
Abstract
Loop-mediated isothermal amplification (LAMP) is a novel method for the rapid and sensitive detection of DNA without the need for expensive equipment. In the present study, LAMP assays were developed for the specific detection of Equid herpesvirus 1 and 4 (EHV-1 and EHV-4, respectively) and for the differentiation of glycoprotein E (gE)-deleted EHV-1 (ΔgE) strain, a candidate strain for a live vaccine, from field EHV-1 strains. Specific primer sets were designed for the gC and gE genes of EHV-1 and for the gC gene of EHV-4. The analytical sensitivities of the LAMP assays were compared with those of polymerase chain reaction (PCR). The detection limits of LAMP for EHV-1 gC and gE and PCR for EHV-1 gC were 1 plaque-forming unit (PFU)/tube, and those of LAMP and PCR for EHV-4 gC were 0.1 PFU/tube. The DgE strain could be differentiated from wild-type EHV-1 strains based on the reactivity in the LAMP for EHV-1 gC in combination with the LAMP for EHV-1 gE. The analytical specificities of the LAMP for EHV-1 and EHV-4 were examined by using several equine pathogens, and no cross-reactions were observed. The LAMP detection abilities for EHV-1 and EHV-4 on nasal swab samples collected from experimentally infected horses were in good agreement with that of PCR for EHV-1 and EHV-4, respectively. The LAMP assays developed in the current study were sensitive and specific for EHV-1 and EHV-4, and should provide a valuable alternative to PCR for use in clinical laboratories in the field.
Introduction
Equid herpesvirus 1 and 4 (EHV-1 and EHV-4, respectively; family Herpesviridae, subfamily Alphaherpesvirinae, genus Varicellovirus) are major causative agents of respiratory disease in horses. Equid herpesvirus 1 also causes abortion and neurologic disease. 1 Respiratory disease among racehorses and abortions in pregnant mares caused by EHV-1 have a great economic impact on the horse industry in Japan. 11 Because EHV-1 and EHV-4 are antigenically and genetically related, 1 diagnostic methods are required that can distinguish between EHV-1 and EHV-4 infections. Virus isolation (VI), serologic typing, and polymerase chain reaction (PCR) are usually used to diagnose and distinguish between infections with these 2 related viruses. Virus isolation and PCR are useful for diagnosis in the acute phase of an infection. Because VI is time consuming and laborious, several groups have developed sensitive and rapid PCR tests that detect EHV-1 and EHV-4 DNA in a one-step reaction. 3,7,9,20 However, PCR testing requires expensive equipment, and this precludes the use of PCR tests for diagnosis in clinical laboratories in the field.
A glycoprotein E (gE)-deleted EHV-1 (ΔgE) strain was generated in the authors' laboratory to develop a new live attenuated EHV-1 vaccine. The ΔgE strain lacks almost the entire coding region of gE. 19 An inactivated vaccine is inoculated to regulate EHV-1 in Japan, but this inactivated vaccine does not completely control EHV-1. To develop more effective vaccine, the ΔgE strain was developed. Tests were undertaken to guarantee the safety, quality, and efficacy of the ΔgE strain. It is expected that a live vaccine that consists of the ΔgE strain will soon become commercially available in Japan. Therefore, a method for differentiating a live vaccine strain from wild-type EHV-1 strains is needed.
Recently, a technique called loop-mediated isothermal amplification (LAMP) was developed as a novel nucleic acid amplification technique. 14,16,18 The LAMP method uses at least 4 or 6 primers that recognize 6 or 8 distinct regions of the target nucleotides. 13,16,18 The LAMP reaction can be carried out in 30–60 min under isothermal conditions (60–65°C) in a water bath or a heat block. Because the LAMP reaction produces a large amount of amplified product, the result can be judged with the naked eye, based on the turbidity or fluorescence of the reaction mixture. 12,18 The rapidity, high specificity, and simplicity of the LAMP method make it possible to introduce this method for use in clinical laboratories in the field. In the current study, LAMP assays were used for the specific diagnosis of EHV-1 and EHV-4 infections in horses and for differentiating between a gE-deleted EHV-1 candidate vaccine and wild-type EHV-1.
Primers used for loop-mediated isothermal amplification in the current study. *

EHV-1 and EHV-4 = Equid herpesvirus 1 and 4, respectively.
GenBank accession numbers for EHV-1 glycoprotein C and E are S57839 and AB279608; the GenBank accession number for EHV-4 glycoprotein C is A21044.
Materials and methods
Viruses and bacteria
The EHV-1 Ab4p a , 5 and EHV-4 02c21 strains, which were propagated in fetal horse kidney (FHK) cells, were used to determine the analytical sensitivities of the LAMP assays. The FHK cells were grown in Eagle minimum essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. EHV-1 89c25p 10 and EHV-4 TH20p strains 6 were used in the experimental challenge study. To determine the analytical specificity of the LAMP assays, the following common pathogens of equine respiratory disease in Japan were used: 8 wild-type EHV-1 strains, 5 EHV-4 strains, each strain of Equid herpesvirus 2 (08c3 strain), Equine adenovirus 1 (05c3), Equine rhinitis A virus (NM11), Getah virus (MI-110), Equine influenza virus (A/equine/South Africa/4/03, H3N8 subtype), Streptococcus equi subsp. zooepidemicus (W60, 1b and 7a), Streptococcus equi subsp. equi (CF32, Lex and Hidaka 95/2), and Rhodococcus equi (ATCC-33701, ATCC-33703, and ATCC-33704). The EHV-1 ΔgE strain, deleted 1,149 of 1,653 base pairs (nucleotide location: 21-1,169) of the gE gene, was established in the authors' laboratory. 19
Experimental challenge study and virus isolation
Six Thoroughbred foals, ranging from 49 to 129 days of age, were used in the current study. All the foals were born in the authors' facilities and did not receive colostrum for 24 hr after birth. All the foals were clinically healthy and did not possess a neutralizing antibody against EHV-1 and EHV-4 on the day of the EHV-1 or EHV-4 inoculation. The 6 foals were divided into 2 groups of 3 animals, and the foals in each group were inoculated intranasally with 1 × 106 plaque-forming units (PFU) of EHV-1 (89c25p) or EHV-4 (TH20p) with a nebulizer. Nasal samples were collected by using absorbent cotton swabs on days 0, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, and 14 postinfection (PI) from the foals infected with EHV-1. In contrast, nasal samples were collected on days 0, 2, 5, 7, 9, 12 and 14 PI from the foals infected with EHV-4. The nasal swabs were immersed in 2.5 ml of EMEM supplemented with 2% FBS, and aliquots (0.5 ml) of the nasal sample filtrates were cocultured with FHK cells to isolate EHV-1 and EHV-4, and with Madin–Darby bovine kidney cells to isolate EHV-1 in 25-cm 2 flasks. The cells were incubated at 37°C, and cytopathic effects were observed for 1 week. The animal experiment in the present study was approved by the Animal Care Committee of the Epizootic Research Center, Equine Research Institute.
DNA and RNA extraction
Viral DNA and RNA were extracted from 100 μl of culture supernatants and nasal swab samples with a nucleic acid isolation kit, b and bacterial DNA was extracted with a nucleic acid purification kit c according to the manufacturer's instructions. To confirm that the extraction of viral and bacterial nucleic acids was performed properly, PCR or reverse transcription PCR was carried out by using specific primers for each pathogen. 2,4,8,9,15,17,21
Polymerase chain reaction
To compare the analytical sensitivity of the LAMP assays with that of conventional PCR assays, the PCR assays were performed with the primer sets specific for the detection of gC of EHV-1 and EHV-4 described previously 9 by using a fast cycling PCR, d according to the manufacturer's instructions, in a thermal cycler. c The PCR conditions were as follows: 95°C for 5 min, 35 cycles of denaturation at 96°C for 5 sec, annealing at 60°C for 5 sec, and extension at 68°C for 20 sec, and then a final extension at 72°C for 1 min. The PCR products were kept at 4°C until the samples were analyzed by 2.2% agarose gel electrophoresis. e
Loop-mediated isothermal amplification
The specific primer sets for the gC and gE genes of EHV-1 and for the gC gene of EHV-4 were designed by using PrimerExplorer V4 software. f Each primer set includes 6 primers that consist of 2 outer primers (F3 and B3), 2 inner primers (FIP and BIP), and 2 loop primers (loopF and loopB). The sequences and locations of the primers for the LAMP assays used in the current study are shown in Table 1. The LAMP reaction was carried out with a DNA amplification kit g and a fluorescent detection reagent g according to the manufacturer's instructions. In brief, 25 μl of reaction mixture that contained 12.5 μl of 2 × reaction mix buffer (40 mM Tris–HCl [pH 8.8], 20 mM KCl, 16 mM MgSO4, 20 mM [NH4]2SO4, 0.2% Tween-20, 1.6 M betaine, and 2.8 mM each of 4 deoxyribonucleotide triphosphate), 0.2 μM each of 2 outer primers, 1.6 μM each of 2 inner primers, 0.8 μM each of 2 loop primers, 1 μl of Bst DNA polymerase, 1 μl of a fluorescent detection reagent, g and 2 μl of extracted samples. The LAMP reaction was performed at 63°C for 60 min and then was terminated by heating the mixture at 95°C for 2 min. A portion of each LAMP product was analyzed by electrophoresis on a 2.2% agarose gel. e The LAMP reaction was also judged by visual observation. The color of the mixture turned green when the LAMP reaction was positive in the presence of calcein, which was contained in the fluorescent detection reagent, g whereas the color of the mixture remained orange when the LAMP assay result was negative. The products of LAMP for EHV-1 gC, EHV-1 gE, and EHV-4 gC were digested by restriction enzymes, Ava I, h Ban II, h and Msp I, h respectively. The digested LAMP products with expected length were observed by gel electrophoresis.
Results
Analytical sensitivities of LAMP assays for EHV-1 and EHV-4
A positive LAMP reaction was confirmed by observing the green color of the reaction mixture and a ladder pattern of many bands with different sizes on the agarose gel. Because the judgment of a positive reaction by visual observation was identical to that provided by gel electrophoresis (Figs. 1A, 1B, 2A), the LAMP assay results were judged by visual observation in all of the following experiments. Serial 10-fold dilutions of EHV-1 and EHV-4 DNA were amplified by the LAMP and PCR methods. The detection limits of the LAMP assays for the gC and gE genes of EHV-1 and for the gC gene of EHV-4 were examined and compared with those of conventional PCR assays for the gC genes of EHV-1 and EHV-4, respectively. The analytical sensitivities of the LAMP assays for EHV-1 gC and gE were 1 PFU/tube, which was the same as the analytical sensitivity of the PCR assay for EHV-1 gC (Fig. 1A–C). The analytical sensitivities of the LAMP and PCR assays for EHV-4 gC were 0.1 PFU/tube (Fig. 2A, B).
Analytical specificities of LAMP assays for EHV-1 and EHV-4
LAMP assays were performed with 8 wild-type EHV-1 strains and 5 wild-type EHV-4 strains with different origins, the EHV-1 °gE strain, and several other viral and bacterial pathogens in horses (Table 2) to evaluate analytical specificities. The LAMP assays for EHV-1 gC and gE amplified the DNA of all the EHV-1 strains that were tested except for the ΔgE strain in the EHV-1 gE LAMP assay but did not amplify the DNA of all of the EHV-4 strains tested. In contrast, the LAMP assay for EHV-4 gC amplified the DNA of the EHV-4 strains but did not amplify the DNA of the EHV-1 strains that were tested. None of the LAMP assays amplified any nucleic acids of other viruses and bacteria (data not shown).
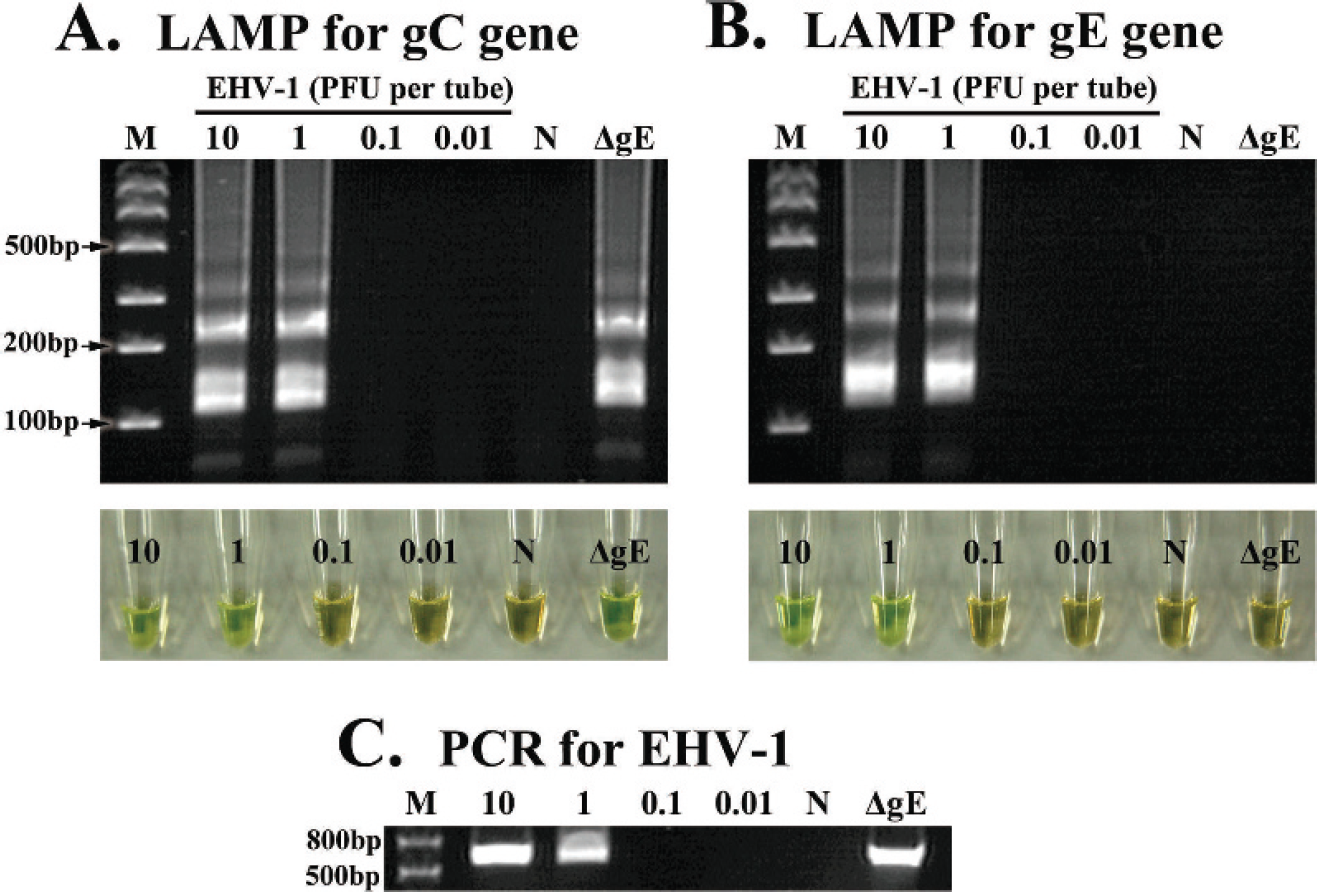
Analytical sensitivity of the loop-mediated isothermal amplification (LAMP) assays for Equid herpesvirus 1 (EHV-1) glycoprotein C (gC) and E (gE), and polymerase chain reaction (PCR) for EHV-1 gC. The LAMP reactions for EHV-1 and conventional PCR were carried out with 10-fold serial dilutions of EHV-1 Ab4p strain and gE-deleted EHV-1 (ΔgE) strain.
Evaluation of LAMP assays for detection of DNA in nasal swab specimens from horses experimentally infected with EHV-1 or EHV-4
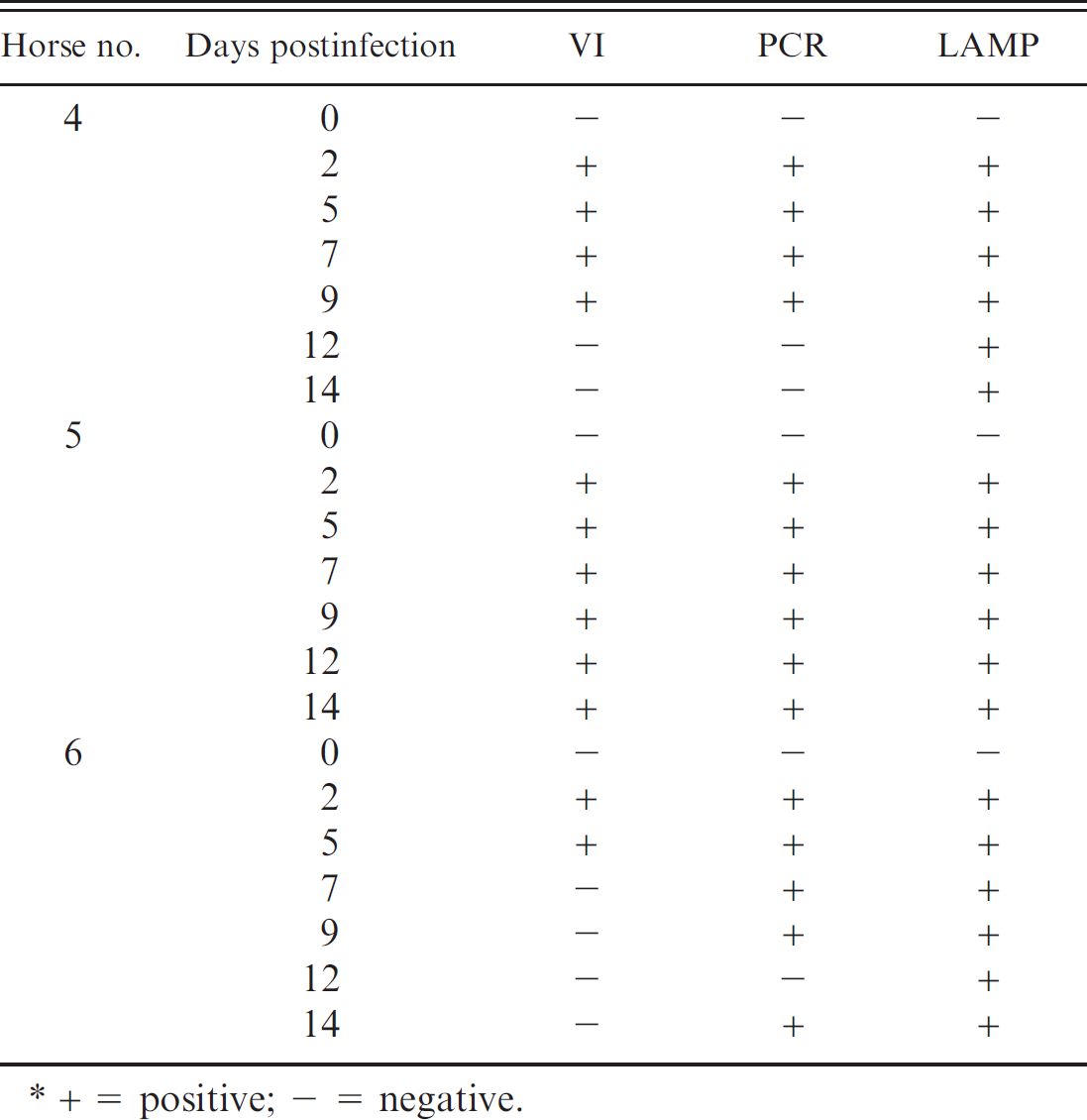
To evaluate the efficacy of LAMP assays of clinical specimens, LAMP assays were compared with VI and conventional PCR assays for the detection of EHV-1 and EHV-4 from nasal swabs collected from foals experimentally infected with EHV-1 or EHV-4 (Tables 3, 4). Nasal swabs, collected from 3 foals infected intranasally with EHV-1, were examined for EHV-1. Equid herpesvirus 1 was detected in horse no. 1 for a longer period with PCR and LAMP assays than with VI. However, similar results were obtained in horse nos. 2 and 3 by VI, PCR, and LAMP assays. No apparent difference in the ability to detect EHV-1 was observed among VI, PCR, and LAMP assays (Table 3).
Nasal swabs, collected from 3 foals infected intranasally with EHV-4, were examined for EHV-4 (Table 4). For EHV-4 detection, similar results were obtained in horse nos. 5 and 6 by PCR and LAMP assays. Furthermore, EHV-4 was detected in horse no. 4 for a longer period with the LAMP assay than with the other assay techniques.
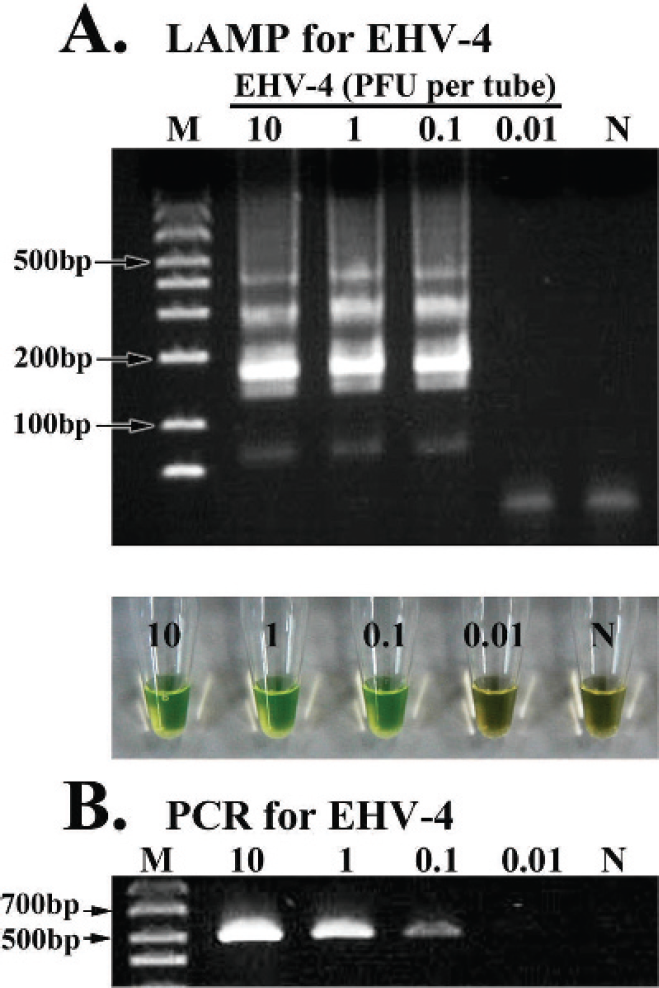
Analytical sensitivity of the loop-mediated isothermal amplification (LAMP) assays for Equid herpesvirus 4 (EHV-4) glycoprotein C (gC) and polymerase chain reaction (PCR) for EHV-4 gC. The LAMP reactions and conventional PCR for EHV-4 were carried out with 10-fold serial dilutions of EHV-4 02c21 strain.
Discussion
Infections of EHV-1 and EHV-4 in horses are a major economic concern throughout the world. 1 The rapid diagnosis of EHV-1 and EHV-4 infections is essential to enable control measures to be implemented. Several PCR methods were described for one-step detection and identification of EHV-1 and EHV-4. 3,7,9,20 However, it is difficult to use PCR methods in clinical laboratories in the field, because they demand expensive equipment and skilled technicians.
Virus and bacteria used in the current study and the loop-mediated isothermal amplification (LAMP) results. *

EHV-1 and EHV-4 = Equid herpesvirus 1 and 4, respectively; gC and gE = glycoprotein C and E, respectively; + = positive; — = negative; ΔgE = glycoprotein E–deleted EHV-1 strain.
In contrast, a LAMP reaction can be carried out at a constant temperature (60–65°C) within 1 hr in a water bath or a heat block. The result of a LAMP assay can be judged by the real-time monitoring of the turbidity of the reaction mixture, visual observation of fluorescence, or gel electrophoresis of the LAMP products. Real-time detection requires specific equipment, such as a real-time turbidimeter to monitor increments in turbidity. Amplified DNA can also be analyzed by gel electrophoresis with which a typical ladder of many DNA bands of different sizes is observed. However, this method could greatly increase the risk of contamination, because the tubes have to be opened, and the reaction mixture, which contains a large amount of amplified DNA, has to be applied onto an agarose gel. Therefore, the fluorescence detection method has been chosen for judging the result of LAMP assays. A positive reaction in which the color of the reaction mixture turns green can be observed with the naked eye without expensive equipment or time-consuming postamplification operations such as gel electrophoresis. In the experiments described in the current study, the detection limits judged by gel electrophoresis of the LAMP products were the same as those judged by visual observation (Figs. 1, 2).
In the present study, LAMP assays were developed for the specific detection of EHV-1 and EHV-4, and for the discrimination of a gE-deleted candidate vaccine strain, ΔgE, and wild-type strains of EHV-1. The analytical sensitivities of the LAMP assays were compared with those of PCR assays. The detection limits of LAMP for EHV-1 gC and gE as well as that of PCR for EHV-1 were 1 PFU/tube (Fig. 1) and that of LAMP and PCR assays for EHV-4 gC were 0.1 PFU/tube (Fig. 2). It was found that the analytical sensitivities of the LAMP assays for EHV-1 and EHV-4 developed in the current study were identical to those of PCR assays for EHV-1 and EHV-4, respectively.
The LAMP assays developed in the present study provided specificity in the amplification of the targeted DNA. With the exception of the ΔgE strain in the LAMP assay for EHV-1 gE, LAMP assays for EHV-1 amplified the DNA of all the EHV-1 strains tested but did not amplify the DNA of all the EHV-4 strains tested and vice versa. As shown in Table 2, the ΔgE strain can be differentiated from wild-type EHV-1 strains based on the reactivity in the LAMP for EHV-1 gC in combination with the LAMP for EHV-1 gE. No nucleic acids extracted from common pathogens of equine respiratory disease other than EHV-1 and EHV-4 showed cross-reactivity when using the LAMP assays developed in the current study (Table 2).
The potential usefulness of the LAMP assays for clinical specimens was evaluated by using nasal swabs of horses experimentally infected with EHV-1 and EHV-4. Although the result for VI on day 1 PI was positive in all 3 horses infected with EHV-1, the results of PCR and/or LAMP assays on the same day were negative. The exact reason for this is not clear, but one possible explanation is that a loss of DNA during the DNA extraction step could affect the amount of DNA extracted from the virus in the nasal swabs owing to a relatively low virus load in early phase of infection. The detection ability of LAMP assays for EHV-1 and EHV-4 in clinical samples was in good overall agreement with that of PCR for EHV-1 and EHV-4, respectively.
Detection of Equid herpesvirus 1 (EHV-1) by virus isolation (VI), polymerase chain reaction (PCR), and loop-mediated isothermal amplification (LAMP) for glycoprotein C (gC) or E (gE) gene in foals infected experimentally with EHV-1 89c25p strain. *

+ = positive; — = negative.
LAMP assays were developed for the specific detection of EHV-1 and EHV-4, and for the differentiation of the gE-deleted EHV-1 strain from field EHV-1 strains. The advantages of LAMP assay are that, compared with PCR, a LAMP assay can be undertaken with simple and cost-effective equipment in a short time by using visual detection. The LAMP assays developed in the current study are sensitive and specific, and should provide a valuable alternative to PCR for use in clinical laboratories in the field.
Detection of Equid herpesvirus 4 (EHV-4) by virus isolation (VI), polymerase chain reaction (PCR), and loop-mediated isothermal amplification (LAMP) in horses experimentally infected with EHV-4 TH20p strain. *
+ = positive; — = negative.
Acknowledgements
The authors thank Akira Kokubun and Junko Tsujimura for invaluable technical assistance.
Footnotes
a.
Kindly provided by Dr. A. J. Davison, Glasgow University, Scotland, UK.
b.
MagNA Pure LC Total Nucleic Acid Isolation Kit, Roche Diagnostics GmbH, Mannheim, Germany.
c.
InstaGene™ Matrix, iCycler™ Thermal Cycler; Bio-Rad Laboratories, Hercules, CA.
d.
Qiagen GmBH, Hilden, Germany.
e.
FlashGel® System for DNA, Lonza Rockland Inc., Rockland, ME.
f.
Fujitsu Ltd, Tokyo, Japan.
g.
Loopamp, Eiken Chemical Co. Ltd., Tokyo, Japan.
h.
Takara Bio Inc., Shiga, Japan.