
Abstract
Mycoplasma gallisepticum infections impose a significant economic burden on the poultry industry. In the current study, a loop-mediated isothermal amplification (LAMP) assay was developed and optimized to detect M. gallisepticum based on a gene within the pyruvate dehydrogenase complex, the pdhA gene, which codes for the major subunit (E1α) in the complex. The reaction conditions were optimized, and the specificity was confirmed by successful amplification of several M. gallisepticum strains, while no amplification was detected with 20 other major bacterial and viral pathogens of poultry. Additionally, the LAMP assay achieved 10-fold higher sensitivity than an existing polymerase chain reaction (PCR) method. The LAMP assay was applied to swab samples collected from poultry farms and compared with PCR. The positive detection rate was 20.2% (37/183) by LAMP and 13.1% (24/183) by PCR. The LAMP assay could provide a cost-effective, quick, and sensitive method for the detection of M. gallisepticum.
Introduction
Mycoplasmas belonging to the class Mollicutes are the most widespread conditionally pathogenic microorganisms in nature. 21 Characteristic traits of this class include small genomes and the absence of a cell wall. 22 Mycoplasma gallisepticum is the primary pathogenic agent of avian mycoplasmosis, which is characterized by chronic respiratory disease in chickens, sinusitis in turkeys, and conjunctivitis in certain other species such as finches. 13 The economic impact of M. gallisepticum on the poultry industry is significant, causing reduction in weight gain, low feed conversation, and a drop in egg production in layer flocks.7,14 Crucially, coinfection with other bacterial and/or viral pathogens dramatically increases morbidity and mortality, including Escherichia coli, Newcastle disease virus, avian Influenza A virus, and infectious bronchitis virus (Avian coronavirus).11,23,26
Although the culture-based procedure is considered to be the gold standard for definitive diagnosis, M. gallisepticum culture is time-consuming and laborious, usually requiring 7–30 days to obtain the result. 24 Furthermore, the results of culture are often obscured by overgrowth or nutrient depletion by other bacteria. 27 Serological methods are the most commonly applied method for the detection of M. gallisepticum in poultry (Standard test procedures for Mycoplasma. National Poultry Improvement Plan and Auxiliary Provisions Animals and Animal Products section 147.7 2004;Part 147. Available from: https://www.law.cornell.edu/cfr/text/9/147.7). However, these serological assays are often complicated by cross-reactions with different species of Mycoplasmas spp., such as Mycoplasma synoviae and Mycoplasma meleagridis.1,6,8
Polymerase chain reaction (PCR) assays, which are characterized by high sensitivity and specificity, are increasingly used in disease diagnosis, with many PCR methods having been established to detect M. gallisepticum.9,12,24 Real-time PCR methods for the diagnosis of M. gallisepticum have been described.17,25 However, PCR-based methods have practical problems such as a high capital cost for the instrumentation, complex procedures, and a need for experienced personnel. 20 Therefore, in order to detect M. gallisepticum infection with limited resources, the development of a cost-efficient, simple, and rapid detection method is required.
Loop-mediated isothermal amplification (LAMP), a novel nucleic acid amplification method, was developed in 2000. 19 LAMP relies on an auto-cycling strand displacement DNA synthesis performed by the Bst DNA polymerase large fragment under isothermal conditions of 60–65°C. The assay is highly specific, and the amplification efficiency is equivalent to that of PCR-based methods. 18 Furthermore, gel electrophoresis is not required because the LAMP method synthesizes large amounts of DNA that can be easily detected by turbidity or fluorescence. Although LAMP assays have been developed for detecting a range of avian pathogens,4,15,16 their application in the diagnosis of M. gallisepticum infection has not been reported, to our knowledge.
In the current study, a LAMP method targeting a gene within the pyruvate dehydrogenase complex, the pdhA gene, for detecting M. gallisepticum was developed and then applied to clinical samples. The pdhA gene was selected as the target because it is a highly conserved gene among M. gallisepticum based on an analysis of the sequences in GenBank.
Materials and methods
Bacteria and viruses
The bacteria, including Mycoplasma spp., and viruses used to evaluate the specificity of the assay are shown in Table 1. The virulent M. gallisepticum strain Rlow, the classical moderately virulent strain S6, and the low virulence strain F were provided by the China Veterinary Culture Collection Center (Beijing, China). The live attenuated vaccine strains—Vaxsafe M. gallisepticum ts-11 and Nobilis M. gallisepticum 6/85—were both purchased commercially.a,b Strain DC9604 was isolated from air sac samples of chickens with chronic respiratory symptoms in China. 5 These strains were cultured in a complete American Type Culture Collection (ATCC; Manassas, VA) broth medium containing bovine heart extract,c yeast extract solution,c 10% horse serum,d and 10% swine serum.d Single clones were grown on plates of ATCC broth medium with 1% Noble agare as described previously.2,3
Bacteria and viruses used in the current study.*
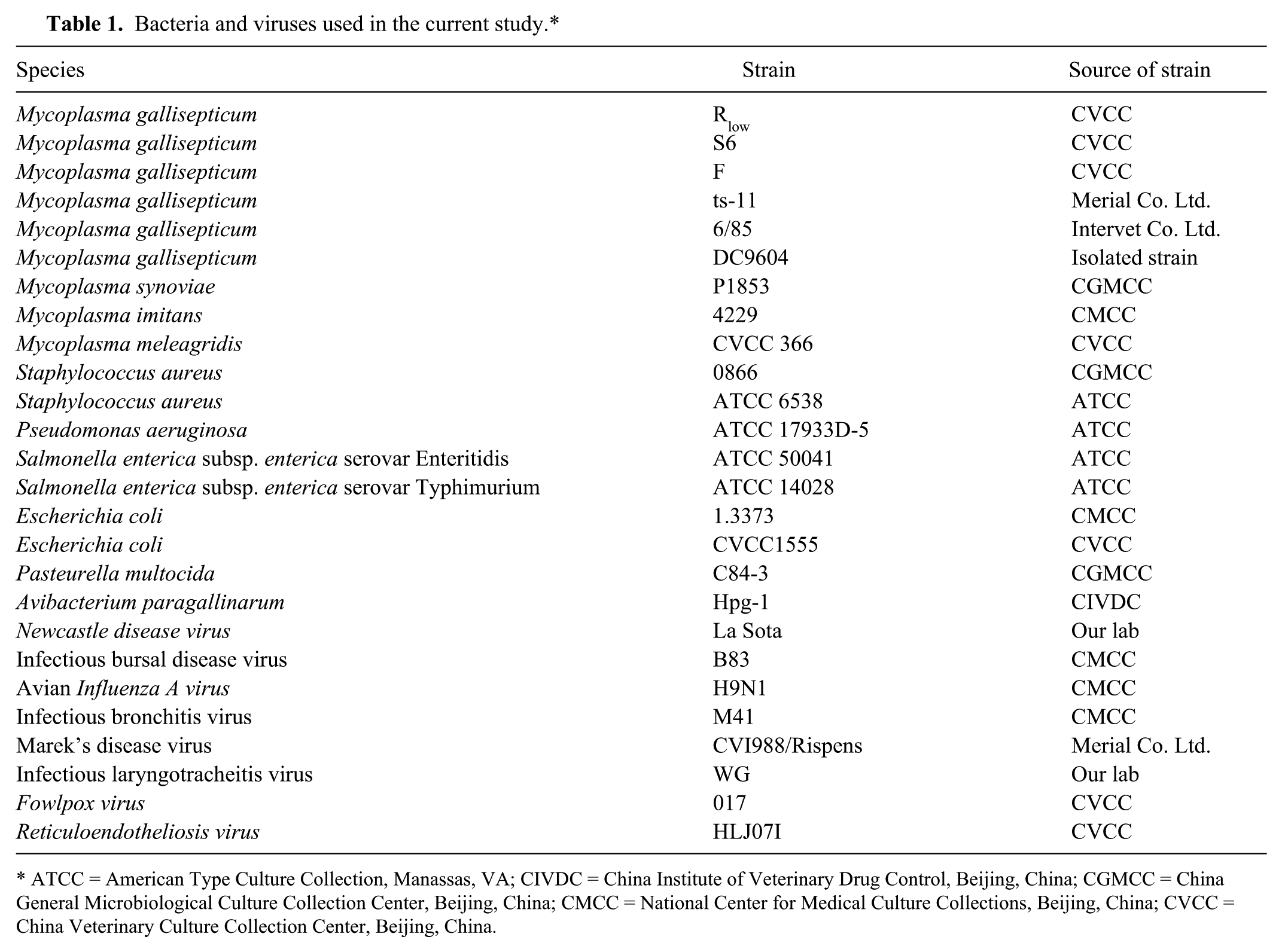
ATCC = American Type Culture Collection, Manassas, VA; CIVDC = China Institute of Veterinary Drug Control, Beijing, China; CGMCC = China General Microbiological Culture Collection Center, Beijing, China; CMCC = National Center for Medical Culture Collections, Beijing, China; CVCC = China Veterinary Culture Collection Center, Beijing, China.
DNA and RNA extraction
Genomic (g)DNA of the Mycoplasma strains were extracted as previously described, 3 while that of the other bacteria was obtained using a commercial extraction kitf according to the manufacturer’s protocol. Genomic DNA of infectious laryngotracheitis virus (ILTV; Gallid herpesvirus 1) and Fowlpox virus (FPV) were also extracted using a commercial DNA extraction kit.f Viral gRNA was extracted from virus-infected samples (cells, tissues, or allantoic fluid) using a commercial RNA extraction reagentd in accordance with the manufacturer’s protocol. First-strand complementary (c)DNA was synthesized using Moloney murine leukemia virus (M-MLV) reverse transcriptase.g The reaction mix, containing viral 10 μL of RNA, 1 μL of 10 mM random hexamer primer, and 9 μL of water, was incubated at 70°C for 10 min. Subsequently, 6 μL of 5× reaction buffer, 0.5 μL RNase inhibitor, 0.5 μL M-MLV reverse transcriptase, and 1 μL of 10 mM deoxynucleoside triphosphate (dNTP) mixture were added. The mixture was incubated at 37°C for 2 hr, and then at 75°C for 5 min. Genomic DNA or cDNA of each sample was quantified by spectrophotometery,h and stored at −20°C to be used as a template for the subsequent LAMP and PCR assays.
Primer design
A set of LAMP primers were designed targeting a region located within the pyruvate dehydrogenase complex, the pdhA gene, which codes for the major subunit (E1α) in the complex. 10 This region had been selected based on an alignment analysis that involved 6 pdhA sequences from M. gallisepticum strains, retrieved from the GenBank database and aligned by a commercial software packagei to identity the conserved region. The LAMP primers consisting of 2 outer primers (forward primer F3 and reverse primer B3) and 2 inner primers (forward inner primer FIP and reverse inner primer BIP) were designed with Primer Explorer V4 (http://primerexplorer.jp/elamp4.0.0/index.html). All of the primers used in this study were analyzed with BLASTn (http://www.ncbi.nlm.nih.gov/BLAST/) for specificity and then commercially synthesized.j The primer sequences are listed in Table 2, and their specific binding regions are shown in Figure 1.
The primers used in the loop-mediated isothermal amplification (LAMP) and polymerase chain reaction (PCR) assays.*
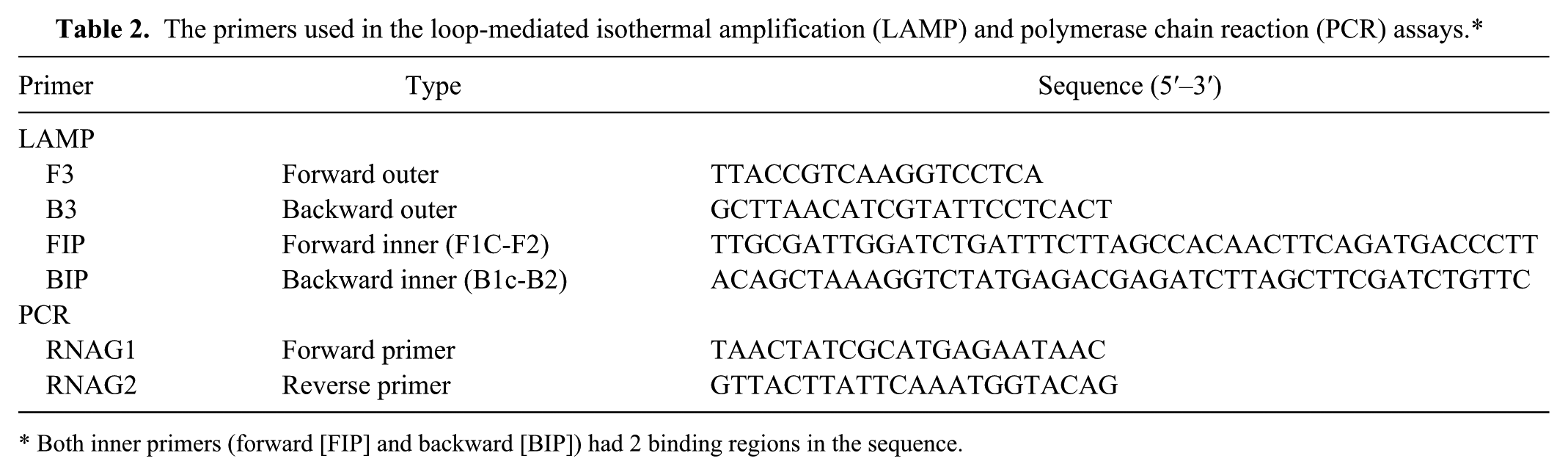
Both inner primers (forward [FIP] and backward [BIP]) had 2 binding regions in the sequence.
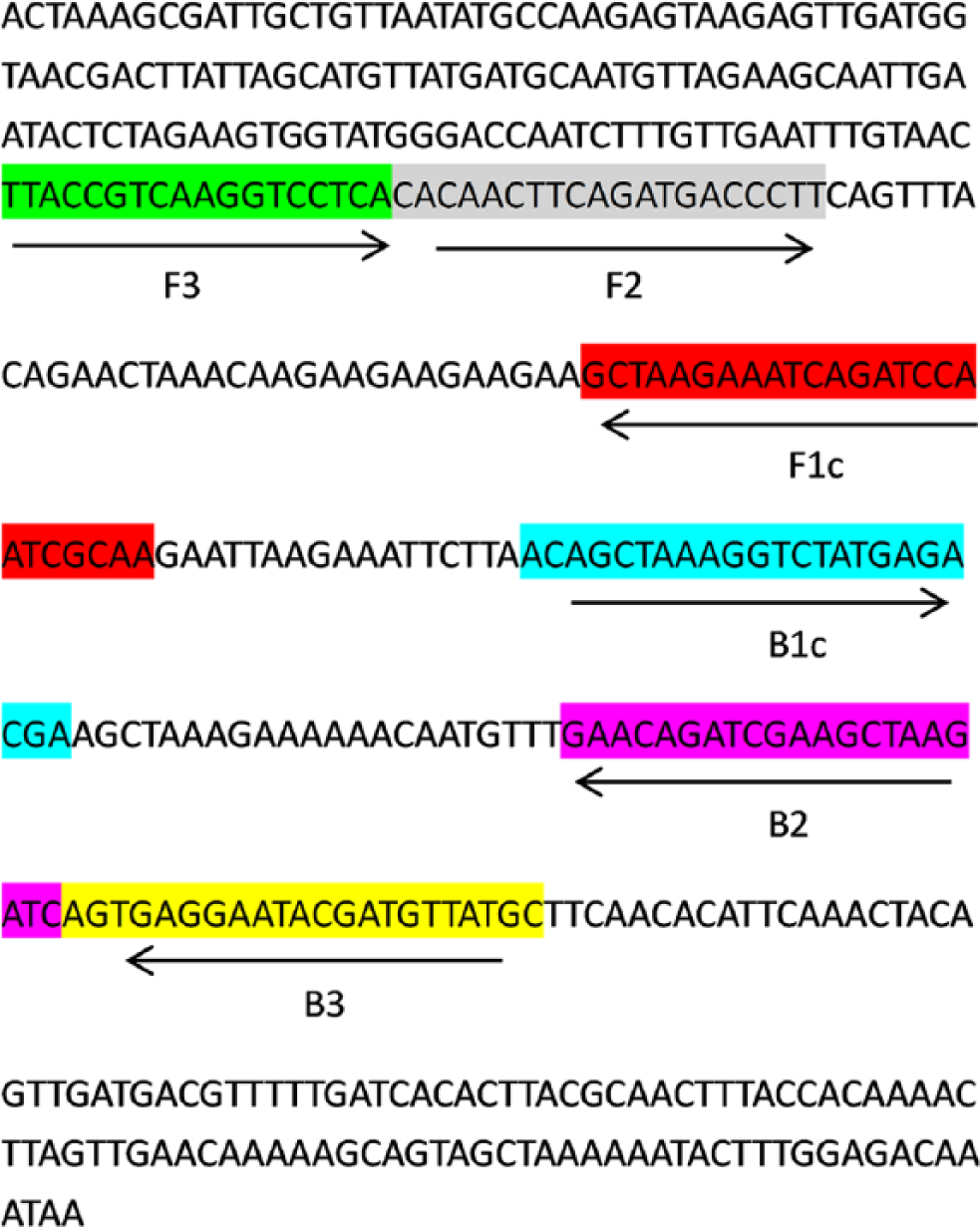
Location of loop-mediated isothermal amplification primers in pdhA gene of Mycoplasma gallisepticum. Forward inner primers (FIP) contained complementary sequence of F1 (F1c) and the F2 sequence. Backward inner primers (BIP) consisted of the complementary sequence of B1 (B1c) and B2 sequence.
Optimization of LAMP reaction conditions
The LAMP reaction conditions were optimized by incubating the mixture for 10, 20, 30, 40, 50, and 60 min. The reaction temperatures used were 55°C, 57°C, 59°C, 61°C, 63°C, 65°C, and 67°C. The concentration of MgSO4k (0, 2, 4, 6, 8, and 10 mM), betainee (0, 0.2, 0.4, 0.6, 0.8, and 1 M), and dNTPk (0.0, 0.4, 0.8, 1.2, 1.6, and 2.0 mM) was also optimized. The inner/outer primer ratio (1:2, 1:4, 1:6, 1:8, 1:10, 1:12) was also tested. The LAMP products were electrophoresed on 2% agarose gels and stained with a commercial agentl to determine the optimal reaction conditions.
Reaction conditions for LAMP
The LAMP reaction was performed in a final volume of 25 μL. The reaction mixture contained 2.5 μL of 10× LAMP reaction buffer, 1.6 mM of each dNTP,k 0.8 M of betaine,e 8 mM MgSO4,k 0.5 μL of Bst DNA polymerase large fragment,m 30 pmol each of inner primers FIP and BIP, 5 pmol each of outer primers F3 and B3, and 1 μL of target template. The mixture was incubated at 65°C for 30 min using a water bath and then heated at 80°C for 10 min to inactivate the Bst DNA polymerase.
Polymerase chain reaction
For performance evaluation of the LAMP assay, an existing PCR detection method was employed for the detection of M. gallisepticum as reported previously. 12 This PCR method, termed the MG PCR, was compared, in terms of sensitivity and specificity, with the LAMP assay. The primers for the MG PCR assay are shown in Table 2. The PCR was carried out using a commercial PCR reagent.n The cycling conditions for the MG PCR were as follows: 95°C for 4 min; followed by 40 cycles of 95°C for 30 sec, 60°C for 20 sec, and 72°C for 30 sec; and a final extension step at 72°C for 10 min. The PCR products were then analyzed by 2% agarose gel electrophoresis, and the target bands were visualized by staining with a commercial agent.l As required, the PCR products were purified with a commercial purification kitk and sequenced.d
Detection of LAMP products
The LAMP results were confirmed by 3 methods. The first method was gel electrophoresis and the observation of the typical ladder pattern of bands under ultraviolet light.o The second method was to stain the LAMP products with a commercial dye,d which changes in color from orange to yellow on binding to double-stranded DNA. The third method was to purify the lowest bands from LAMP amplicons using a commercial PCR purification kitk and a commercial vector,g with subsequent sequencing.d The outer primers were used as the sequencing primers. The resultant sequences were aligned using BLAST.
Sensitivity and specificity of the LAMP assay
Six M. gallisepticum strains were used to evaluate the specificity of the LAMP assay. In addition, other avian Mycoplasma spp., other bacteria, and some viruses were used as negative controls (see Table 1). Each organism was examined at least twice. Genomic DNA from M. gallisepticum Rlow was 10-fold serial diluted from 1 × 108 fg to 0.01 fg DNA/μL. The detection limit of the LAMP assay was compared with that of the MG PCR using the same diluted gDNA as template in triplicate. The LAMP assays were performed using the optimal reaction conditions described above.
Application of LAMP to clinical samples
One-day-old specific pathogen–free (SPF) chickens were purchased commerciallya and raised under controlled condition at 30°C. The chickens were housed in isolators under a 12-hr light and dark cycle with continuous access to food and water. Twenty 4-week-old chickens were divided into 2 equal groups. Ten chickens were challenged with ~1.0 × 106 colony forming units of M. gallisepticum strain Rlow (48-hr culture in broth) by intratracheal inoculation, and the other 10 chickens were treated with 1 mL of phosphate buffered saline (PBS). Three weeks postchallenge, tracheal swabs from both groups were collected as previously described. 29 Each swab was placed into a 1.5-mL microfuge tube containing 400 μL of sterile PBS, and vortexed for 2 min. The gDNA from these samples was then extracted by a commercial kit,n following manufacturer’s protocol. The MG PCR and the LAMP were then used to test for the presence of M. gallisepticum. Each sample was tested in triplicate. Animal experiments were approved by guidelines set by the Institutional Animal Care and Use Committee of the Shanghai Veterinary Research Institute. In order to confirm the reproducibility of the LAMP assay, gDNA extracts from 5 tracheal swabs of experimentally infected chickens were tested by the LAMP assay every 10 days for 40 days, meaning a total of 5 sets of tests were performed. For each test, 1 negative control without DNA template was included.
From May 2013 to April 2014, a total of 183 field tracheal swabs were randomly sampled from chickens from 13 different poultry farms in Anhui, Jiangsu, and Zhejiang provinces, China. These clinical samples were used to validate the applicability of the LAMP assay under field conditions. The DNA extractions were performed as described above and analyzed using both the LAMP and MG PCR assays. Tracheal swab samples collected from 10 healthy SPF chickens were used as negative control. Each sample was performed in triplicate.
Statistical analysis
Statistical significance was calculated using the chi-square test.p A P value <0.05 was considered statistically significant.
Results
Optimization of the LAMP method
In order to determine the optimum conditions of LAMP assays, 10 ng gDNA from M. gallisepticum Rlow was used as a target template and the results assessed by gel electrophoresis. The results showed that ladder-like bands appeared at betaine concentrations of 0 M–1 M (Supplementary Fig. 1A; Supplementary Figures 1–5, available at http://vdi.sagepub.com/content/by/supplemental-data). While amplification was detectable at all tested ratios of inner/outer primers, the ratio of 1:6 was chosen (Supplementary Fig. 1B). Amplification was detected at all the tested temperatures, and 65°C was chosen as the reaction temperature for all applications (Supplementary Fig. 1C). The minimal reaction time was 30 min (Supplementary Fig. 1D). According to these observations, the optimal LAMP reaction conditions were determined as the following: a reaction temperature of 65°C for 30 min, 8 mM concentration of Mg2+, Bst polymerase content of 0.5 μL, 1.6 mM of dNTP, 0.8 M of betaine, and an inner/outer primer ratio of 1:6. The tubes were heated at 80°C for 10 min to terminate the reaction.
Specificity of LAMP and PCR assays
Genomic DNA samples of 6 M. gallisepticum strains Rlow, ts-11, S6, F36, 6/85, and DC9604, were used to verify the specificity of the LAMP assay. All of the samples exhibited the typical ladder patterns in electrophoresis (Fig. 2). No cross-reactions were observed when the bacteria and virus listed in Table 1 were tested, even though the time of reaction was extended to 1 hr (Fig. 3). The lowest bands from the LAMP products from the M. gallisepticum Rlow strain were purified, and the nucleotide sequence was determined and found to match the pdhA sequence of M. gallisepticum in GenBank, confirming the specificity of the LAMP primers for M. gallisepticum. The specificity of the LAMP assay was also confirmed by observing a color change after adding a fluorescent dye.l A positive LAMP reaction exhibited a yellow color while a negative reaction corresponded to the original orange color (Fig. 2). Positive amplification in the MG PCR was determined by the presence of a 330-bp band and confirmed by DNA sequencing.
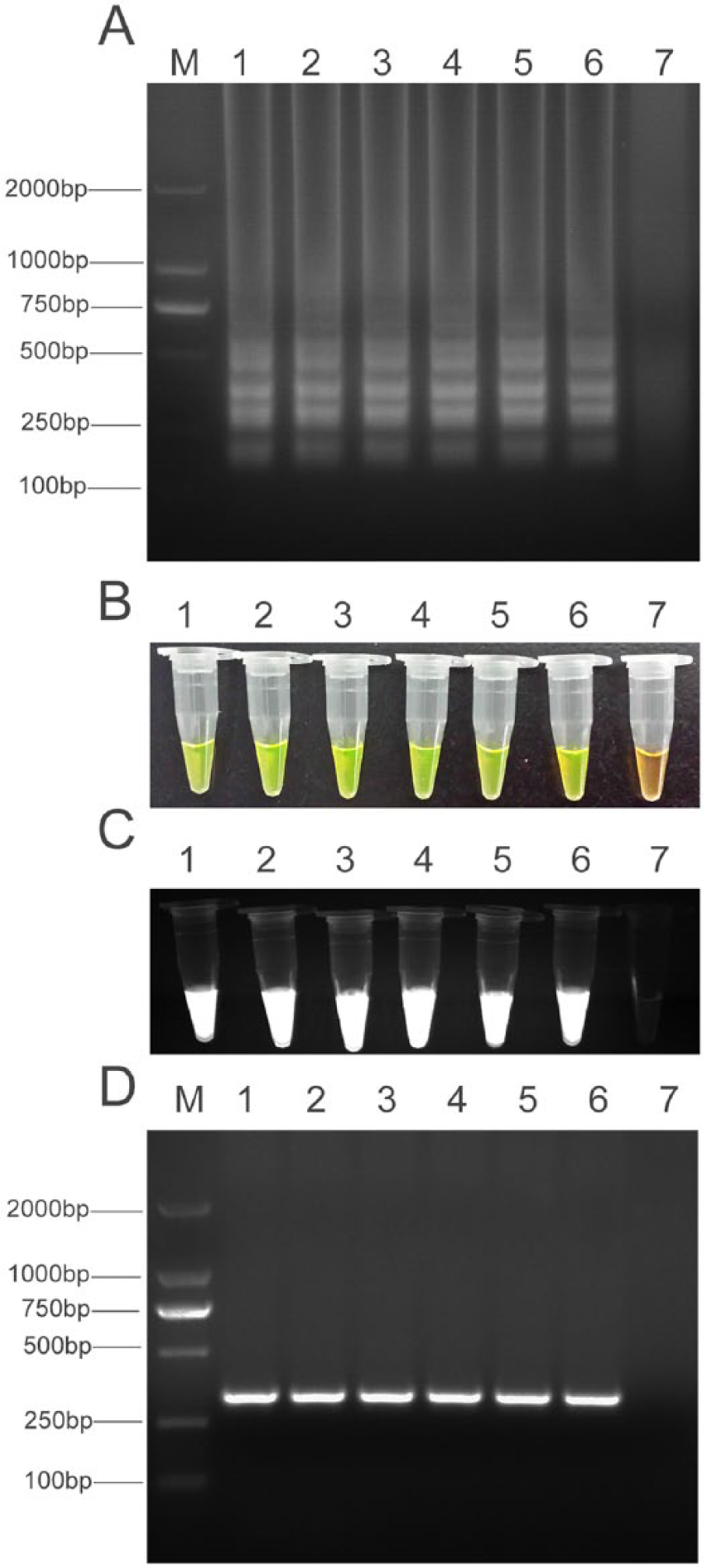
Detection of Mycoplasma gallisepticum by loop-mediated isothermal amplification (LAMP) assay.

Specificity of loop-mediated isothermal amplification (LAMP) assay. Lane M: DNA marker DL-2000.
Sensitivity of LAMP and PCR assays
To compare the detection limits of the LAMP and MG PCR assays, gDNA from M. gallisepticum Rlow strain was tested in a 10-fold serial dilution from 100 ng/μL to 0.1 fg/μL. Using gel visualization, the detection limit of the LAMP assay was found to be 10 fg gDNA, while that of conventional PCR was 100 fg, indicating that the LAMP assay established in this study was 10-fold more sensitive than the PCR method (Fig. 4). Moreover, positive amplification of LAMP was achieved in less than 30 min, while PCR required over 1 hr.
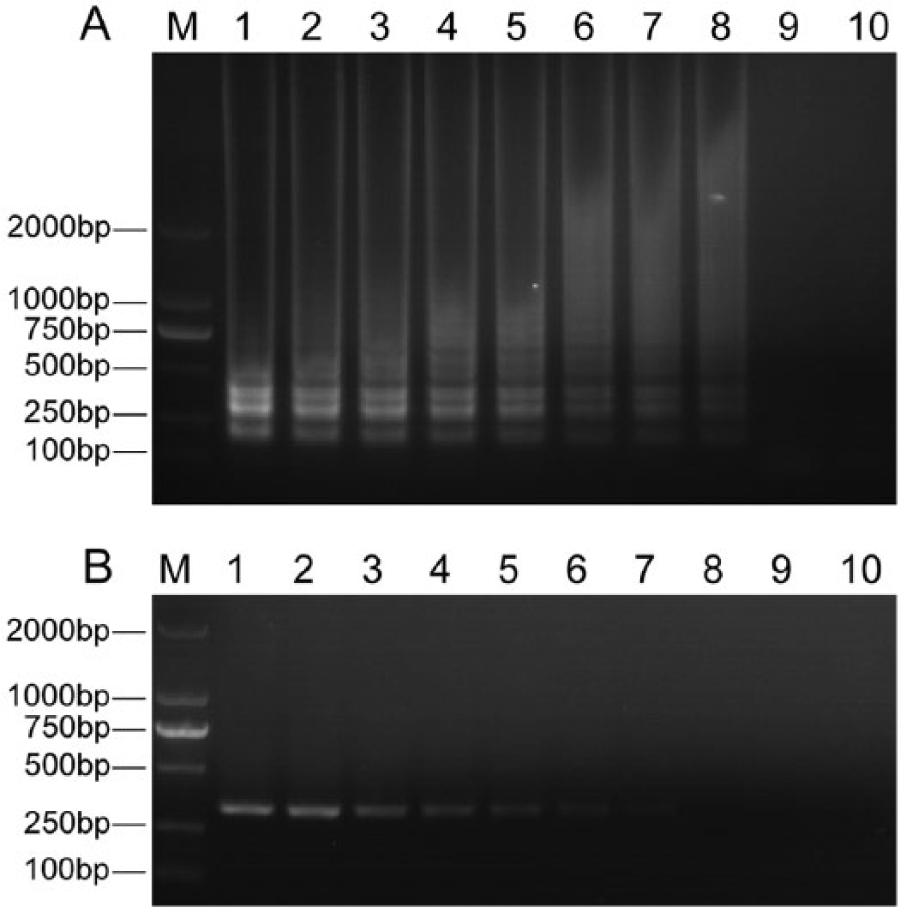
Sensitivity of loop-mediated isothermal amplification (LAMP) and conventional polymerase chain reaction (PCR) methods.
Detection of laboratory and field samples using LAMP and PCR assays
To demonstrate the utility of LAMP as a diagnostic tool for epidemiological and diagnostic studies, experimentally infected samples were collected and screened using both the LAMP and the MG PCR methods. Both the LAMP and PCR methods detected M. gallisepticum DNA in all 10 tracheal samples from the infected chickens while all 10 negative control chickens were negative in both assays, indicating 100% agreement between 2 methods. Typical results are shown in Supplementary Figure 2. The reproducibility of the LAMP assay was also tested (Supplementary Fig. 3). All of the 5 extracts, when tested at the 5 time intervals, gave positive results, which showed that the LAMP assay had good reproducibility. In addition, LAMP was also applied to the 183 clinical tracheal samples and compared with the MG PCR. A positive result, by either LAMP or PCR, for M. gallisepticum was detected on 58.3% (7/12) of the poultry farms. The distribution of positive samples on these 7 farms is listed in Table 3. The proportion of LAMP positives ranged from 6.2% to 37.5%, with a total positive detection rate of 20.2% (37/183). The MG PCR assay detected 24 positive samples and 159 negative samples with a positive detection rate of 13.1% (24/183). A correlation of 64.9% (24/37) was observed between LAMP and PCR, not significantly different (P > 0.05). Furthermore, all of the PCR-positive samples were also detected by LAMP, while the LAMP assay detected an additional 13 positive samples compared to PCR.
Detection of Mycoplasma gallisepticum in clinical samples by loop-mediated isothermal amplification (LAMP) and polymerase chain reaction (PCR).*
All samples were collected from poultry farms in Anhui (A, C), Jiangsu (D, F, H), and Zhejiang (I, K) provinces, China. Each farm had ~6,000 chickens. Numbers in parentheses are percentages.
Discussion
Our study reports on the development of a novel method for the rapid detection of M. gallisepticum. The primers for the LAMP assay were designed based on the alignment analysis of pdhA gene sequences of 6 M. gallisepticum strains listed in GenBank. A BLASTn analysis showed that the 750–1,000-bp sequence of pdhA gene was highly conserved within M. gallisepticum, but not for other species of Mycoplasma spp., other bacterial pathogens, and relevant viral pathogens. Therefore, this region was chosen as the target to develop the LAMP assay. Two sets of inner primers and outer primers that recognize 6 distinct regions in the pdhA gene, ensuring high specificity for target amplification, were developed. In the current study, the reaction condition was found to be optimal at 65°C for 30 min. The nontarget pathogens tested, including other Mycoplasma spp., as well as bacteria and viruses, gave no positive results, indicating the specificity of the current detection method. The LAMP assay was found to be 10-fold more sensitive than the MG PCR assay, with LAMP having a detection limit of 10 fg DNA. In the examination of samples from experimental infections and natural infections, the LAMP assay either gave identical results (experimental infection samples) or more positive results (natural infection samples).
Primer design is the key to a successful LAMP reaction. Factors such as guanine–cytosine (GC) content, primer distance, and amplification length should be strictly controlled. 28 Because of the characteristic feature of low GC content (30–40%) in mycoplasmas, 23 finding appropriate primer sets can be quite difficult. Loop primers can increase the DNA synthesis site during reaction process, thus shortening the reaction time and increasing the efficiency of amplification. 19 However, loop primers were not employed in the current study because the software did not produce any acceptable loop primers due to the low GC content of the sequence. Using the LAMP primer sets designed in our study, amplified gene sequences matched the sequences of M. gallisepticum, but no other pathogens in GenBank, indicating that the primers designed are feasible for detection of M. gallisepticum.
In summary, the LAMP assay developed in our study presents a new, inexpensive, and rapid method for detection of M. gallisepticum with high sensitivity and specificity. It does not require any specific and expensive instruments, only a simple water bath is necessary. The LAMP assay appears to be a potential diagnostic tool for the specific detection of M. gallisepticum infection in chickens under laboratory and field conditions.
Footnotes
Acknowledgements
We thank Wang Yongshan (Institute of Veterinary Medicine, Jiangsu Academy of Agricultural Sciences, China) and Li Zejun (Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences, China) for providing viral and bacterial strains.
Sources and manufacturers
Merial Vital Beijing Animal Health Co. Ltd., Beijing, China. Intervet Shanghai Center for Animal Health, Shanghai, China. Becton and Dickinson Co., Franklin Lakes, NJ. Life Technologies, Grand Island, NY. Sigma-Aldrich, St. Louis, MO. Tiangen Biotech Co. Ltd., Beijing, China. Promega Corp., Madison, WI. NanoDrop 2000, Thermo Fisher Scientific Inc., Wilmington, DE. Lasergene 7.1 software, DNASTAR Inc., Madison, WI. Sangon Biotech Co. Ltd., Shanghai, China. Takara, Dalian, China. SBSgene, Shanghai, China. New England Biolabs Inc., Ipswich, MA. Qiagen, Shanghai, China. GelDoc XR, Bio-Rad Laboratories, Hercules, CA. SAS Institute Inc., Cary, NC.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by Chinese Natural Science Foundation (grant 30871883 and 31001077), the Project of Basic Research for the National Non-profit Institute of China (2012JB11), and Special Fund for Agro-scientific Research in the Public Interest (grant 201303044 and 201303038).