
Abstract
Rhodococcus equi is the most important causative bacterium of severe pneumonia in foals. We report herein the development of a specific loop-mediated isothermal amplification (LAMP) assay, which targets a gene encoding vapA for detecting virulent R. equi. The detection limit of the LAMP assay was 104 colony forming units (CFU)/mL, which was equal to 10 CFU/reaction. The clinical efficacy of the LAMP assay was compared with those of 2 published PCR-based methods: nested PCR and quantitative real-time (q)PCR. Agreements between bacterial culture, which is the gold standard for detection of R. equi, and each of the 3 molecular tests were measured by calculating a kappa coefficient. The kappa coefficients of the LAMP (0.760), nested PCR (0.583), and qPCR (0.888) indicated substantial agreement, moderate agreement, and almost perfect agreement, respectively. Although the clinical efficacy of LAMP was not the best among the 3 methods tested, LAMP could be more easily introduced into less well-equipped clinics because it does not require special equipment (such as a thermocycler) for gene amplification. Veterinary practitioners could diagnose R. equi pneumonia more quickly by using LAMP and could use the results to select an appropriate initial treatment.
Rhodococcus equi is a gram-positive, facultative, and intracellular bacterium that is an important pathogen in foals, causing severe pneumonia characterized by pulmonary abscesses.4,7 Virulence of R. equi is attributed to an 85- to 90-kb plasmid that encodes the virulence-associated protein A (VapA). This protein must be present in order for an R. equi isolate to cause disease in foals, and R. equi isolates without vapA are avirulent. 5 Definitive diagnosis of R. equi pneumonia in foals with signs of lower respiratory tract disease is based on bacterial culture or polymerase chain reaction (PCR) amplification of DNA from a tracheal wash (TW) or tracheobronchial aspirate, in combination with cytologic evidence of septic pneumonia in the fluid.7,20 Although some PCR-based methods have been reported for detecting vapA,1,8,19,22 it is difficult to use these PCR methods in less well-equipped clinics because special equipment (such as a thermocycler) is required.
The loop-mediated isothermal amplification (LAMP) method was developed in 2000 18 and amplifies nucleic acids with high speed, specificity, and efficiency. A LAMP assay can be performed under isothermal conditions with simple heating equipment (e.g., a water bath). In recent years, LAMP has been applied clinically as a method for rapid detection of various equine pathogens.11,12,17 Herein we report on the development of a LAMP method specific to R. equi that targets vapA to diagnose R. equi pneumonia more easily and quickly than can PCR methods.
The LAMP assay primers were designed on the basis of the published sequences of vapA by using commercial software a (Table 1). The GenBank accession numbers of the vapA sequences used were JN990991, AP001204, and NC002576. The reaction mixture for the LAMP assay was prepared by using a DNA amplification kit b in accordance with the manufacturer’s instructions. The LAMP reactions were performed for 60 min at 65°C, and the LAMP products were detected by monitoring the turbidity with a real-time turbidimeter. c Two published PCR-based assays, namely a nested PCR (nPCR) 22 and a quantitative real-time PCR (qPCR), 19 were used to compare analytical sensitivities. The nPCR was performed with a commercial master mix, d whereas the qPCR assay was performed with a different one. e The remaining steps used in each method followed those used in the previous reports.19,22 The DNA volumes used for the above-described LAMP, nPCR, and qPCR assays were 2 μL, 5 μL, and 1 μL, respectively. Rhodococcus equi American Type Culture Collection (ATCC) 33701 and distilled water were used as positive and negative control in each reaction, respectively.
The loop-mediated isothermal amplification (LAMP) primer set used in the current study to detect vapA in Rhodococcus equi.
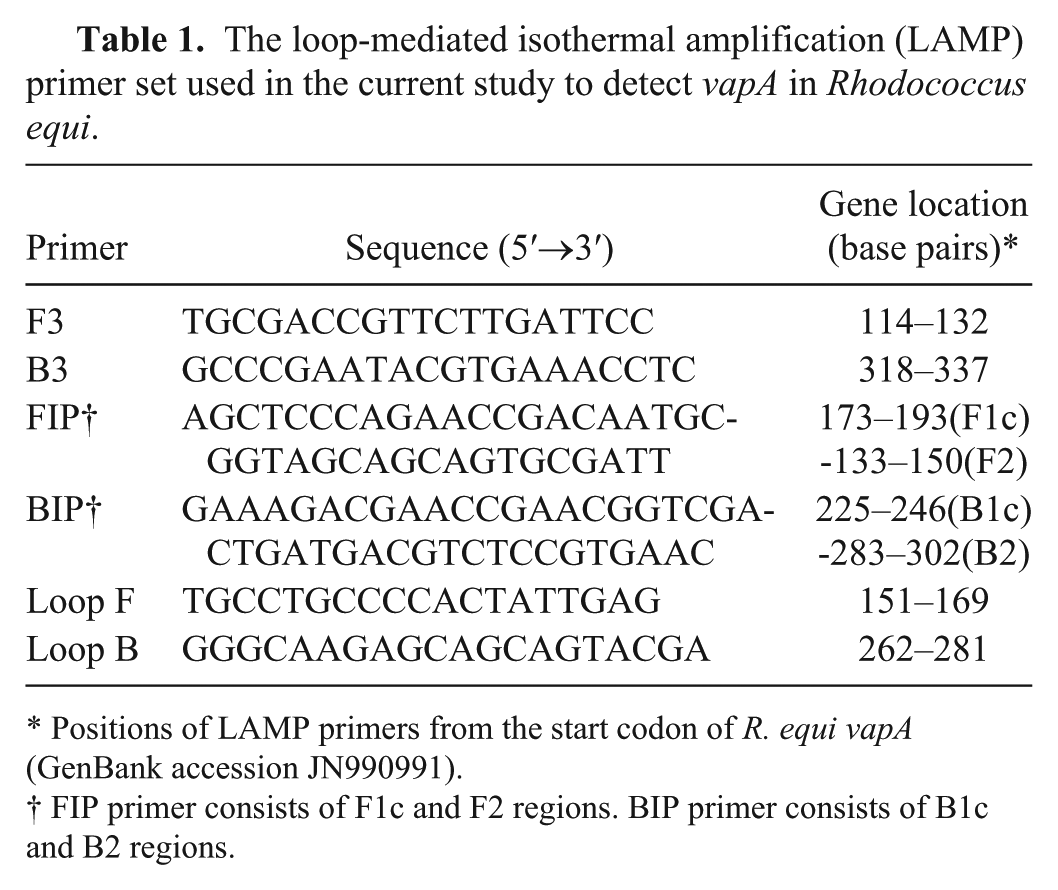
Positions of LAMP primers from the start codon of R. equi vapA (GenBank accession JN990991).
FIP primer consists of F1c and F2 regions. BIP primer consists of B1c and B2 regions.
A total of 106 strains were used to confirm specificity (Table 2). The 53 strains of Rhodococcus equi harboring the vapA gene used consisted of ATCC 33701 and 52 clinical strains isolated from ill foals between 1984 and 2010 in Japan. The 20 strains of Rhodococcus equi without the vapA gene used were ATCC 33702, ATCC 33703, ATCC 33704, ATCC 33705, ATCC 33706, ATCC 33707, 2 equine clinical strains, and 12 swine strains. Another 33 clinical strains, belonging to 11 other species, were isolated from ill horses. Bacterial DNA was extracted by using a commercial DNA extraction kit f in accordance with the manufacturer’s instructions.
Bacterial strains and isolates used in the current study.
To measure the detection limits of the LAMP, nPCR, and qPCR, 10-fold serial dilutions of a suspension of R. equi ATCC 33701 were prepared, and the colony forming units (CFU) in the suspensions were counted. Bacterial DNA in the suspensions was then extracted by using a commercial DNA extraction kit f in accordance with the manufacturer’s instructions. Because the DNA volumes used for each genetic test were different, we used the lowest volume of the template (1 µL) for qPCR and diluted the template to 2 µL for LAMP and 5 µL for nPCR to contain the same quantity of the template. Analytical sensitivity tests were performed 3 times for each genetic test; the lowest bacterial concentrations that yielded positive results at least twice were regarded as the detection limits (CFU/mL).
Positive amplification by LAMP products from the 53 R. equi strains possessing vapA was confirmed. By contrast, no positive LAMP amplification occurred with the R. equi strains without vapA and non–R. equi strains. In the negative control, no LAMP products occurred when the reaction time was extended from 60 min to 120 min.
The detection limit of the LAMP assay was 104 CFU/mL, which was equal to 10 CFU/reaction (Fig. 1). The matching results for the nPCR were 104 CFU/mL and 10 CFU/reaction, and those for the qPCR were 103 CFU/mL and 1 CFU/reaction.
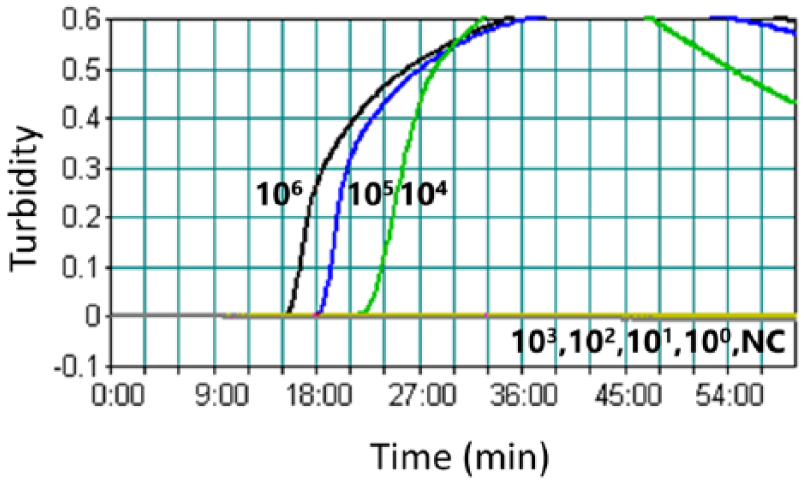
Detection limits, expressed as colony forming units/mL of the loop-mediated isothermal amplification (LAMP) assay. Analytical sensitivity of the LAMP was monitored by a real-time measurement of turbidity. A turbidity of >0.1 was considered to be positive for LAMP. NC = negative control without target DNA.
To compare the clinical efficacies of the LAMP, nPCR, and qPCR assays, we used 86 TWs from Thoroughbred foals with lower respiratory tract infection. All TWs were collected, as described in a previous study, 21 between February and June 2014 in the Hidaka district in Japan. A 100-μL aliquot of the TWs was incubated aerobically on nalidixic acid–novobiocin–Acti-dione (cycloheximide)–potassium tellurite (NANAT) selective agar plates, as described previously 24 at 30°C for 48 h. Each TW was boiled at 100°C for 10 min and then centrifuged at 12,000 × g for 3 min. The aliquots were then subjected to the LAMP assay and the 2 PCR-based assays.
The sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) of the 3 diagnostic tests were determined by comparing with bacterial culture, which is the gold standard test of R. equi.2,9,16 Agreement between bacterial culture and each molecular test was measured by calculating the kappa coefficient with a commercial software program. g The values of the kappa coefficients were interpreted according to the criteria of a previous report 14 : poor agreement, <0; slight agreement, 0.01–0.20; fair agreement, 0.21–0.40; moderate agreement, 0.41–0.60; substantial agreement, 0.61–0.80; almost perfect agreement, 0.81–1.00.
Out of the 86 clinical specimens, 70 were positive by bacterial culture and 16 showed negative results. The efficiencies of the 3 molecular methods, in terms of sensitivity, specificity, PPV, NPV, and kappa coefficient are presented in Table 3. Although the specificity and PPV of the 3 tests were the same (or almost the same), the sensitivity and NPV values of the nPCR (81.4% and 53.6%, respectively) were lower than those of the LAMP (91.4% and 71.4%) and qPCR (97.1% and 88.2%). The kappa coefficient between bacterial culture and the nPCR (0.583) showed moderate agreement, and those for the LAMP (0.760) and qPCR (0.888) showed substantial agreement and almost perfect agreement, respectively.
Comparison of clinical efficacies among the 3 molecular assays compared in the current study: loop-mediated isothermal amplification (LAMP), nested (n)PCR, and quantitative real-time PCR (qPCR).
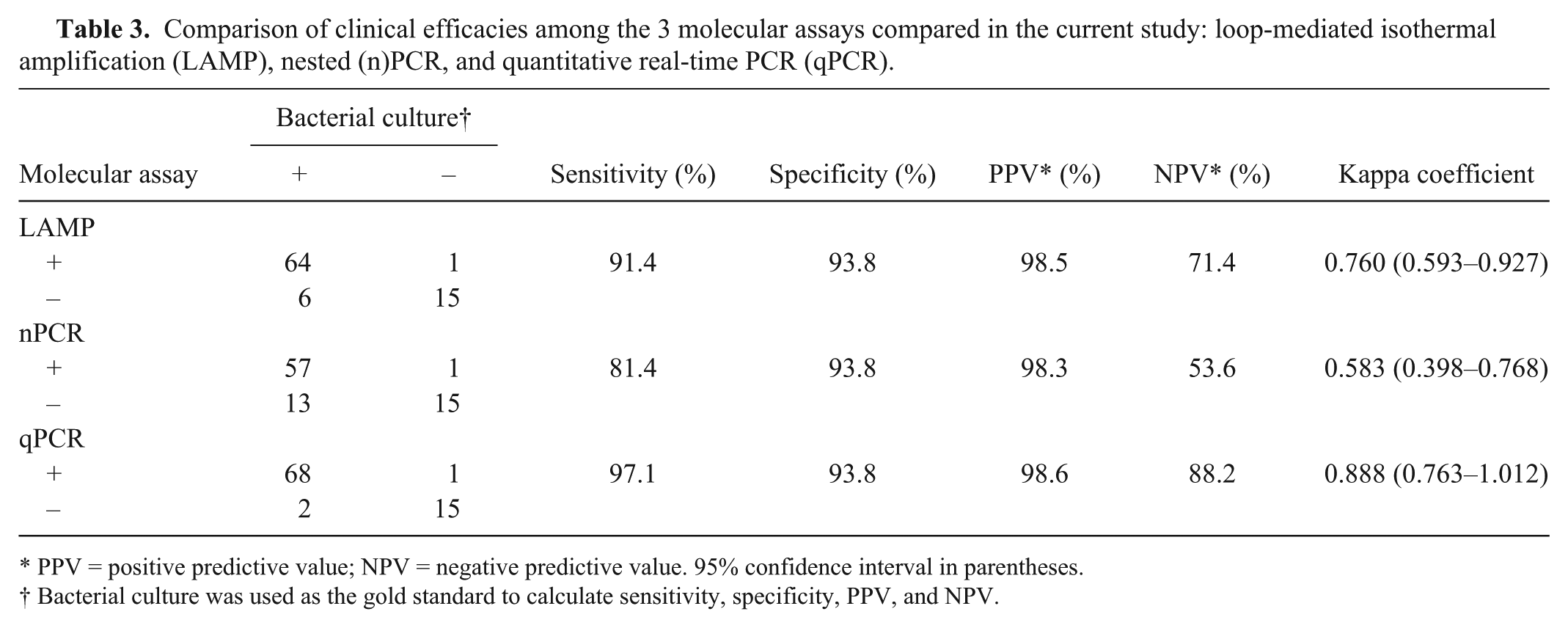
PPV = positive predictive value; NPV = negative predictive value. 95% confidence interval in parentheses.
Bacterial culture was used as the gold standard to calculate sensitivity, specificity, PPV, and NPV.
Many pathogens cause respiratory tract infections in foals, including beta-hemolytic Streptococcus spp., 13 Pneumocystis carinii,6,23 Equid herpesvirus,3,15 and R. equi.4,7 Because the treatments for each pathogen are different, it is very important, even in less well-equipped clinics, to have a way of determining the etiological agent. In the current study, we have developed a specific LAMP assay for detecting virulent R. equi, which is an important pathogen of foals and causes severe pneumonia. The LAMP assay could specifically detect virulent R. equi practically as well as the PCR-based assays.
Of the 16 clinical specimens that were negative by culture, 1 was positive by all the molecular tests. Although the reason is not clear, it is possible that administration of antimicrobials might have caused the negative culture result. As for the other clinical specimens that yielded R. equi, the LAMP could detect the vapA gene more efficiently in the specimens than could nPCR. The kappa coefficients indicated that the agreement between bacterial culture and the LAMP was stronger than that of culture with the nPCR.
A previous study reported that the use of a LAMP assay was advantageous in DNA templates that were simply prepared. 10 Although excessive mucous in TW samples might negatively influence DNA amplification, the simple preparation method for DNA template used in our study (boiling) performed well, as shown by the fact that the LAMP and qPCR assays obtained substantial and almost perfect agreement with bacterial culture, respectively. Although the boiling method proved useful for the LAMP assay in the current study, other commercially available DNA extraction methodsf,h have decreased the false-negative results more than the boiling method in a prior study of a LAMP assay for Streptococcus equi. 11 Therefore, the other commercially available methods might be more efficient depending on certain sample types (e.g., fecal samples and abscess material).
The qPCR was the most sensitive method for detecting R. equi in terms of both analytical sensitivity and clinical sensitivity. However, qPCR assay requires expensive equipment. In contrast, LAMP needs no special equipment, and the results can be judged visually by using a fluorescent detection reagent. Moreover, the reaction time for LAMP (60 min) was shorter than that for the PCR-based assays (a few hours). We therefore conclude that LAMP could be easily introduced into less well-equipped clinics as a sensitive method of detecting R. equi in clinical specimens. Veterinary practitioners can diagnose R. equi pneumonia in foals more quickly with the LAMP assay, even in clinics with only basic equipment, and can select the appropriate treatment in the early phase of infection.
Footnotes
Authors’ contributions
Y Kinoshita contributed to conception and design of the study; contributed to analysis and interpretation of data; and drafted the manuscript. H Niwa contributed to conception and design of the study; contributed to interpretation of data; and critically revised the manuscript. T Higuchi contributed to acquisition of data. Y Katayama gave final approval and agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
PrimerExplorer V4 software, Fujitsu Limited, Tokyo, Japan.
b.
Loopamp DNA amplification kit, Eiken Chemical Co. Ltd., Tokyo, Japan.
c.
LA-320C, Eiken Chemical Co. Ltd., Tokyo, Japan.
d.
EmeraldAmp PCR master mix, Takara Bio Inc., Kyoto, Japan.
e.
EagleTaq master mix with ROX, Roche Diagnostics GmbH, Mannheim, Germany.
f.
InstaGene matrix, Bio-Rad Laboratories, Tokyo, Japan.
g.
Excel Statistics version 7.0, Esumi Co. Ltd., Tokyo, Japan.
h.
Loopamp PURE DNA extraction kit, Eiken Chemical Co. Ltd., Tokyo, Japan.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) declared that they received no financial support for their research and/or authorship of this article.