way to identify species, especially when fibers are processed and dyed. The detection limit of this LAMP method, a well-matched counterpart of Taqman qPCR, was as low as 1 pg for vulpes and 10 ng for felis. More importantly, this method alleviates the need for laborious post-PCR processing, such as electrophoresis and notorious ethidium bromide dying, making it more convenient and faster than traditional PCR analysis. Even without costly specialized equipment, one water bath is sufficient for the genetic distinction. This method could be applicable to the genetic identification in fur products.
Supplemental Material
Supplementary_materials – Supplemental material for Species identification of felis and vulpes by a novel loop-mediated isothermal amplification assay in fur products
Supplemental material, Supplementary_materials for Species identification of felis and vulpes by a novel loop-mediated isothermal amplification assay in fur products by Shunji Yu, Wenjia Gu, Yi Yu, Qinfeng Qu and Yi’nan Zhang in Journal of Engineered Fibers and Fabrics
Footnotes
Acknowledgements
Many thanks to Yuanyuan YU and Beini Pang for their generous technical and data support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research is supported by Technical Standard Project of Shanghai “Scientific and Technological Innovation Action Plan” to Yi’nan Zhang (16DZ0501700).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.