
Abstract
Keywords
Introduction
Multiple sclerosis (MS) is a chronic, immune-mediated disease of the central nervous system, characterized by demyelinating white matter lesions and neurodegeneration. The relapsing-remitting (RR)-MS phenotype usually presents a highly variable clinical course, characterized by periods of clinical stability interrupted by acute relapses. The ability to minimize the clinical manifestation of new brain lesions represents a crucial factor influencing disease course in MS. 1
Synaptic long-term potentiation (LTP) represents a possible substrate of clinical recovery after brain damage and refers to the strengthening of synaptic efficacy induced by neural activity at existing synapses. 2 It has been shown that the amount of LTP inducible with different transcranial magnetic stimulation (TMS) protocols after brain damage, the so-called LTP reserve, correlates with the ability to compensate neuronal denervation and minimize clinical symptoms. 3 Accordingly, in RR-MS patients LTP-like plasticity elicited by the paired associative stimulation (PAS) protocol correlated with the degree of clinical recovery 3 months after a relapse. 4
A number of findings suggest that both in animal models (ie, experimental autoimmune encephalomyelitis) and in MS patients, inflammation can alter synaptic transmission and plasticity.5-9 In particular, different proinflammatory and anti-inflammatory molecules inducing and maintaining the inflammatory process also modulate the expression of synaptic plasticity, influencing the disease course. 10 Previous studies have highlighted that the proinflammatory cytokine interleukin (IL)-1β alters synaptic plasticity in the hippocampus of control mice, reproducing the effect observed in mice with experimental autoimmune encephalomyelitis.11,12 Moreover, also in MS patients, higher cerebrospinal fluid (CSF) levels of this cytokine have been associated with altered TMS-induced synaptic plasticity 9 and enhanced neuronal damage and clinical disability.13,14 Conversely, it has been suggested that some neurotrophins may promote LTP induction in vitro 15 and in MS patients,16,17 favoring a stable disease course in RR-MS. 18
IL-6 represents a major proinflammatory cytokine that has been associated to different neurological conditions.19,20 Preclinical investigations have shown that IL-6 acts by interfering with neuronal and synaptic functions in different experimental conditions (neurons culture, transgenic model mice, acute and chronic exposure) and in different brain areas (cortex, cerebellum, striatum, hippocampus).21-25 In particular, it has been previously suggested that elevated concentrations of this cytokine may negatively affect MS disease course.20,26-28 In the present study, we explored whether IL-6 alters the expression of LTP and affects the ability to compensate the clinical impact of ongoing brain damage in early RR-MS patients.
Material and Methods
Study Design
The effect of IL-6 on synaptic plasticity was first examined in vitro in mice hippocampal slices. We assessed whether IL-6 affects synaptic function at the concentrations of 100 ng/mL, considered relevant to pathological conditions. 29 Extracellular field recordings were performed to analyze changes in slope of field excitatory postsynaptic potentials (fEPSPs) after LTP induction by a high-frequency stimulation protocol. We compared the magnitude of LTP 60 minutes after induction in both IL-6 and vehicle groups (vehicle, n = 3; IL-6, n = 4). In addition, to explore whether IL-6 could influence the probability of neurotransmitter release, paired pulse facilitation was tested in mice hippocampal slices (n = 4) comparing data obtained before and after perfusion with IL-6.
The impact of IL-6 CSF levels on prospective disease course was explored in a group of 150 consecutive RR-MS patients admitted to the neurological clinic of either University Tor Vergata Hospital or Neuromed Hospital. RR-MS diagnosis was made on the basis of clinical, laboratory, and magnetic resonance imaging (MRI) parameters. 30 Clinical evaluation and brain and spine MRI were performed at the time of CSF collection, during hospitalization, and during the follow-up period (36 months) every 6 months after diagnosis. When clinical relapse was suspected, unscheduled clinical evaluation and MRI were performed. First-line disease-modifying therapy was initiated after diagnosis, including interferon (IFN)-β1a (Rebif 22 = 3%; Rebif 44 = 40%; Avonex = 6%), IFN-β1b (Betaferon = 21%), glatiramer acetate (14%), dimethylfumarate (7%), fingolimod (6%), natalizumab (2%), mitoxantrone (1%), and siponimod (1%). In patients presenting a subsequent clinical relapse, second-line treatments were adopted (including fingolimod, mitoxantrone, natalizumab, or alemtuzumab).
Finally, we assessed in a subgroup of 36 MS patients the correlation between IL-6 CSF levels and both motor cortex excitability and TMS-induced LTP-like plasticity. These measures were collected within 24 hours from CSF withdrawal.
In Vitro Hippocampal Plasticity
All experiments were conducted in accordance with the Internal Institution Review Committee, the European Directive 2010/63EU and the European Recommendations 526/2007, and the Italian D.Lgs 26/2014. All efforts were made to minimize the number of animals used.
C57BL6J mice (4-6 weeks old) were killed by cervical dislocation, and their brains were quickly removed from the skull and placed in chilled artificial CSF (aCSF) gassed with a mixture of 95% O2 and 5% CO2 (pH = 7.4). aCSF composition was (in mM) as follows: NaCl 126, KCl 2.5, NaH2PO4 1.25, CaCl2 2.4, MgCl 1, NaHCO3 26, D-glucose 1.
Sagittal hippocampal slices (250 µm) were prepared using a Leica VT1200S automatic slicer and left to recover for 1 hour in oxygenated aCSF at room temperature before recordings. After recovering, slices were transferred onto the recording chamber, continuously perfused with warm oxygenated aCSF (32°C, ~3 mL/min). fEPSPs were evoked by means of an insulated bipolar electrode placed in stratum radiatum and recorded by placing aCSF filled recording microelectrodes (2-4 MΩ) in the CA1 area.
LTP was induced by a high-frequency stimulation protocol (a single 100-Hz train of 1 s duration). Signals were amplified and low-pass filtered at 0.3 kHz. After digitization, potentials were sampled at 1 kHz, and the resulting data were analyzed offline using the software pClamp 10.6 (Axon Instruments).
Mouse IL-6 was purchased from Miltenyi Biotec, prepared as concentrated stock solution (200 µg/mL) and stored at −20°C. To explore whether acute incubation with IL-6 may affect neurotransmitter release at the presynaptic level, we also tested paired pulse facilitation at Schaffer collateral and CA1 synapses. Paired stimuli with interstimulus intervals of 50, 100, 150, 200, 300, and 500 ms were applied.
Clinical and Radiological Evaluation of MS Patients
The study involving human subjects was approved by the Ethics Committees of the University Tor Vergata Hospital in Rome and Neuromed Research Institute in Pozzilli, Italy, according to the Declaration of Helsinki. All patients gave written informed consent to the study.
The following clinical variables were considered. Disease onset was defined as the first episode of focal neurological dysfunction suggestive of MS deduced by medical history. Disease duration was calculated as the number of months from disease onset to the time of diagnosis. Clinical relapses were defined as the development of new or recurrent neurological symptoms not associated with fever or infection, lasting at least 24 hours. Clinical activity at the time of CSF collection was defined as the presence of a concomitant clinical relapse. Disability was assessed using the Expanded Disability Status Scale (EDSS). 31 Worsening of the EDSS score was defined as a change from baseline of at least 1 point (for baseline EDSS ≥ 1) or >1.5 points (for baseline EDSS = 0). The progression index was defined as the ratio between the EDSS score and disease duration.
MRI examination (1.5 or 3 T) consisted of dual-echo proton density, fast fluid-attenuated inversion recovery, T2-weighted spin-echo images and precontrast and postcontrast T1-weighted spin-echo images. The presence of gadolinium (0.2 mL/kg intravenous)-enhancing lesions was evaluated by a neuroradiologist who was unaware of the patient’s clinical details. A new gadolinium-enhancing lesion was defined as a typical area of hyperintense signaling on postcontrast T1-weighted images. An active MRI was defined as one showing new or enlarging T2 lesions and/or postcontrast enhanced T1-weighted lesion. Radiological activity at the time of CSF collection was defined as the presence of gadolinium-enhancing lesions at MRI performed during hospitalization.
CSF Collection and Analysis
Lumbar puncture was performed during hospitalization, at the time of diagnosis. No patient was treated with corticosteroids or immunoactive therapies before CSF collection. The presence of oligoclonal bands was assessed in all patients.
The CSF was centrifuged and immediately stored at −80°C until analyzed using a Bio-Plex multiplex cytokine assay (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s instructions. CSF concentrations of IL-6 were calculated according to a standard curve generated for the specific target and expressed in picograms per milliliter. When the concentrations were below the detection threshold (assay sensitivity = 2.6 pg/mL), they were assumed to be 0 pg/mL.
Biomarkers of neurodegeneration and axonal damage as amyloid-β 1-42 and neurofilament light protein were also measured in the CSF. For the analysis of amyloid-β 1-42, standard procedures were adopted as detailed elsewhere. 32 The levels of neurofilament light protein in the CSF were measured by fitting data to a 4-parameter standard curve using GraphPad Prism Software Package (San Diego, CA).
Transcranial Magnetic Stimulation
All patients gave written informed consent to TMS and were asymptomatic in the right upper limb. Corticosteroids or other MS-specific immunoactive therapies were initiated later when appropriate. Motor-evoked potentials (MEPs) were elicited through a figure-of-eight coil with external loop diameter of 70 mm connected to a Magstim 2002 magnetic stimulator (The Magstim Company Ltd, Whitland, Dyfed, UK). The coil was held tangentially to the scalp, with the handle pointing backward and away from the midline at about 45°, in the optimal scalp site (hot spot) to evoke MEPs in the contralateral target muscles. Raw electromyographic signals were recorded with surface electrodes placed in a belly-tendon fashion on the right first dorsal interosseous muscle for testing cortical excitability and on the right abductor pollicis brevis muscle for evaluating cortical plasticity. Responses, sampled at 5 kHz with a CED 1401 A/D laboratory interface (Cambridge Electronic Design, Cambridge, UK), were amplified and filtered (bandpass 20 Hz to 2 kHz) with a Digitimer D360 amplifier (Digitimer Ltd, Welwyn Garden City, Hertfordshire, UK), then recorded by a computer with Signal software (Cambridge Electronic Design).
Motor thresholds were calculated at rest (resting motor threshold), as the lowest stimulus intensity able to evoke MEPs of 50 µV in 5 of 10 consecutive trials, and during a slight voluntary contraction (active motor threshold) of the target muscle (20%-30% of the maximum voluntary contraction), as the lowest intensity able to evoke MEPs of 200 µV in 5 of 10 consecutive trials.
Intracortical excitability was assessed with paired-pulse TMS measuring the short-interval intracortical inhibition and the intracortical facilitation. For paired-pulse TMS, 2 separate Magstim 2002 stimulators were connected to a Bistim2 module (The Magstim Company). According to established techniques,33,34 the intensity of the first (conditioning) stimulus was set at 80% of the active motor threshold, whereas for the second (test) stimulus, the intensity was set to obtain unconditioned MEPs of about 0.5 to 1 mV peak-to-peak in amplitude. Paired pulses were given at interstimulus intervals of 3 ms for short-interval intracortical inhibition and 10 ms for intracortical facilitation. Three experimental conditions of 30 trials (10 test pulses given alone and 10 conditioned pulses for each interstimulus interval) were randomly tested. The mean peak-to-peak amplitude of the conditioned MEP, at each interstimulus interval, was expressed as a percentage of the mean peak-to-peak amplitude of the unconditioned MEP.
Cortical plasticity was explored with PAS. 35 Pairs of median nerve electric shocks and single TMS pulses over the abductor pollicis brevis hot spot were delivered at an interstimulus interval of 25 ms. The median nerve was stimulated at the wrist through a pair of surface electrodes connected to a constant current stimulator (model DS7A, Digitimer Ltd). The intensity of the TMS pulses was set to evoke MEPs of about 0.5 to 1 mV peak-to-peak amplitude in the abductor pollicis brevis muscle at baseline. The same intensity was used to obtain MEPs after PAS. The intensity of the median nerve stimulation (0.2 ms duration) was set at 300% of the perceptual threshold. In all, 200 pairs of stimuli were delivered at a rate of 0.25 Hz, and 25 MEPs were recorded from the median-innervated abductor pollicis brevis muscle before and 5, 15, 30, 45, and 60 minutes after PAS. MEP amplitudes were averaged at each time point after PAS and then normalized to the mean baseline amplitude.
Statistical Analysis
Normality distribution of continuous variables was assessed by the Shapiro-Wilk test. Data were expressed as mean (SD) or, if they did not follow a normal distribution, median (interquartile range [IQR]). Categorical variables are given in terms of absolute (n) and relative frequency (%). Logarithmic transformation was applied to reduce the skewness of data distribution and to better approximate the normal distribution. The Pearson correlation or, if data were not normally distributed, Spearman nonparametric correlation were applied to evaluate possible association between continuous variables.
The relationship between 2 continuous variables was depicted by a scatter plot. Association between categorical variables was examined by applying the χ2 or, when necessary, Fisher exact test. A logistic regression model with escalation to second-line therapy as dependent variable was used to test whether disease duration could influence the association between IL-6 groups and escalation to second-line therapy.
Difference in continuous variables between the IL-6 groups was evaluated using the nonparametric Mann-Whitney test for independent samples. Difference between IL-6 groups in EDSS and in progression index observed at 3 years after lumbar puncture (on a logarithmic scale) was corrected for disease duration effect using analysis of covariance (ANCOVA).
The Poisson model was applied to evaluate the effect of IL-6 group on the number of clinical relapses in the 3 years of follow-up (the dependent variable), adjusting for the number of radiological relapses during the follow-up, clinical and radiological activity at the time of lumbar puncture, age, EDSS, and disease duration. A stepwise selection method was applied to obtain the best set of factors associated with the number of clinical relapses. The final model included the IL-6 groups and the number of radiological relapses. The results are presented in terms of incidence rate ratio (IRR) and the corresponding 95% CI. For multiple testing, the Benjamini-Hochberg (B-H) false discovery rate controlling procedure was applied. A P value <.05 was regarded as statistically significant.
Statistical analysis of in vitro electrophysiological recordings was performed using the unpaired Student t-test, and data were expressed as mean ± standard error of the mean, with the significance level being set at P <.05. N refers to the number of slices. Data were analyzed normalizing the fEPSP slope (mV/ms) and comparing the 2 experimental conditions (vehicle, IL-6) by evaluating differences in the last 10 minutes, 50 to 60 minutes after high-frequency stimulation.
All statistical analyses were performed using IBM SPSS Statistics for Windows (IBM Corp, Armonk, NY).
Results
IL-6 Disrupts Hippocampal Synaptic Plasticity In Vitro
To characterize the impact of IL-6 on synaptic plasticity, we explored whether LTP induction could be influenced by acute application of IL-6 on mouse hippocampal slices. Bath application of IL-6 (100 ng/mL, 10 minutes, n = 4) before high-frequency stimulation protocol was able to alter the late-phase of LTP compared with the vehicle group (n = 3) by significantly decreasing the fEPSP slope parameter [t(5), P = .042; Figure 1A]. No significant differences were observed at all interstimulus intervals tested in paired pulse ratio before and during (10 minutes) IL-6 application (Figure 2B). These findings suggest that IL-6 may specifically interfere with experimentally induced LTP, without influencing basal synaptic excitability.
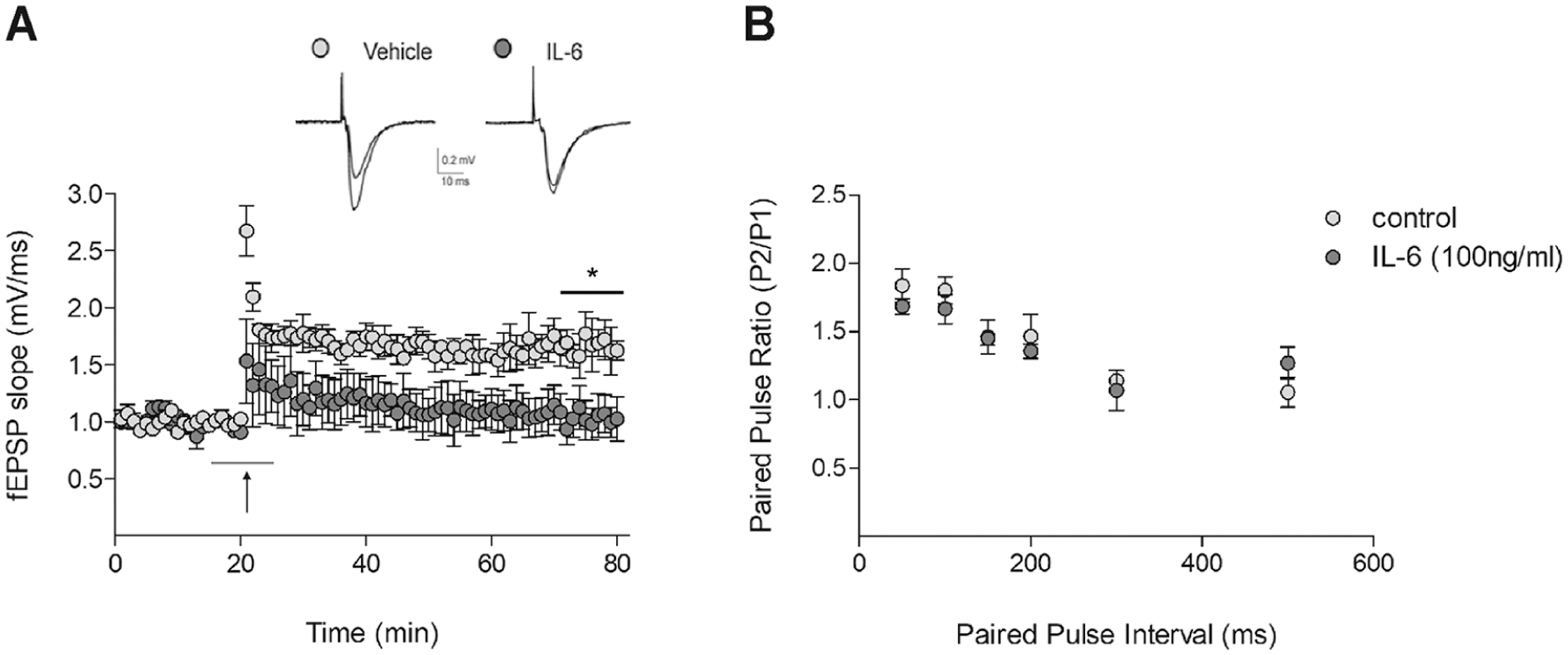
Effect of IL-6 incubation on LTP induction and PPF in mice hippocampal slices. A. Averaged time course of LTP (vehicle, n = 3; IL-6, n = 4) induced by HFS. Black arrow indicates HFS (100 Hz, 1 s). Horizontal gray line indicates 10-minute bath application of IL-6 (100 ng/mL). Statistical significance in the late phase of LTP was evaluated between 50 and 60 minutes following delivery of conditioning trains in both conditions (vehicle, n = 3; IL-6, n = 4; *P < .05, unpaired t-test). Above sample traces show fEPSPs during a baseline interval and 60 minutes after conditioning train delivery. B. IL-6 does not affect PPF in the hippocampal CA1 area. Data are represented as mean ± standard error of the mean of the slope ratio obtained by dividing the slope of second pulse (P2) by the slope of first pulse (P1). Data were obtained by comparing paired pulse stimulation before (control) and after 10 minutes of bath perfusion with IL-6 100 ng/mL (n = 4, all P > .05, paired t-test).
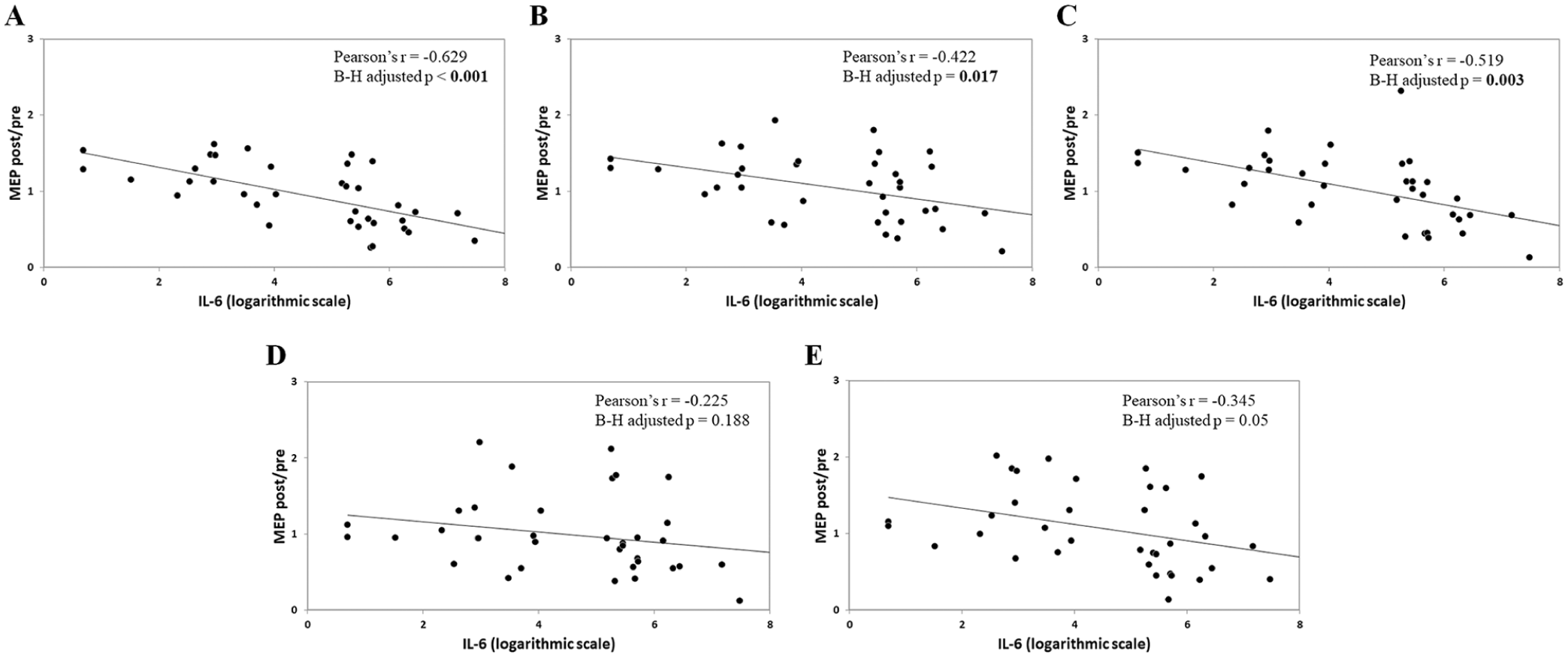
IL-6 CSF levels and PAS-induced effects in MS patients: Correlations between IL-6 CSF concentrations and PAS-induced effects at different time points. A. 5 Minutes. B. 15 Minutes. C. 30 Minutes. D. 45 Minutes. E. 60 Minutes. Data are expressed as mean MEP amplitudes (MEP post) normalized to the mean baseline MEP (MEP pre). To obtain a better graphical representation, IL-6 concentrations are shown on a logarithmic scale.
IL-6 CSF Levels Correlate With Altered LTP-Like Plasticity Expression in RR-MS Patients
To further address the potential association between CSF IL-6 and synaptic plasticity expression, we analyzed the correlation between the IL-6 levels and the PAS-induced effect in a subgroup of 36 RR-MS patients (mean age = 35 years; sex, 22 female; EDSS median = 1.5; disease duration median = 14 months). TMS was well tolerated in all patients, and no adverse effects were reported.
A significant negative correlation emerged between IL-6 CSF concentrations and changes in cortical excitability induced by the PAS protocol after 5 minutes [Pearson r(36) = −0.629, P < .001, B-H adjusted P < .001], 15 minutes [Pearson r(36) = −0.422, P = .010, B-H adjusted P = .017], and 30 minutes [Pearson r(36) = −0.519, P = .001, B-H adjusted P = .003]. Conversely, after correcting for multiple comparisons, correlations after 45 [Pearson r(36) = −0.225, P = .188, B-H adjusted P = .188] and 60 minutes [Pearson r(36) = −0.345, P = .039, B-H adjusted P = .05] were not significant (Figure 2). TMS exploring possible correlations between CSF IL-6 levels and basic physiological properties of the motor cortex showed that neither the resting nor the active motor threshold and neither the short-interval intracortical inhibition nor the intracortical facilitation were affected by IL-6 CSF concentrations (all P > .2; Supplementary Figure).
These findings suggest that, in RR-MS patients, higher IL-6 CSF levels could interfere with the early phases of LTP induction after PAS. This effect was not mediated by changes in basal cortical excitability, as seen in mouse hippocampal slices.
IL-6 CSF Levels and Prospective Disease Activity
Because LTP represents one of the physiological mechanisms of recovery after brain damage2,3 and the substrate favoring the clinical compensation of new brain lesions in MS, 1 we investigated whether IL-6 CSF levels could differ in patients with variable ability to compensate the negative clinical impact of new brain lesions.
With this aim, we explored whether IL-6 could differentially influence the clinical and radiological manifestations of the disease in a cohort of 150 RR-MS patients. Clinical and demographic characteristics of MS patients are shown in Table 1. IL-6 levels were not associated with patient’s age, sex, EDSS, and clinical/radiological disease activity at the time of CSF collection (all P > .1). Patients were divided into 2 groups (high IL-6 and low IL-6) according to the IL-6 median value (13.11 pg/mL; IQR = 2.93-234.64). At the time of lumbar puncture, although the 2 groups did not differ in demographic and clinical characteristics (Table 2), disease duration was significantly longer in the high IL-6 group compared with the low IL-6 group (high IL-6 group median = 10.2, IQR = 2.56-33.78, vs low IL-6 group median = 3.3, IQR = 0.93-17.8; Mann-Whitney P = .02) according to a previous report. 28 To exclude the potential confounding effect of disease duration, we corrected all the analyses for the disease duration effect.
Clinical and Demographic Characteristics at the Time of CSF Collection.
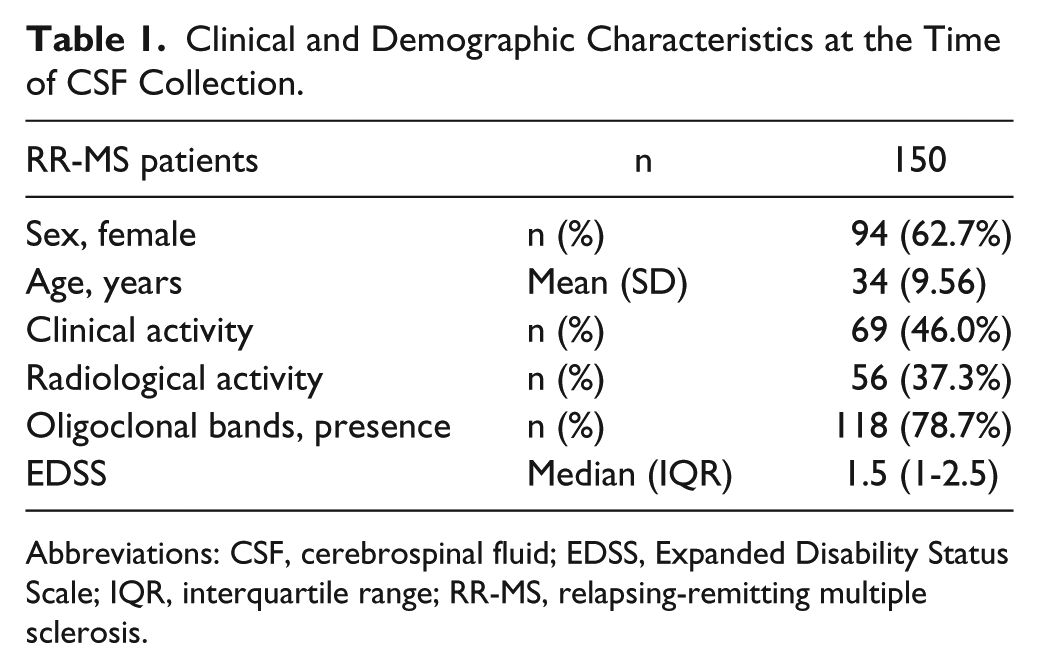
Abbreviations: CSF, cerebrospinal fluid; EDSS, Expanded Disability Status Scale; IQR, interquartile range; RR-MS, relapsing-remitting multiple sclerosis.
Clinical and Demographic Characteristics According to IL-6 Group.
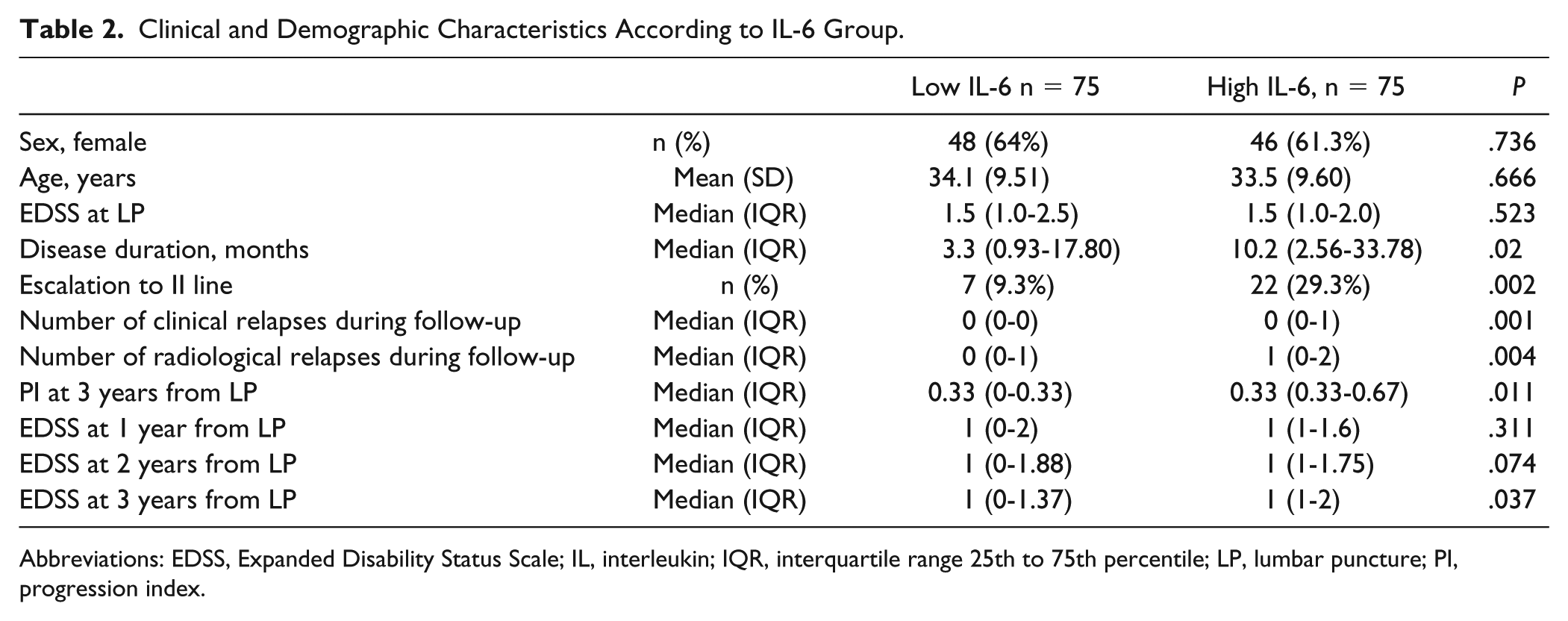
Abbreviations: EDSS, Expanded Disability Status Scale; IL, interleukin; IQR, interquartile range 25th to 75th percentile; LP, lumbar puncture; PI, progression index.
In the high IL-6 group, an increased number of both clinical (high IL-6 group median = 0; IQR = 0-1 vs low IL-6 group median = 0; IQR = 0-0; Mann-Whitney P = .001) and radiological (high IL-6 group median = 1; IQR = 0-2 vs low IL-6 group median = 0; IQR = 0-1; Mann-Whitney P = .004) new events were observed in the follow-up period after CSF withdrawal (36 months). According to the greater disease severity in the high IL-6 group, the proportion of patients who needed an escalation to second-line therapies during the same period significantly differed in the 2 groups [high IL-6 group, 29.3%; low IL-6 group, 9.3%; χ2(2, n = 150) = 9.618; P = .002]. These differences remained significant after correcting for the disease duration in the 2 groups (P = .005).
To quantify the impact of IL-6 concentrations on the number of clinical relapses, we applied a Poisson model adjusting for the other clinical parameters considered (clinical activity and radiological activity at the time of lumbar puncture, age, EDSS, disease duration, and number of prospective radiological relapses). By the stepwise selection method (see statistic session), the final model included the IL-6 group and the number of radiological relapses. Notably, the results showed that during the follow-up, higher values of IL-6 were associated with a greater number of clinical relapses, adjusting for the number of radiological relapses: the adjusted rate was 2.28 times higher for patients with high IL-6 compared with patients with low IL-6 (adjusted IRR = 2.28, 95% CI = 1.34-3.86, P = .002; Figure 3).
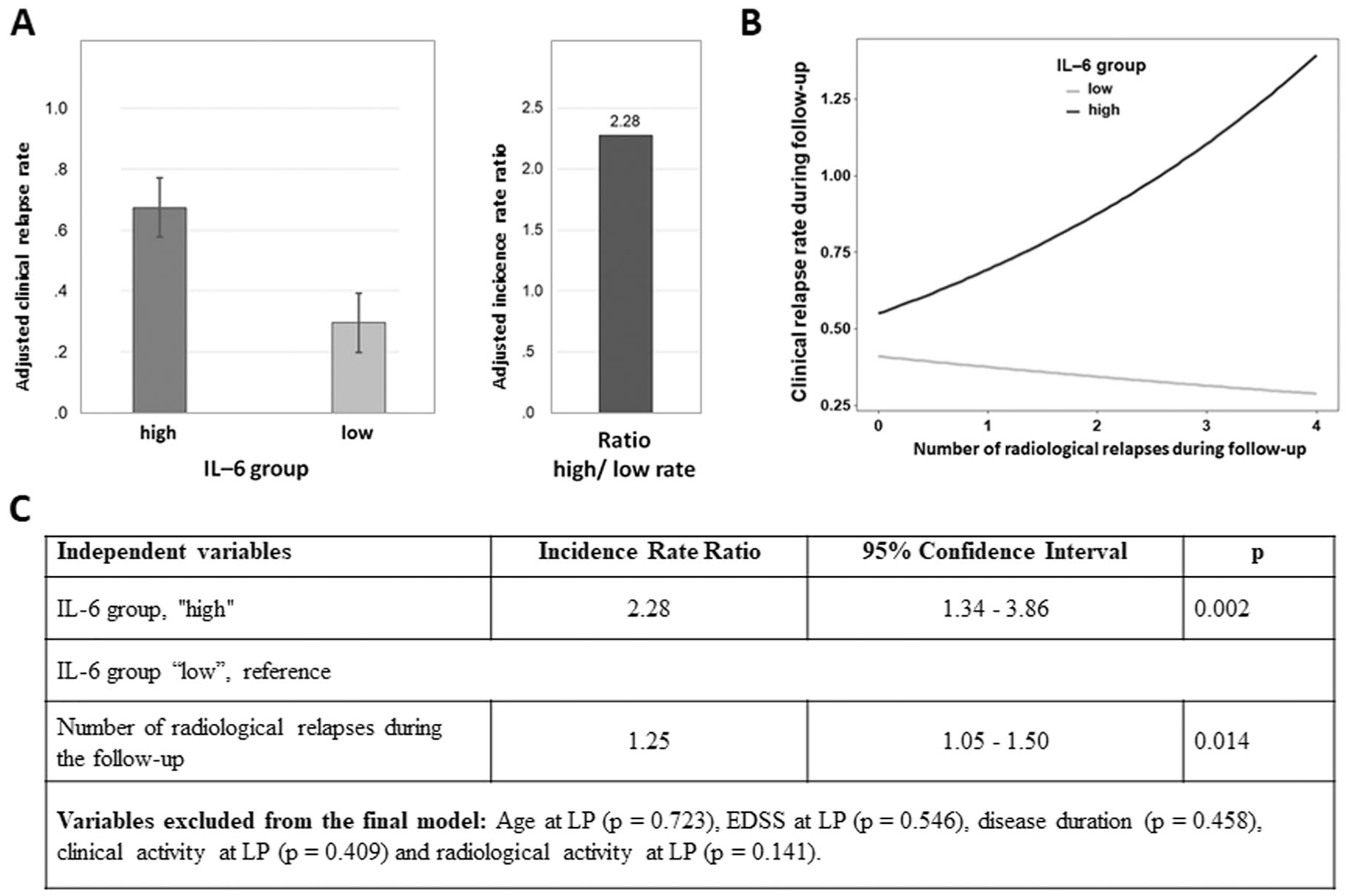
Impact of IL-6 group on clinical and radiological prospective disease activity. A. Adjusted clinical relapse rate in the 2 groups of high and low IL-6. B. Clinical relapse rate according to the number of radiological relapses. The steeper slope of the high IL-6 group indicates poor ability to compensate new radiological brain lesions. C. Results from a multivariate Poisson regression model.
Finally, both EDSS and progression index calculated 3 years after lumbar puncture were significantly higher in the high IL-6 group: EDSS (high IL-6 group median = 1, IQR = 1-2; low IL-6 group median = 1, IQR = 0-1.37; Mann-Whitney P = .032); progression index (high IL-6 group median = 0.33, IQR = 0.33-0.67; low IL-6 group median = 0.33, IQR = 0-0.33; Mann-Whitney P = .014). These results remained significant after correcting for the disease duration effect [EDSS: F(1, 147) = 5.13, P = .025; progression index: F(1, 143) = 5.36, P = .022].
Overall, these data are consistent with the idea that higher central concentrations of IL-6 are associated with worse clinical course in RR-MS patients and that at least part of IL-6–associated poor prognoses likely reflect the disruption of synaptic plasticity produced by this cytokine. In particular, the present results indicate that IL-6 CSF levels may specifically affect the clinical expression of the disease, increasing the probability that new inflammatory MRI lesion appearance could be associated with clinical symptoms.
IL-6 CSF Levels and Biomarkers of Neurodegeneration
To further explore the effect of IL-6 on inflammation-driven tissue damage, we examined the correlation between the CSF concentration of this cytokine and the CSF levels of different biomarkers of neuronal and axonal damage.
The CSF biomarkers explored included the following: amyloid-β 1-42 (median = 293.50; IQR = 187.5-454.5) and neurofilament light protein (median = 1053.5; IQR = 488.75-2462.5). IL-6 concentrations negatively correlated with amyloid-β 1-42 [Spearman ρ(150) = −0.402; P = .001] CSF levels. Conversely, no significant correlation emerged between IL-6 and neurofilament light protein CSF concentrations [Spearman ρ(150) = −0.191, P = .204; Figure 4].
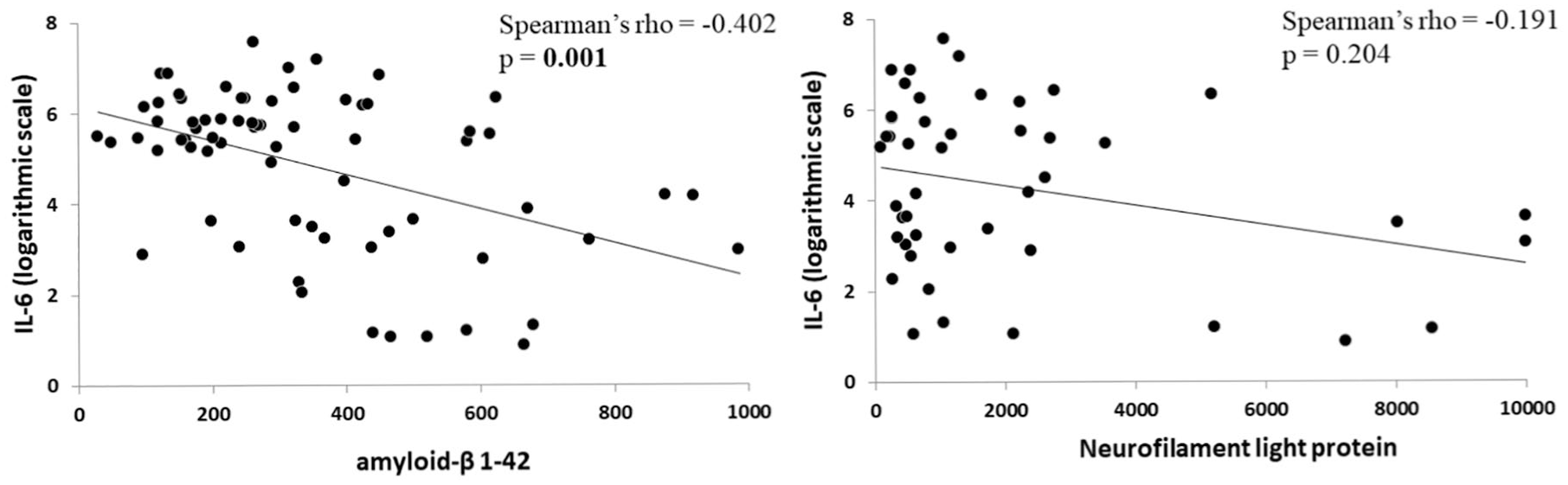
IL-6 CSF levels and biomarkers of neurodegeneration and neuronal damage: Correlations between IL-6 CSF concentrations and biomarkers of neurodegeneration and neuronal damage. To obtain a better graphical representation, IL-6 concentrations are shown on a logarithmic scale. Amyloid-β 1-42 and neurofilament light protein concentrations are expressed as picograms per milliliter.
These results show that CSF levels of biomarkers of neuronal damage are not associated with CSF IL-6 concentrations at the time of diagnosis. Conversely, because altered amyloid-β 1-42 homeostasis has been previously identified as a factor negatively influencing synaptic plasticity in RR-MS, 36 the possibility that impaired synaptic plasticity could partly depend on changes in amyloid-β 1-42 metabolism cannot be ruled out.
Discussion
Central inflammation is emerging as a crucial factor influencing the disease course of MS. In particular, different proinflammatory molecules enhance neurodegeneration and interfere with synaptic plasticity mechanisms, promoting a worse disease course. In the present study, we have found that elevated IL-6 CSF levels measured at the time of diagnosis are associated with worse disease course, as supported by higher clinical disability and increased measures of disease progression. In particular, enhanced prospective disease activity in the follow-up has been revealed in patients with higher IL-6 CSF levels, as shown by increased number of both clinical and radiological relapses. These results are in line with previous studies evidencing the role of this proinflammatory molecule in inducing and maintaining the inflammatory response in MS 26 and its negative impact on prospective disease course. 28 In addition, it has been previously reported that elevated serum concentrations of IL-6 are associated with increased levels of disability in MS. 37 However, the precise role of IL-6 signaling in MS pathogenesis has not been fully understood and multiple effects of this cytokine have been described.38,39
In a recent study, we have shown that in 150 RR-MS patients followed up for 3 years, those who had clinical or radiological relapses during follow-up had higher CSF IL-6 levels at the time of diagnosis. 28 In the present study, we found increased number of clinical relapses in the high IL-6 group after correcting for the number of radiological relapses during follow-up. In addition, our data support the hypothesis that elevated IL-6 levels in MS may interfere with LTP-like plasticity mechanisms because a negative correlation was observed between IL-6 CSF levels and the effects of the PAS protocol.
Experimental studies pointed out that LTP could be an important mechanism involved in the clinical compensation of new brain lesions. Animal studies have shown that after acute focal ischemic brain damage, clinical recovery was related to the ability of surviving neurons to increase their synaptic activity. 2 Also in humans, the efficiency of LTP-like plasticity mechanisms explored with TMS was positively correlated with clinical recovery both after stroke 3 and after a MS relapse. 4 Our data could, therefore, suggest that in patients with higher CSF levels of IL-6, worse clinical manifestations may depend on the reduced ability to minimize the clinical effect of structural damage.
It has been demonstrated that inflammation alters synaptic transmission and plasticity both in vitro and in MS patients.8-10 Preclinical studies have highlighted that different proinflammatory molecules, including IL-1β and IL-6, alter synaptic functioning. Previous studies from our group have demonstrated that IL-1β from the CSF of MS patients with active MRI lesions was able to alter synaptic transmission when incubated in mice hippocampal slices and that synaptic function could be restored after blocking IL-1β signaling.9,13 In addition, it has been shown that IL-1β alters synaptic plasticity expression in vitro.11,12 Accordingly, both decreased efficacy of GABAergic transmission and enhanced glutamatergic transmission could lead to synaptic dysfunction, neuronal hyperexcitability, and altered LTP. 11 Similarly, in RR-MS patients, elevated CSF levels of IL-1β have been associated with paradoxical TMS-induced plasticity. 9 IL-6 is a proinflammatory cytokine playing important roles both in physiological and pathological conditions, although its functions have not been completely elucidated. In particular, our results and previous studies 29 could suggest that, differently from IL-1β, acute exposure to IL-6 may disrupt LTP without affecting basal synaptic glutamate release.
Inflammation promotes neurodegeneration both in mice with experimental autoimmune encephalomyelitis and in MS patients.5,14 In particular, elevated CSF concentrations of specific proinflammatory molecules measured at the time of diagnosis negatively influenced the clinical course.14,28,40 Persisting detectable CSF levels of IL-1β in stable MS patients were associated with increased neuronal atrophy and worse disease course. 14 Conversely, anti-inflammatory molecules and neurotrophic factors also influence synaptic transmission and plasticity, being able to counteract the deleterious impact of neuroinflammation. In particular, it has been demonstrated that higher CSF levels of the platelet-derived growth factor are associated with enhanced LTP-like plasticity, 16 better clinical compensation of brain damage, 17 and stable clinical disease course. 18
Because central inflammation is associated with increased measures of neurodegeneration in MS patients, we explored whether IL-6 central concentrations were correlated with the CSF levels of established biomarkers of neuronal and axonal damage. Increased levels of axonal cytoskeletal proteins have been found in the CSF of MS patients throughout the disease course,41,42 representing not only a marker of neurodegeneration, 43 but also of acute demyelinating lesions. 44 In addition, it has been previously reported that acute inflammation in MS is associated with altered amyloid-β metabolism.36,45 We found a negative correlation between IL-6 and amyloid-β 1-42 CSF concentrations; conversely, no associations emerged with neurofilament light protein CSF levels. These results confirm that increased central inflammation is associated with altered amyloid-β homeostasis. Accordingly, it has been previously reported that in the CSF of RR-MS patients, the levels of different proinflammatory molecules, including IL-8 and IFNγ, were negatively correlated to amyloid-β 1-42 levels. 45 Moreover, the lack of association between IL-6 and neurofilament light protein could suggest that the enhanced clinical disability observed in the high IL-6 group, unlikely depends on different degree of axonal damage.
Previous evidence showed that reduced CSF levels of amyloid-β 1-42 were correlated with impaired LTP-like plasticity in relapsing MS patients. 36 To clarify whether the detrimental effect of high IL-6 CSF levels on synaptic plasticity could be the result of a direct effect on synaptic functioning or indirectly mediated by an altered amyloid-β homeostasis, we explored the acute effects (10 minutes bath application) of IL-6 on hippocampal synaptic plasticity in vitro. The finding that hippocampal plasticity in vitro was abolished after a short-lasting incubation of IL-6 makes unlikely the possibility that the effect could be mediated by complex cascades of events involving altered amyloid-β metabolism and rather suggests a direct pharmacological action of IL-6 at the synaptic level. This observation is in line with previous studies showing a negative role of IL-6 in synaptic plasticity. In mice overexpressing IL-6, the amount of hippocampal LTP was found to be reduced. 46 Furthermore, incubating recombinant human IL-6 in rat hippocampal slices blocked LTP.22,23 Intriguingly, the negative impact of IL-6 on LTP was prevented by the incubation with an anti–IL-6 receptor antibody. 23 The finding that exogenous application of IL-6 also reduced post-tetanic potentiation likely explains that IL-6 acts at both the presynaptic and postsynaptic levels. 23
Only IL-6 CSF levels have been investigated in the present study; therefore, to better define the impact of CSF inflammatory milieu on MS course, further investigations are required to examine the role of a wider panel of proinflammatory and anti-inflammatory cytokines and growth factors. Moreover, the lack of prospective measures of neuronal damage represents another limitation of the present findings, and further studies are needed to exclude the possibility that different degrees of neuronal atrophy could enhance the effect of IL-6 in RR-MS shown here. Finally, it would be helpful to clarify whether the association between CSF IL-6 concentrations and altered LTP expression could be significant also in the more advanced disease phases.
Conclusions
Neuroinflammation alters LTP expression, impairing the ability to compensate the negative clinical impact of new brain lesions in RR-MS, thus favoring clinical worsening after each relapse. Our findings suggest that resolving central inflammation should be mandatory to hinder the progressive accumulation of disability over time in this severe neurological condition.
Supplemental Material
Spplementary_figure – Supplemental material for Interleukin-6 Disrupts Synaptic Plasticity and Impairs Tissue Damage Compensation in Multiple Sclerosis
Supplemental material, Spplementary_figure for Interleukin-6 Disrupts Synaptic Plasticity and Impairs Tissue Damage Compensation in Multiple Sclerosis by Mario Stampanoni Bassi, Ennio Iezzi, Francesco Mori, Ilaria Simonelli, Luana Gilio, Fabio Buttari, Francesco Sica, Nicla De Paolis, Georgia Mandolesi, Alessandra Musella, Francesca De Vito, Ettore Dolcetti, Antonio Bruno, Roberto Furlan, Annamaria Finardi, Girolama A. Marfia, Diego Centonze and Francesca Romana Rizzo in Neurorehabilitation and Neural Repair
Footnotes
Supplementary material for this article is available on the Neurorehabilitation & Neural Repair website along with online version of this article.
Authors’ Note
Roberto Furlan and Annamaria Finardi are now to San Raffaele Scientific Institute, Milan, Italy.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The present study was supported by the Italian Ministry of Health (Ricerca Corrente), by the 5 × 1000 grant to IRCCS Neuromed and by FISM-Fondazione Italiana Sclerosi Multipla-cod. 2019/s/1 to DC.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.