
Abstract
Background. Mortality predictions following traumatic brain injury (TBI), and our understanding of TBI pathology, may be improved by including genetic risk in addition to traditional prognostic variables. One promising target is the gene coding for brain-derived neurotrophic factor (BDNF), a ubiquitous neurotrophin important for neuronal survival and neurogenesis. Objective. We hypothesized the addition of BDNF genetic variation would improve mortality prediction models and that BDNF Met-carriers (rs6265) and C-carriers (rs7124442) would have the highest mortality rates post-TBI. Methods. This study examined BDNF functional single nucleotide polymorphisms rs6265 (val66met) and rs7124442 (T>C) in relation to mortality in a prospective, longitudinal cohort with severe TBI. We examined 315 individuals receiving care for a closed head injury within the University of Pittsburgh Medical Center, aged 16 to 74 years. Mortality was examined acutely (0-7 days postinjury) and postacutely (8-365 days postinjury). A gene risk score (GRS) was developed to examine both BDNF loci. Cox proportional hazards models were used to calculate hazard ratios for survivability post-TBI while controlling for covariates. Results. BDNF GRS was significantly associated with acute mortality, regardless of age. Interestingly, subjects in the hypothesized no-risk allele group had the lowest survival probability. Postacutely, BDNF-GRS interacted with age such that younger participants in the no-risk group had the highest survival probability, while older participants in the hypothesized no-risk group had the lowest probability of survival. Conclusions. These data suggest complex relationships between BDNF and TBI mortality that interact with age to influence survival predictions beyond clinical variables alone. Evidence supporting dynamic, temporal balances of pro-survival/pro-apoptotic target receptors may explain injury and age-related gene associations.
Introduction
In the United States, ~52,000 deaths are attributed to traumatic brain injury (TBI) yearly. 1 As TBI is a highly heterogeneous disease, it is difficult to predict immediate and long-term outcomes. Understanding factors that predict mortality post-TBI may improve treatment. With the recent focus on personalized medicine, this study uses a Rehabilomics 2 approach, examining genetic factors that capture innate heterogeneity across recovery to improve mortality predictions beyond traditional prognostic factors.
The International Mission on Prognosis and Analysis of randomized Controlled Trials in TBI (IMPACT) has demonstrated high predictability of post-TBI mortality/outcome using a core model of age, injury severity (motor subscale of the Glasgow Coma Scale [GCS] 3 ), and pupillary reactivity. 4 Further studies support extending this model by adding neurological findings (evidence of midline shift or presence of subarachnoid hemorrhage 5 ). However, these studies have not examined genetic factors, nor have they addressed the possibility of evolving or dynamic predictors of mortality. As influences on mortality likely change across recovery, it is important to examine mortality predictors over time.
Age is a consistent determinant of TBI survival. Older adults comprise a large segment of the population sustaining TBI, with comparatively worse outcomes and higher mortality rates despite similar injury parameters. 6 Beyond an increased premorbidity burden with age, this phenomenon may be due to adverse age effects on secondary injury cascades. For example, older age leads to greater susceptibility to glutamate-mediated oxidative stress and damage. 7 In experimental models, older animals show decreased neuronal survival postinjury. 8
A ubiquitous neurotrophin in the brain, brain-derived neurotrophic factor (BDNF), may interact with age to influence TBI pathology. BDNF is important for synaptic plasticity, neurogenesis, and neuronal survival. 9 Yet in areas like the hypothalamus, BDNF can regulate metabolism.10,11 Evidence suggests BDNF may also affect brainstem control of cardiovascular function.12 -14 Thus, BDNF may affect autonomic regulation and reflect the current energy state, 15 suggesting multiple mechanisms through which BDNF signaling could influence TBI mortality and recovery. While BDNF may affect mortality in other populations,16 -18 no study has examined BDNF relationships with mortality post-TBI.
BDNF signals through the tyrosine-related kinase-B (TrkB) receptor, full length (TrkB.FL) and truncated (TrkB.T), as well as the p75NTR receptor, activating antagonistic biochemical cascades that are dependent on receptor milieu. Studies suggest TrkB.FL/TrkB.T/p75NTR expression ratios may vary across the lifespan 19 and following ischemia. 20 Similarly, there are dynamic receptor expression changes following experimental TBI. 21 Thus, BDNF’s role in TBI recovery may be dependent on the relative balance of these target receptors.
The BDNF gene has a common, functional, single nucleotide polymorphism (SNP), Val66Met (rs6265), that alters activity-dependent secretion of BDNF in vitro 22 and shows an age-dependent relationship in cognition. 23 Importantly, rs6265 is in linkage disequilibrium (LD) with another BDNF SNP, rs7124442. The rs7124442 variant reportedly affects neuronal BDNF mRNA trafficking. 24 While rs6265 25 and rs7124442 26 have been associated with TBI cognitive recovery, no studies have examined how these variants influence other aspects of TBI recovery, specifically mortality. As rs6265 can influence hypothalamus–pituitary–adrenal (HPA) axis reactivity27,28 and autonomic control of heart rate, 29 there may be important contributions for BDNF variants in TBI recovery outside of cognitive outcomes.
We examined BDNF variation in survivorship post-TBI. We hypothesized that the Met-allele (rs6265), with its relatively decreased activity-dependent BDNF secretion, and the C-allele (rs7124442), with its relatively impaired BDNF mRNA trafficking, would be risk alleles in mortality predictions. We chose to examine a cumulative gene risk score (GRS) incorporating variation at both loci when developing mortality prediction models. In this report, we demonstrate a dynamic, temporal relationship between BDNF GRS and post-TBI survivorship. Our data show a BDNF gene risk relationship with survivorship 0 to 7 days postinjury that is contrary to our hypothesized relationships. Yet, when evaluating mortality models 8 to 365 days postinjury, we show a BDNF age * GRS interaction with survivorship. These findings underscore the importance of understanding age and BDNF signaling relationships following TBI.
Methods
Demographics
This prospective cohort study was approved by the University of Pittsburgh Institutional Review Board. Enrollment criteria for this study included age ≥16 and <75 years and an admission GCS score ≤8 indicating severe TBI. Exclusion criteria included documented prolonged hypoxia prior to admission or penetrating head injury. Subjects were consecutively recruited and consent obtained by appropriate proxy. To minimize genetic stratification effects, 30 associations are reported in Caucasians only (n = 284, reported findings). Important for generalizability of findings, reported analyses in Caucasians-only were similar to results in the total population (n = 315, data not shown).
The analyzed cohort (284 participants) was aged 16 to 74 years (mean = 35.96 ± 15.46; median = 33) with closed head injury receiving care within the University of Pittsburgh Medical Center. GCS scores 3 ranged from 3 to 15 (mean GCS = 6.20 ± 2.54; median = 6) when using the best GCS obtained within 24 hours postinjury. Demographic information (age, race, sex, and mechanism of injury) was collected through clinical chart review and subject/caregiver interviews. Age was treated as a continuous and categorical variable, split at the 75% quartile (Q3) of our population (above and below, Q3 = 45 years). By 365 days postinjury, 25.8% of subjects had died, and distribution of Glasgow Outcome Scale 31 scores for survivors was as follows: 2, n = 13; 3, n = 66; 4, n = 49; 5, n = 22.
The University of Pittsburgh Trauma Registry provided abstracted information from the acute care medical record regarding postinjury complications. These complications were categorized as pulmonary, infection, cardiovascular, musculoskeletal, hematological, renal, wound, gastrointestinal, and neurological complications (Table 1).
Complications Categories.
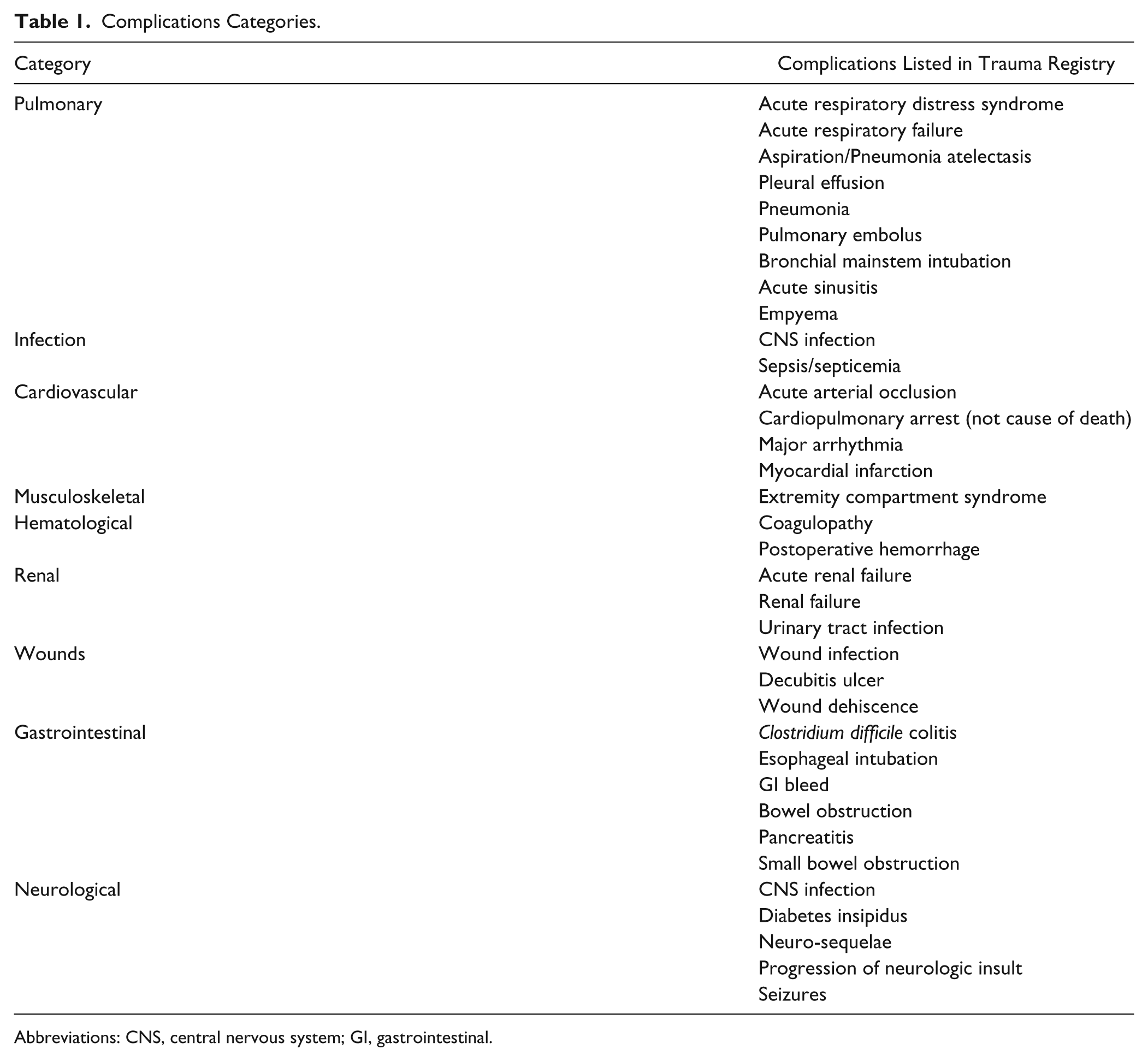
Abbreviations: CNS, central nervous system; GI, gastrointestinal.
Neurological injury assessments were abstracted from admission head computed tomography reports and were categorized by the following injury subtypes: contusion, subdural hematoma (SDH), subarachnoid hemorrhage (SAH), intraventricular hemorrhage (IVH), intracranial hemorrhage (ICH), epidural hematoma (EDH), and diffuse axonal injury (DAI). A neurological burden score (NBS) was calculated by summing injury types that significantly affected survival for a given mortality group. NBS was only used in the acute mortality analysis and consisted of SAH, EDH, and contusion. For the postacute mortality group, only the ICH category survived correction for age and GCS in Cox model predictions of survivorship.
Seizure information was abstracted from available medical records and coded as time to first posttraumatic seizure (PTS), up to 365 days postinjury. Time to first seizure was divided into 2 groups, consistent with mortality cohorts and standard PTS nomenclature: acute (0-7 days) and postacute PTS (8-365 days). 32 Notation in medical records referring to convulsions, seizures, status epilepticus, or seizure disorder was documented as a PTS episode.
Mortality
Time until death was recorded in days postinjury, up to 1 year postinjury, using the Social Security Death Index. 33 Mortality was evaluated over 2 time-epochs, 0 to 7 days postinjury (acute) and 8 to 365 days postinjury (postacute). For 0 to 7 days, survivorship was right censored at 7 days postinjury. For 8 to 365 days, subjects were only included if they survived greater than 7 days, and survivorship was right censored at 365 days. For logistic regression and receiver operating curve (ROC) analysis, mortality was examined as a binary outcome at 7 days postinjury (acute) and 365 days postinjury (postacute, excluding subjects who died before 7 days). Acutely, 11.62% of subjects died (time until death: median = 3 days, min = 0 days, max = 7 days, Q1 = 2 days, Q3 = 6 days). An additional 14.16% died postacutely (time until death: median = 19 days, min = 8 days, max = 301 days, Q1 = 11.5 days, Q3 = 31 days).
Genotyping and SNP Selection
DNA was isolated from blood using a simple salting out procedure 34 or from cerebrospinal fluid using the Qiamp protocol from Qiagen. BDNF rs6269 and rs7124442 were genotyped by TaqMan allele discrimination assay using Assay on Demand reagents (Applied Biosystems). This assay used fluorescent labeled probes to detect allele(s) present for DNA sample. All allele frequencies were in Hardy–Weinberg equilibrium.
Selected SNPs (rs6265, rs7124442) have been reported as functional. Both SNPs have a minor allele frequency >20%. Each SNP represents a different haplotype block of BDNF covering variation corresponding to BDNF isoform a. A cumulative BDNF GRS was developed using rs6265 Met (Val/Met or Met/Met) and rs7124442 C (T/C or C/C) carrier status as hypothesized risk alleles based on the literature.22,24 Thus, a GRS of 0 was the hypothesized no risk group (Val/Val, T/T); a GRS of 1 included carriers for 1 risk allele (Val/Val, C-carriers or Met-carriers, T/T); and a GRS of 2 included carriers of both risk alleles (Met-carriers, C-carriers).
Statistical Analysis
Analysis was conducted using Statistical Analysis Software (version 9.2; SAS Institute) and the Statistical Package for Social Sciences (version 21.0; SPSS). Descriptive analysis included mean ± standard deviation (SD) for continuous variables. Frequencies were calculated for categorical variables. Genetic analysis used categorizations based on allele carrier status, and the BDNF GRS was used to examine cumulative genetic risk associations with mortality. Demographic and clinical information was compared with mortality status and genotype using Student’s t tests and ANOVA (Mann–Whitney or Kruskal–Wallis where appropriate) to compare means, and χ2 or Fisher’s exact test to compare frequencies.
Demographic, clinical, and genotype information was examined for survivorship associations using either a Kaplan–Meier or Cox proportional hazards model. 35 The log-rank test was used to determine significant differences between 2 survival curves (significant if P < .05). To control for covariates, we used multivariate Cox proportional hazards regression. Demographic and clinical variables were included in the final Cox models if they survived correction for age and GCS (P ≤ .2). The proportionality of hazards assumption was tested and confirmed for relevant variables.
Mortality status was examined using multivariate ROC analysis to quantify model prediction capacity and to relate to published studies. 5 Mortality was assessed at 7 days and 365 days postinjury. Using area under the curve (AUC), ROCs estimated incremental increases in model sensitivity and specificity gained when including GRS and/or GRS interactions, compared to base models of relevant clinical variables. Base models were compared to final models using a χ2 test for significant differences in AUC (P < .05 considered significant).
Results
Genetic Associations With Demographics
Demographic distributions were examined for both BDNF loci (Table 2). The mean age for T/T homozygotes (rs7124442) was higher versus C-carriers (38.0 ± 16.3 vs 33.4 ± 14.0, P = .011). DAI was less common among T/T homozygotes versus C-carriers (24.7% vs 35.7%, P = .043). However, subjects with DAI were significantly younger compared to subjects without DAI (28.7 ± 11.9 vs 39.0 ± 15.8, P = .038). There were no demographic variable differences by rs6265 Met-carrier status.
Demographic Variables by Genotypes.
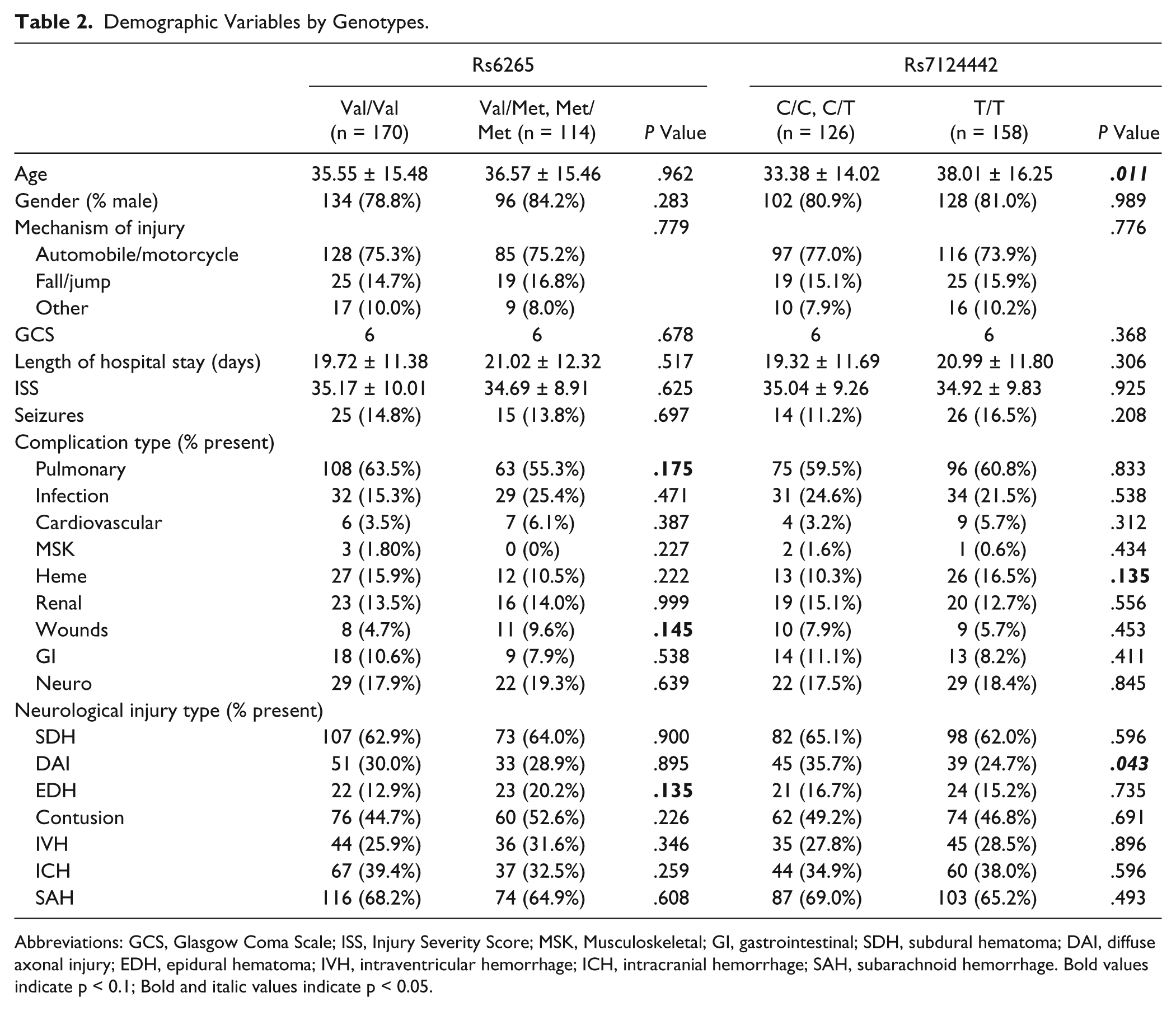
Abbreviations: GCS, Glasgow Coma Scale; ISS, Injury Severity Score; MSK, Musculoskeletal; GI, gastrointestinal; SDH, subdural hematoma; DAI, diffuse axonal injury; EDH, epidural hematoma; IVH, intraventricular hemorrhage; ICH, intracranial hemorrhage; SAH, subarachnoid hemorrhage. Bold values indicate p < 0.1; Bold and italic values indicate p < 0.05.
Mortality Associations With Demographics
Demographic associations with acute/postacute mortality and survivorship were examined (Table 3). Survivors had lower mean age (all group, P < .001) and higher GCS scores (all group, P = .002) compared to nonsurvivors. In Cox proportional hazards regression, age and GCS predicted survival probability for both 0 to 7 days and 8 to 365 days models (P < .03, all comparisons). Pulmonary (adjusted for age, GCS: P < .001, hazard ratio [HR] = 0.156) and cardiac (adjusted for age, GCS: P = .196, HR = 2.065) complications survived correction for age and GCS (P < .2) and were included in the overall models for acute survival. Pulmonary complications were less frequent in subjects who died acutely (Table 3), but subjects with pulmonary complications were significantly younger than those without pulmonary complications (34.1 ± 15.1 vs 38.8 ± 15.6, P = .009). Cardiac complications were more frequent in subjects who died acutely compared to survivors (12.1% vs 2.8%, all group, P = .026). For the 8 to 365 days model, a wound complication (adjusted for age, GCS: P = .020, HR = 2.857) was the only complication to significantly predict survival probability.
Demographic Information Across Mortality Groups (Acute, Postacute, and Survivor) (Kruskal–Wallis, Fischer’s Exact, Significance at P < .05).
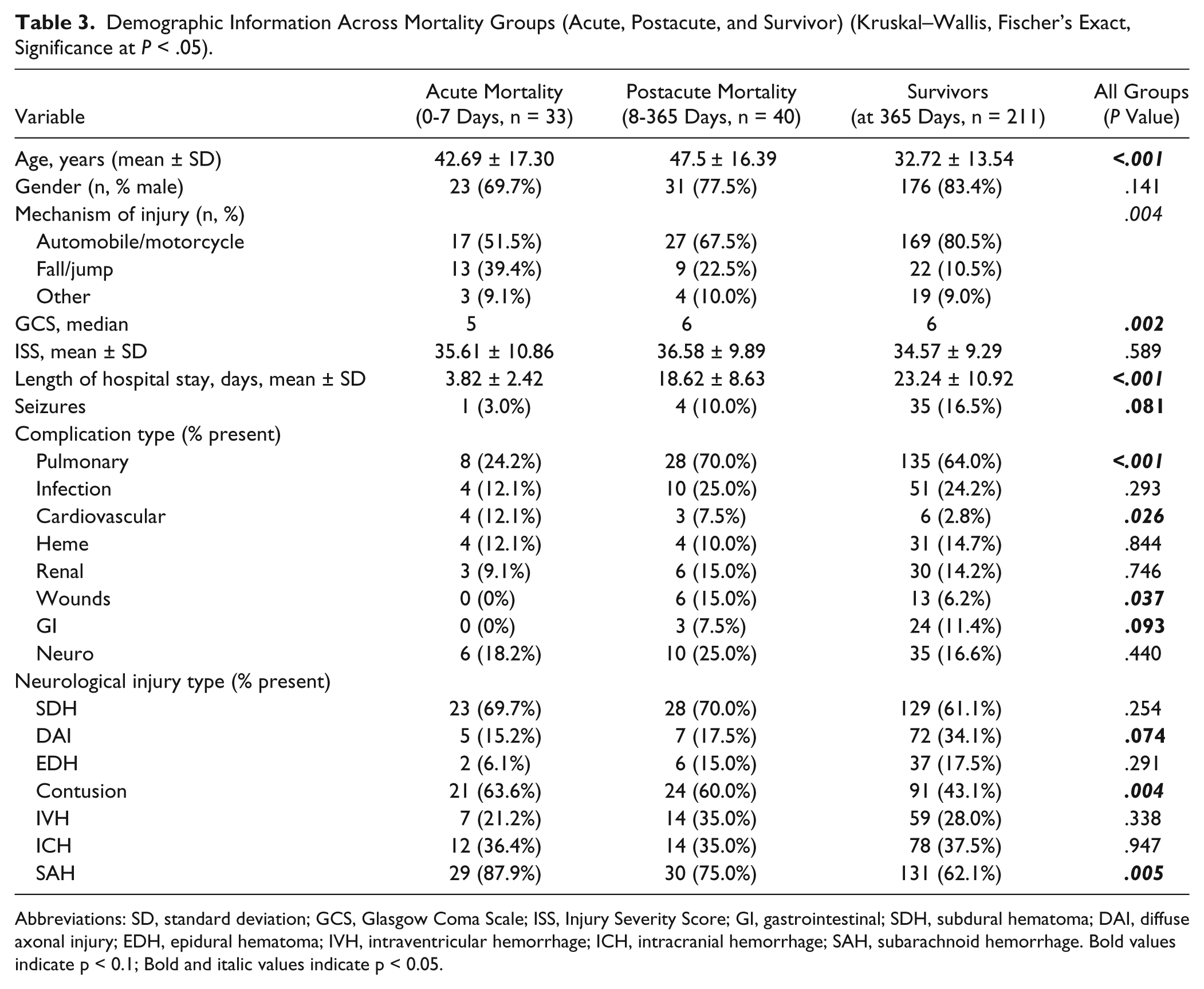
Abbreviations: SD, standard deviation; GCS, Glasgow Coma Scale; ISS, Injury Severity Score; GI, gastrointestinal; SDH, subdural hematoma; DAI, diffuse axonal injury; EDH, epidural hematoma; IVH, intraventricular hemorrhage; ICH, intracranial hemorrhage; SAH, subarachnoid hemorrhage. Bold values indicate p < 0.1; Bold and italic values indicate p < 0.05.
While there were no differences in frequency of radiological findings between acute and postacute mortality groups, there were significant associations with survivorship. For the 0 to 7 days model, the NBS (including SAH, epidural hematoma, and contusion; range of 0-3) predicted survivorship (corrected for age, GCS: P = .046, HR = 1.379). SAH and contusions were more frequent in subjects who died (0-7 days or 8-365 days) versus survivors (Table 3). For the 8 to 365 days model, those with ICH tended to have higher survival frequencies (age and GCS corrected, P = .079, HR = 0.541).
Genetic Associations With Mortality
Kaplan–Meier curves reflecting mortality 0 to 7 days postinjury tended to have different survivorship probabilities based on BDNF GRS (P = .144, Figure 1A). Those with a GRS = 0 hypothesized risk alleles had the lowest probability of survival. Multivariate Cox regression predicting survivorship 0 to 7 days postinjury showed BDNF GRS became significant after adjusting for age, GCS, NBS, pulmonary complications, and cardiac complications (Table 4A). Those with a GRS of 1 or 2 had a higher probability of survival compared to those with a GRS of 0 (GRS = 1, P = .0107, HR = 0.351, 95% confidence interval [CI] = 0.157-0.784; GRS = 2, P = .0286, HR = 0.349, 95% CI = 0.136-0.895). Eight to 365 days postinjury, there was a trend for different survivorship probabilities based on BDNF GRS (P = .134, Figure 1B), where those with a GRS = 0 had the highest survival probability. In this case, multivariate Cox regression demonstrated that this trend for BDNF GRS was not significant with covariate adjustment (Table 4B).
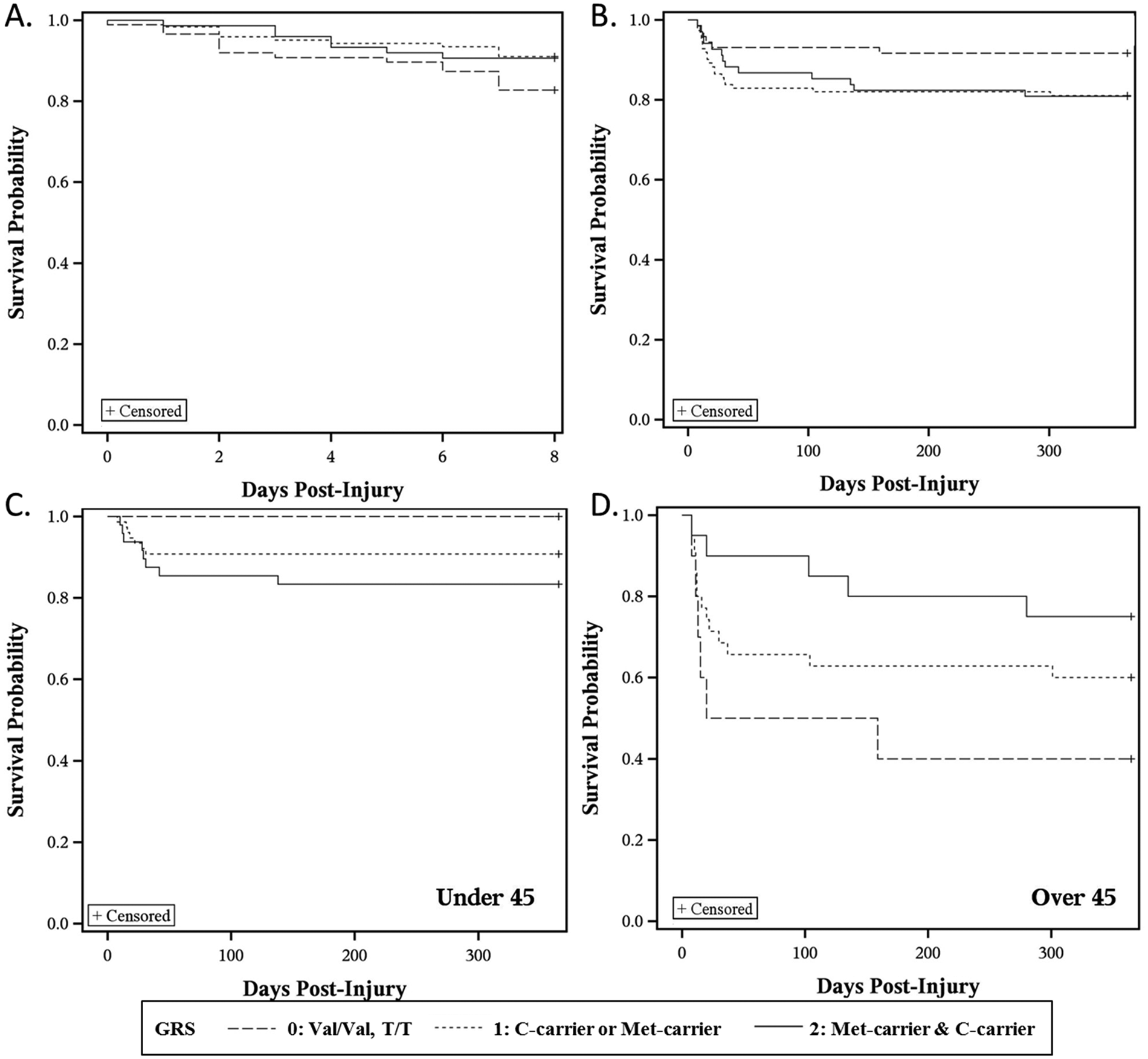
Kaplan–Meier curves show survivorship probabilities stratified by BDNF gene risk score (GRS).
Cox Model of BDNF Gene Risk Score.
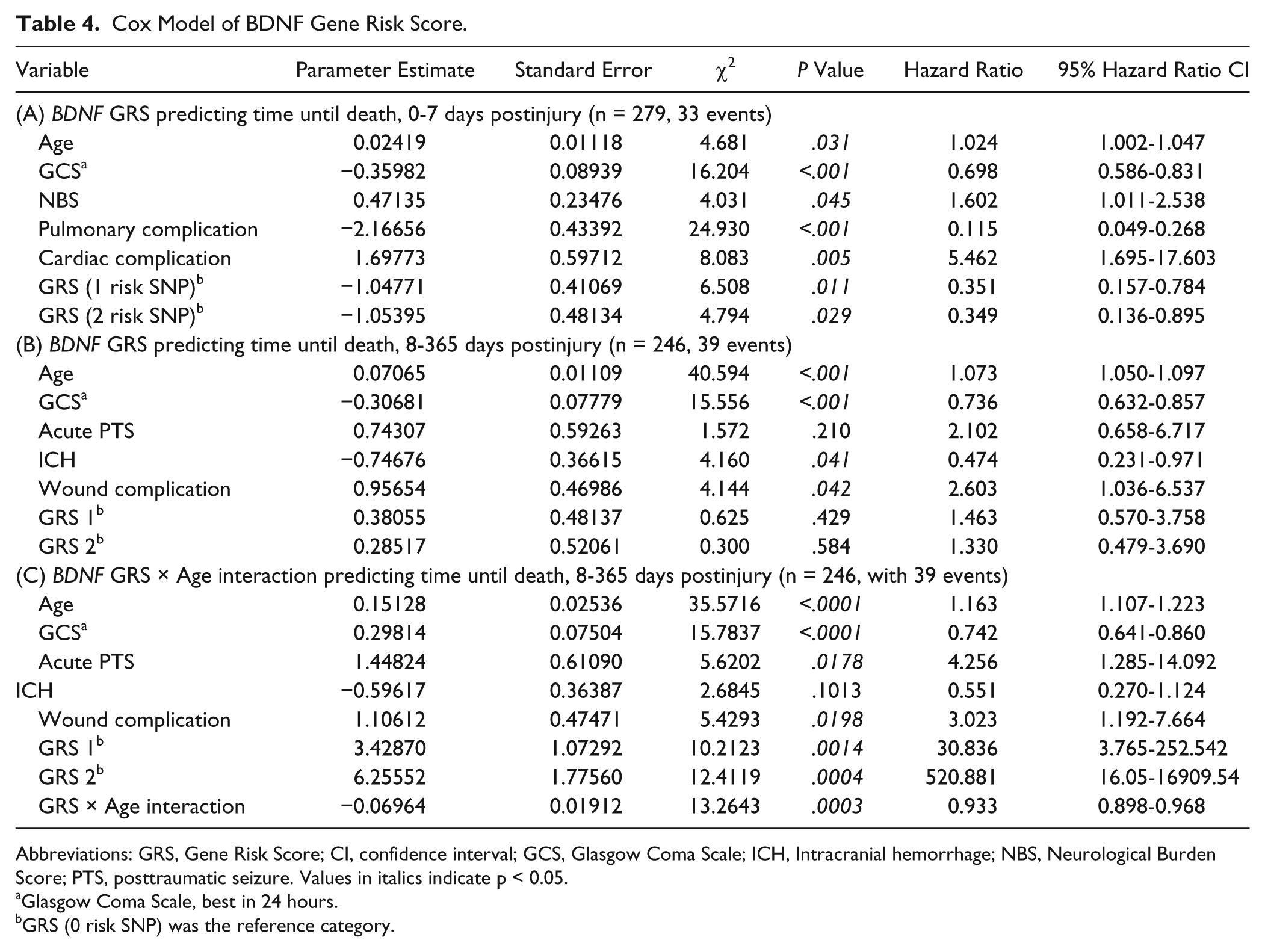
Abbreviations: GRS, Gene Risk Score; CI, confidence interval; GCS, Glasgow Coma Scale; ICH, Intracranial hemorrhage; NBS, Neurological Burden Score; PTS, posttraumatic seizure. Values in italics indicate p < 0.05.
Glasgow Coma Scale, best in 24 hours.
GRS (0 risk SNP) was the reference category.
Next, age * GRS interactions with survival probability were examined. There was no significant age * GRS interaction in 0 to 7 days survivorship probability (data not shown). However, Table 4C shows a significant age * GRS interaction for a 8 to 365 days model predicting survivorship (Age * GRS interaction, P = .0003, HR = 0.933, 95% CI = 0.898-0.968). To increase the interpretability of this interaction, we tested the relationship of GRS with survivorship prediction 8 to 365 days postinjury across different age cut-points. The population was stratified, below and above Q3 (age = 45), and GRS was examined in Kaplan–Meier curves for the 2 age strata. Among participants <45 years old, GRS associations with survivorship demonstrated that those with a GRS = 0 had the highest probability of survival (P = .006), while participants >45 years old with a GRS = 0 had the lowest survivorship probability (P = .106; Figure 1C-D).
We further validated these models in multivariate ROC examining post-TBI mortality status. Using a similar censorship strategy and covariate selection as with our Cox models, we examined mortality status for acute (0-7 days) and postacute (8-365 days) mortality. The 0 to 7 days base model had an AUC = 0.8407. The addition of BDNF GRS and a GRS * age interaction did not significantly improve this model. The 8 to 365 days base model (excluding subjects who died in the acute phase) had an AUC = 0.8359. The addition of the GRS * age interaction increased the AUC above the base model (from 0.8359 to 0.8763, P = .021, Figure 2).
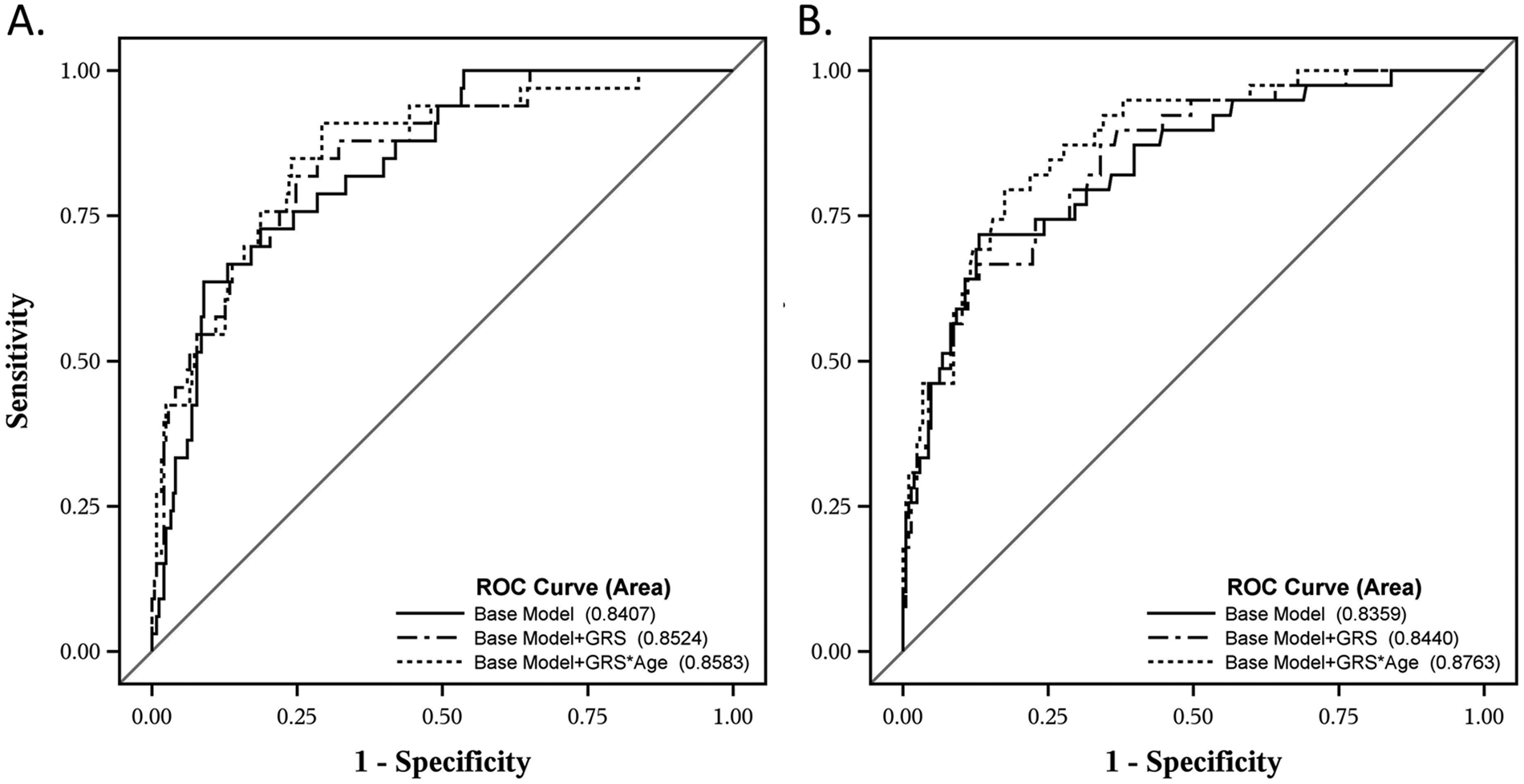
Receiver operating curves of GRS models in acute (0-7 days) and postacute (8-365 days) mortality.
Discussion
This study demonstrates important gene and gene * age interactions with BDNF in post-TBI mortality. First, we identified a dynamic temporal relationship between BDNF and survival. Second, we showed the hypothesized BDNF gene risk relationship to mortality is not supported acutely (0-7 days postinjury), as the hypothesized no risk group had the lowest survival probability. Third, we demonstrated BDNF genetics interact with age to inform survivorship predictions 8 to 365 days postinjury. Here, the hypothesized risk relationship was supported in younger individuals, while older individuals maintained a similar risk pattern to that observed acutely. We speculate dynamic relationships between BDNF and mortality risk post-TBI may be attributable to age and injury effects on BDNF target receptors. Other actions of BDNF in autonomic function and other body systems may also impact its role in TBI-related mortality.
These unique relationships between BDNF and TBI mortality may be related to specific alterations in target receptor milieu with regard to TBI and age. BDNF, first synthesized as proBDNF, is processed either in the soma, extracellular space (after release), or dendrites (after endocytosis). 36 If not cleaved, proBDNF targets the pro-apoptotic p75NTR receptor, 37 while mature BDNF initiates pro-survival signaling through the full length TrkB receptor (TrkB.FL). Following experimental TBI, tissue plasminogen activator (tPA), an enzyme that cleaves proBDNF, shows increased activity ipsilateral to the injury, 38 and mice lacking tPA show reduced edema and cortical lesion volume. 39 Additionally, there are 2 truncated isoforms of TrkB, TrkB.T1 and TrkB.T2, which lack intracellular tyrosine signaling but are implicated in other pathways.20,40 In experimental TBI, there are transient increases in hippocampal TrkB.FL; this study also showed regionally specific p75NTR increases up to 8 weeks post-TBI. 21 Although controversial, the ratio of TrkB.FL/Trk.T expressed has been suggested to influence cell survival during excitotoxic injury.20,40 Other studies report pre-incubation with BDNF prior to excitotoxic conditions/insults may be neuroprotective,41,42 but this effect is likely receptor-dependent. As studies demonstrate transient increases in hippocampal BDNF transcription immediately following experimental TBI,21,43 followed by chronically decreased levels, 31 understanding temporal receptor expression changes may be critical in TBI.
Under the assumption that mature BDNF-TrkB.FL signaling is the primary action in adults, 44 our risk allele assignment was based on the hypothesis that lower BDNF signaling would result in reduced pro-survival signaling and negatively affect survival. Thus, it would be reasonable to suggest lower BDNF signaling prior to/during an injury would exacerbate neuronal death post-TBI via reduced pro-survival signaling. However, the genetic variant hypothesized to increase BDNF activity-dependent secretion (Val/Val, rs6265) was associated with increased acute (0-7 days) mortality risk. Consistent with animal literature, 21 this relationship may be the result of an injury-specific balance of BDNF’s target receptors, with relative increases in TrkB.T or p75NTR compared to uninjured adults.
We show an important age * gene interaction with the BDNF GRS in postacute mortality prediction. One explanation for age-specific risk profiles is differential expression patterns of target receptors across aging.19,45 Romanczyk et al. reported dynamic prefrontal cortex TrkB expression across the lifespan, with a peak in young to middle adulthood that decreases with age. 46 Webster et al reported similar TrkB expression patterns in the hippocampus and temporal lobe. 47 Compared to young adult rats, aged rats have reduced TrkB.FL, but not TrkB.T, 48 altering receptor ratios. Post-TBI, an age-specific shift in the balance of BDNF receptor ratios, from pro-survival to pro-apoptotic, could diminish recovery. Thus, older individuals with BDNF genotypes associated with higher baseline BDNF signaling may have a disadvantage.
This study focuses on 2 variants, rs6265 and rs7124442. While the rs6265 Met-allele impairs secretion and intracellular processing of mature BDNF in hippocampal neurons, 22 there is currently no evidence that it alters proBDNF/BDNF ratios. The rs7124442 C-allele reduces BDNF mRNA trafficking from the soma to dendrites in hippocampal cultures. 20 Given that studies suggest BDNF mRNA translated in dendrites are more likely to be secreted in the proBDNF form, 49 it is possible rs7124442 alters proBDNF/BDNF ratios. While these variants are hypothesized to affect BDNF signaling, it is unclear if this remains true with age or injury. We suggest these variants are indicative of variability in neurotrophic support postinjury and, thus, may interact with receptor expression to produce TBI-specific dynamic risk profiles.
Previous studies with the BDNF gene suggest it interacts with age and environment to affect cognitive function. 50 One study showed Met-carriers, despite theoretically lower activity-dependent BDNF secretion, have greater cognitive recovery following penetrating TBI decades after injury. 25 These data are part of a growing literature demonstrating TBI-specific genetic risk relationships with TBI outcomes.51 -53 In contrast to our findings, BDNF variation has also been examined in stroke, where the Met-allele was linked to poor recovery, regardless of age, 54 though it is unclear if age interactions were explored in this relatively older stroke population compared to our cohort. However, a preclinical study examining Val66Met in a rodent stroke model showed the Met-allele enhanced motor recovery chronically, further supporting the concept of injury-specific associations for this variant. 55 Yet these studies focused on cognitive/plasticity effects of BDNF. It will be important to understand regional and temporal roles for BDNF in recovery or rehabilitation-based interventions compared to mortality risk.
In addition, BDNF can regulate energy metabolism and autonomic function. Evidence suggests that BDNF may be involved in brainstem regulation of cardiovascular function.12 -14 Similarly, BDNF modulates the sympathetic/parasympathetic balance in cardiovascular function. 56 Interestingly, rs6265 is associated with differences in heart rate variability 29 and acute stress heart rate reactivity in healthy populations. 27 In fact, one study showed that local BDNF administration following surgical sympathectomy induced hippocampal vascular changes and edema. 57 This study suggests BDNF effects during a state of compromised autonomic function (e.g., immediately following TBI 58 ) could impact TBI pathology, particularly at early time-points when mortality rates are highest. There is a dearth of research about BDNF function outside of cognition or plasticity post-TBI, limiting speculation about how BDNF and CNS-peripheral modulation of autonomic function post-TBI might occur. It is also not clear how age may interact with BDNF in autonomic regulation.
Our data also suggest temporally specific prognostic factors for mortality across recovery. Many TBI survival studies use a cross-sectional approach to mortality. Our results suggest there are different factors contributing to mortality predictions over time that are not captured within the current literature. Acutely, a higher NBS significantly reduced survivorship probability, consistent with published studies showing the addition of neuroradiological findings improves mortality predictions. 5 While pulmonary complications occur less frequently in survivors, subjects with pulmonary complications also were significantly younger, consistent with published studies in TBI. 59 However, pulmonary complication effects were independent of age. One consideration for this finding is the possibility that there is a delayed onset for pulmonary complications, such as acute respiratory distress syndrome, which may be secondary to TBI-specific ICP management, and thus may not be a large negative factor within acute mortality models. 60 Also, cardiac complications were a negative predictor of survival acutely. GCS was a significant factor in mortality predictions, consistent with previous studies on the relationship of injury severity to mortality post-TBI. 5
Subjects in the postacute survival model had a median time until death of 19 days, suggesting that in this early postacute time frame there are unique factors in mortality prediction. Early posttraumatic seizures were not related to mortality predictions in the acute cohort. Yet acute seizure occurrence negatively affected survivorship postacutely, consistent with epidemiological studies linking seizures to higher mortality risk after TBI, 61 suggesting the pathology associated with early seizures is also relevant to postacute mortality. Subjects with wound complications had reduced survival probability, likely reflecting other important health/recovery factors, such as mobility, as contributing postacute mortality risk. 62 Wounds may also be a surrogate measure for infections or sepsis that could affect mortality post-TBI. Subjects with ICH had a higher probability of survival postacutely compared to those without ICH. As these subjects survived acutely, they were likely monitored closely for surgical intervention, standard care that may have increased survival probability postacutely.
This study also shows that, in our postacute model, our GRS significantly added prognostic capacity to the base mortality model. While different cohorts, assessed over different time-frames, our AUC displays better mortality discrimination ability compared to previously published prediction models that include age, GCS, and pupil dilation only (AUC = 0.7875). Furthermore, the use of Cox models strengthens our survivorship predictions. These data suggest that, while clinical variables (e.g., GCS) inform outcome, genetic factors can influence mortality predictions beyond what standard clinical variables are able to accomplish, particularly for the postacute period when most primary neurological injury effects on mortality outcomes have already occurred. While novel and promising prognostic models, these findings need validation in independent studies with larger populations.
There are some limitations in interpretation and generalizability to consider. While BDNF is primarily expressed in the brain, 33 BDNF is also synthesized and secreted from vascular endothelial cells and may have a peripheral action (see review by Caporali and Emanueli 63 ). Also, there is a substantial peripheral store of BDNF in platelets, 50 yet it is unclear if platelet release is altered in TBI. One study suggests plasma BDNF levels predict mortality in ICU patients without direct brain injury, but the mechanism of this association is unclear. 16 Plasma BDNF levels were also related to all-cause mortality in a cohort of older women. 17 With altered metabolic homeostasis immediately following TBI, there may be a vascular action of BDNF that could influence mortality.
There are some additional considerations in this study as it uses a candidate gene approach. The study findings are specific to a racially homogenous population, though models incorporating our small population with other racial backgrounds were stable, with similar results as reported (data not shown). Future studies should evaluate BDNF risk in populations with diverse racial backgrounds to increase generalizability to the TBI population. The variants studied also had relatively low frequency for some genotypes, warranting a carrier approach, similar to published Val66Met studies.25,28 Similarly, these variants only cover the most well-studied isoform of BDNF, yet emerging research indicates that there are multiple isoforms for BDNF. 64 Future research may require additional genotyping to assess the relevance of these isoforms on TBI pathology and outcome prognostication.
Future studies may examine how these findings relate to other genetic variants such as apolipoprotein E (APOE) that have shown associations to TBI mortality. 65 It was also difficult to examine genetic relationships to injury type given the severity of injury in this cohort, as many subjects showed multiple injury subtypes. As we do not have specific cause of deaths for this study, future studies may examine specific cause of death relationships to genetic risk.
Importantly, this study implicates BDNF signaling in TBI mortality prediction, supporting the need for validation studies and a better understanding of BDNF signaling post-TBI across body systems. This work supports the need for examination of specific regional TrkB.FL/TrkB.T/p75NTR receptor ratios in experimental TBI models and postmortem tissue. As serum BDNF is decreased acutely in clinical TBI, 66 future studies may investigate BDNF as a biomarker, with age/gene variation as possible BDNF modifiers post-TBI. Future studies focusing on the dynamic roles of BDNF in mortality compared to rehabilitation and recovery following TBI are needed and may yield different associations reflective of unique pathology and/or recovery mechanisms.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Department of Defense (W81XWH-07-1-0701, Wagner), National Institute on Disability and Rehabilitation Research (NIDRR H133A120087, Wagner), and National Institute of Health (R01HD048162, Wagner; R01NR008424 and R01NR013342, Conley).