
Abstract
Background. Older adults have higher mortality rates after severe traumatic brain injury (TBI) compared to younger adults. Brain-derived neurotrophic factor (BDNF) signaling is altered in aging and is important to TBI given its role in neuronal survival/plasticity and autonomic function. Following experimental TBI, acute BDNF administration has not been efficacious. Clinically, genetic variation in BDNF (reduced signaling alleles: rs6265, Met-carriers; rs7124442, C-carriers) can be protective against acute mortality. Postacutely, these genotypes carry lower mortality risk in older adults and greater mortality risk among younger adults. Objective. Investigate BDNF levels in mortality/outcome following severe TBI in the context of age and genetic risk. Methods. Cerebrospinal fluid (CSF) and serum BDNF were assessed prospectively during the first week following severe TBI (n = 203) and in controls (n = 10). Age, BDNF genotype, and BDNF levels were assessed as mortality/outcome predictors. Results. CSF BDNF levels tended to be higher post-TBI (P = .061) versus controls and were associated with time until death (P = .042). In contrast, serum BDNF levels were reduced post-TBI versus controls (P < .0001). Both gene * BDNF serum and gene * age interactions were mortality predictors post-TBI in the same multivariate model. CSF and serum BDNF tended to be negatively correlated post-TBI (P = .07). Conclusions. BDNF levels predicted mortality, in addition to gene * age interactions, suggesting levels capture additional mortality risk. Higher CSF BDNF post-TBI may be detrimental due to injury and age-related increases in pro-apoptotic BDNF target receptors. Negative CSF and serum BDNF correlations post-TBI suggest blood–brain barrier transit alterations. Understanding BDNF signaling in neuronal survival, plasticity, and autonomic function may inform treatment.
Introduction
The World Health Organization suggests traumatic brain injury (TBI) will be a leading cause of death and disability by 2020, with about 10 million people affected each year. 1 Advanced age is a consistent determinant of TBI survival. 2 Older adults comprise a large segment of the population sustaining TBI, with comparatively worse outcomes and higher mortality rates despite similar injuries among younger individuals. 3 Recent literature indicates individuals who survive the acute phase following a moderate/severe TBI are still at increased risk for mortality during postacute phases of recovery and, on average, have a shorter life span.2,4
Systemic and central nervous system (CNS) biomarkers can inform acute mortality predictions after moderate/severe TBI.5,6 In addition, biomarkers elucidate TBI-specific pathology relevant to the molecular pathways they represent, identifying new targets for neuroprotective therapeutics or indicating important injury-specific phenomena in well-studied pathways. While some studies have evaluated early biomarker patterns predicting mortality and/or global outcomes,5-8 less is known about how innate biological factors, like genetics, interact with acute biomarkers to influence mortality and/or global outcomes.
Current studies suggest demographic and clinical variables like age, injury severity, and pupillary reactivity influence mortality prediction 9 ; however, our previous work demonstrates genetic variation within the brain-derived neurotrophic factor (BDNF) gene informs mortality prognostication beyond that captured by clinical variables. 10 BDNF, a ubiquitously expressed neurotrophin in the brain, is an intriguing target for TBI intervention research due to its role in neuronal survival, neurogenesis, and plasticity.11,12 However, in experimental TBI, acute infusions of BDNF did not improve motor, cognitive recovery or neuronal survival postinjury. 13
BDNF is also intriguing marker during TBI rehabilitation due to its important effects on the autonomic nervous system through hypothalamic metabolic regulation14,15 and brainstem control of the cardiovascular system.16-18 Variation within the BDNF gene has been associated with hypothalamus–pituitary–adrenal (HPA) axis reactivity19,20 and autonomic regulation of heart rate. 21 Major trauma like TBI results in a sustained stress response,6,22 and individual variation in stress hormone production and response may lead to different outcomes postinjury.7,23 Given BDNF’s role in autonomic function and neuronal survival, BDNF may have multiple actions that could affect TBI mortality and global outcomes, making BDNF an attractive biomarker to explore across recovery where these actions may have dynamic influences.
BDNF is synthesized as pro-BDNF and then cleaved to mature BDNF. 24 Mature BDNF has pro-survival signaling capabilities via the full-length TrkB receptor (TrkB.FL), while pro-BDNF can be pro-apoptotic via p75NTR receptor binding. 25 Age26-28 and experimental TBI 29 can shift regional balances in BDNF receptor ratios from pro-survival to pro-apoptotic. In fact, our previous work demonstrated age and injury-specific associations with the BDNF gene and mortality post-TBI. 10 We examined 2 single-nucleotide polymorphisms (SNP) within BDNF, rs6265, with reduced activity-dependent secretion of BDNF, 30 and rs7124442, which impairs neuronal BDNF mRNA trafficking. 31 Given the effects on BDNF signaling, rs6265 Met-allele and rs7124442 C-allele were identified as possible risk factors, with the hypothesis that reduced BDNF signaling would be detrimental to recovery. Surprisingly, low signaling genotypes were protective in acute TBI-related mortality. Postacutely, low signaling genotypes again carried lower mortality risk in older adults, while these same variants were risk factors among younger adults. Given the effects of age and injury on target receptor milieu and our previous findings with BDNF variation in TBI-related mortality, we hypothesized BDNF may be a temporally dynamic, target receptor dependent, and genetically influenced biomarker for mortality and/or outcome post-TBI.
BDNF has been extensively evaluated as a biomarker in affective disorders, where lower serum levels are associated with depressive episodes. 32 Decreased serum BDNF levels have also been associated with mortality in uninjured populations.33-35 In uninjured humans and rodent models, serum BDNF levels tend to correlate with cerebrospinal fluid (CSF) and brain BDNF.36,37 Studies examining genetic control of BDNF serum levels have been mixed 38 but have not been conducted in TBI populations. Following TBI, Kalish and Phillips 39 reported that serum BDNF levels are acutely decreased, correlating with injury severity.
Given the evidence for genetic, age, and biosusceptibility (biomarker) relevance to mortality post-TBI, we propose a multifaceted role for BDNF involvement in mortality and/or outcome post-TBI, which can be operationalized within a rehabilomics framework.40,41 Rehabilomics integrates biological factors, clinical characteristics, and demographics into a multidimensional approach to evaluate outcome and improve individualized rehabilitation strategies post-TBI. In this study, we measured BDNF levels in serum and CSF across the first week after severe TBI in 203 subjects. We evaluated BDNF levels, interactions with BDNF genetic variation, and mortality associations across the first year post-TBI. While this study focused on mortality, we also examined BDNF levels in global outcome following TBI. Both CSF and serum BDNF demonstrated significant predictive capacity when assessing mortality over a 1-year recovery period, in addition to genetic risk, suggesting BDNF levels as a novel and informative TBI biomarker. The findings support further work understanding BDNF pathophysiology as a contributing factor to mortality following TBI.
Methods
Participants
This prospective cohort study was approved by the University of Pittsburgh Institutional Review Board. Enrollment criteria included age 16 to 74 years old and an admission Glasgow Coma Scale (GCS) score ≤8, indicating severe TBI. Exclusion criteria included documented prolonged hypoxia prior to admission or penetrating head injury. Participants were consecutively recruited while receiving inpatient care within the University of Pittsburgh Medical Center. Consent was obtained from next-of-kin. All subjects sustained a nonpenetrating TBI and had evidence of intracranial injury on computed tomography (CT). Subjects had at least one sample measurement (serum or CSF, at any time point). These subjects are a subset of a larger study investigating possible biomarkers and genetic factors related to individual recovery following TBI.
Injury severity was defined for analysis as the best GCS obtained within the first 24 hours postinjury. Demographics, including age and sex, were collected by chart review and subject or caregiver interviews. Injury severity scores (ISS) were abstracted by trained trauma center registrars. ISS captures the injury severity of the 3 most injured anatomical regions. 42 Table 1 contains a detailed description of the study population.
Participant Description (N = 202).

Abbreviations: SD, standard deviation; GCS, Glasgow Coma Scale; SDH, subdural hematoma; DAI, diffuse axonal injury; EDH, epidural hematoma; IVH, intraventricular hemorrhage; ICH, intracranial hemorrhage; SAH, subarachnoid hemorrhage.
Healthy control participants were Caucasian with an age range of 18 to 60 years, and 40% of the participants were women. All samples collected from controls were obtained at ~7
Sample Collection and Processing
BDNF levels were measured in CSF and serum samples collected for 1 week postinjury. When possible, CSF samples were collected passively up to twice daily via an external ventricular drain placed for clinical care. Serum was collected daily at ~7
CSF and serum samples obtained from the TBI and control cohort were stored at −80°C before batch BDNF analysis using an ELISA kit (RayBiotech, Norcross, GA). To maintain sample integrity for BDNF assessments in TBI and control samples, we used samples not previously thawed for other analyses. Briefly, standards and samples were pipetted onto a 96-well plate precoated with human BDNF antibody. Following shaking for 2.5 hours at room temperature, plates were washed and incubated with biotinylated BDNF antibody for 1 hour. HRP-conjugated streptavidin was then added for an incubation of 45 minutes. The addition of a tetramethylbenzidine substrate allowed for a color reaction. Concentrations were calculated using mean absorbance of each sample at 460 nm to correlate with sample BDNF concentrations present. Samples were diluted within the range of the ELISA kit (no dilution for CSF, 1:250 for serum), with a kit sensitivity of 80 pg/mL, an intra-assay variation of <10%, and an interassay variation of <12%.
Mortality and Outcome
Time until death (TUD) was recorded in days postinjury, up to 1 year postinjury, using the Social Security Death Index. 43 Consistent with previous work, 10 mortality was evaluated over 2 time epochs, 0 to 7 days postinjury (acute) and 8 to 365 days postinjury (postacute), and then across the entire recovery span of 0 to 365 days. For 0 to 7 days, survivorship was right censored at 7 days postinjury. For 8 to 365 days, subjects were included if they survived >7 days, and survivorship was right censored at 365 days. Mortality was also examined as a binary outcome at 365 days postinjury. Acutely, 11.62% of participants died (TUD: median = 3 days, min = 0 days, max = 7 days, Q1 = 2 days, Q3 = 6 days). An additional 14.16% died postacutely (TUD: median = 19 days, min = 8 days, max = 301 days, Q1 = 11.5 days, Q3 = 31 days).
We evaluated whether BDNF levels were indicative of mortality or graded across a range of outcomes using the Glasgow Outcome Scale (GOS; 1 = dead, 2 = vegetative state, 3 = severe disability, 4 = moderate disability, 5 = good recovery) at 6 and 12 months. 44 Research-trained neuropsychometrists, blinded to genetic and biomarker information, obtained GOS scores. GOS was subdivided as favorable (4-5) and unfavorable (2-3) outcome for analysis with BDNF levels. For this study, follow-up rates were >85% for 6 months and >80% at 12 months postinjury.
Genotyping and SNP Selection
DNA was isolated from blood using a salting out procedure 45 or from CSF using the Qiamp protocol from Qiagen. BDNF rs6269 and rs7124442 were genotyped by TaqMan allele discrimination assay using Assay on Demand reagents (Applied Biosystems). This assay utilized fluorescent labeled probes to detect allele(s) present for DNA sample.
Selected SNPs (rs6265, rs7124442) had a minor allele frequency >20%. Each SNP represents a different haplotype block of BDNF covering variation corresponding to “isoform a” of the translated protein. A cumulative BDNF gene risk score (GRS) was used as previously published, 10 where rs6265 Met (Val/Met or Met/Met) status and rs7124442 C (T/C or C/C) carrier status were hypothesized risk alleles due to reportedly reduced BDNF signaling.30,31 Thus, a GRS = 0 was the hypothesized no risk group (Val/Val, T/T); GRS = 1 included carriers for 1 risk allele (Val/Val, C-carriers or Met-carriers, T/T); and GRS = 2 included carriers of both risk alleles (Met-carriers, C-carriers).
Statistical Analysis
Data analysis was conducted using Statistical Analysis Software (SAS), version 9.3. Descriptive analyses included mean and standard deviation and/or median for continuous and ordinal variables and frequencies for categorical variables. Demographic and clinical relationships to BDNF levels were assessed using Mann–Whitney and Kruskal–Wallis tests, due to skewedness of BDNF level data. Genetic analysis utilized categorizations based on allele carrier status using χ2 or Fisher’s exact test where appropriate, and the BDNF GRS was used to examine cumulative genetic risk associations with mortality as previously described. 10 Age was dichotomized at 45 years similar to previous TBI mortality analyses. 10 Due to possible genetic stratification effects, 46 genetic associations are reported in Caucasians only (n = 181). Similarly, due to racial differences in BDNF levels, 47 and analysis with BDNF, analysis was limited to Caucasians only (n = 181). One subject was removed due to sample processing issues.
For consistency with previous work, 10 mortality was examined across 0 to 7 days postinjury (acute), 8 to 365 days postinjury (postacute), and across the entire recovery span of 0 to 365 days. Mortality associations with BDNF levels were examined using TUD in Cox proportional hazards models. 48 The proportionality of hazards assumption was tested and confirmed for all final Cox model variables. Interaction terms were identified by previous studies (in the case of age * GRS) or existing hypotheses (in the case of GRS * serum BDNF) and tested in Cox models. All models were developed using a backwards-stepwise approach, with variables remaining in the final models if P ≤ .2. Final models were then tested to determine the effect of removal of the BDNF levels variable on the remaining associations in the model. A P value <.05 was considered statistically significant.
Results
Description of CSF and Serum BDNF Levels 0 to 6 Days Post-TBI
As depicted in Figure 1A, average CSF BDNF levels tended to be higher for subjects with TBI compared to controls (0.20 ± 0.01 ng/mL vs 0.14 ± 0.02 ng/mL, P = .061), with similar trends noted for days 1 and 5 (P < .10). Average serum BDNF levels in TBI subjects (150.05 ± 4.90 ng/mL) were reduced versus controls (277.86 ± 28.11 ng/mL, P < .0001) beginning day 0 (P = .007) and remained below controls for all time points (P < .0001 for all comparisons, Figure 1B). For subjects with TBI, average CSF and serum BDNF levels tended to be negatively correlated (r = −.209, P = .070, n=76). In controls, serum and CSF BDNF levels were positively correlated but this association was not significant (r = .67, P = .219, n = 5).
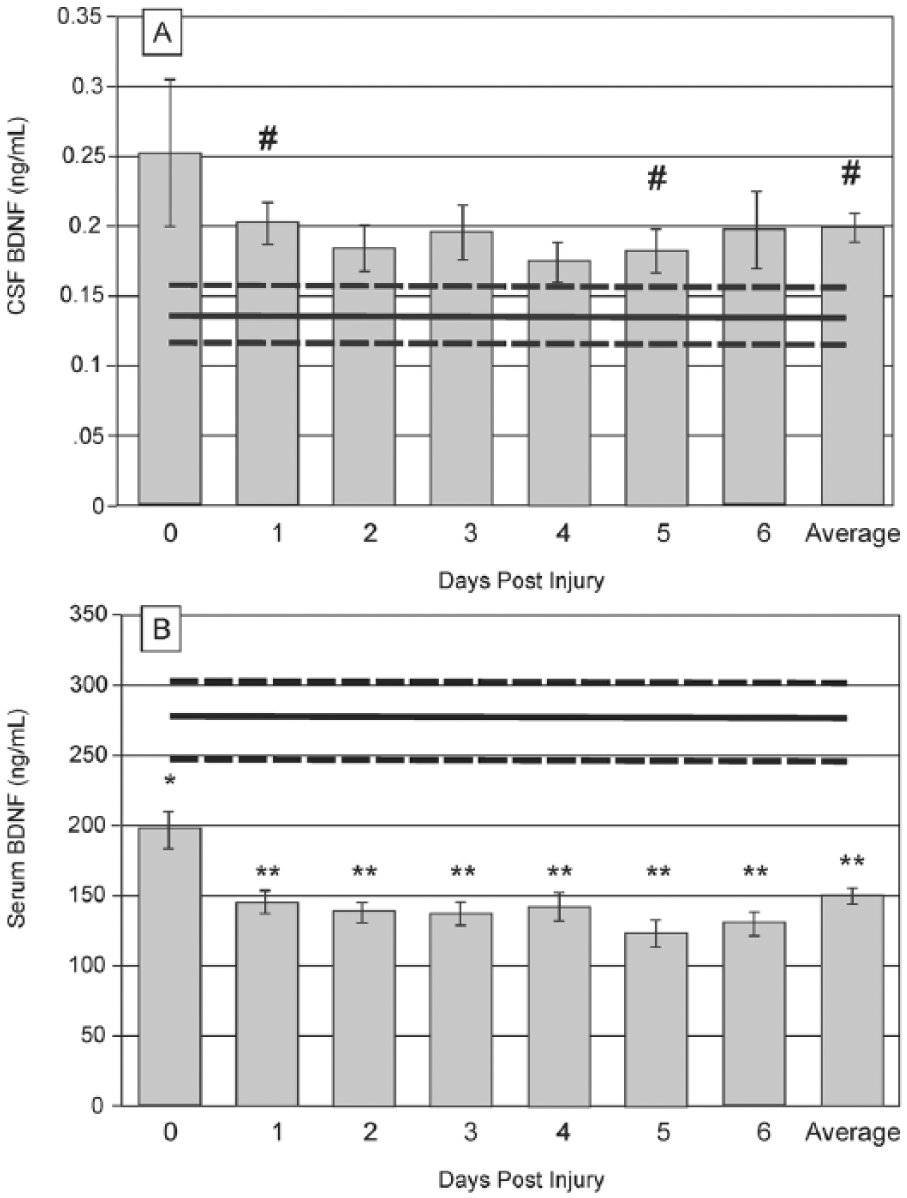
Daily brain-derived neurotrophic factor (BDNF) levels over the first 7 days postinjury, compared to healthy controls (mean represented by gray horizontal line, ± standard error in dashed gray horizontal lines). (A) Daily mean cerebrospinal fluid (CSF) BDNF levels tend to be higher than control levels (trending at days 1 and 5, P < .1 for both). Average CSF levels across the first week postinjury tended to be elevated from control levels (P = .061). (B) Daily mean serum BDNF levels fall below control levels immediately following injury (day 0, P = .007) and remain reduced through day 6 (P < .0001 for all comparisons) (#P < .1, *P < .05, **P < .001).
Associations of CSF and Serum BDNF Levels With Demographic and Clinical Variables
Average (days 0-6) CSF and serum BDNF levels were examined for associations with demographic and clinical variables. Table 2 reports relationships between serum BDNF, CSF BDNF, demographics, clinical variables, and genetic variants (BDNF rs6265 and rs7124442). All genotype frequencies were in Hardy–Weinberg equilibrium. There were no significant associations between genetic variants and BDNF levels. Average CSF levels positively correlated with age (r = .17, P = .045), while serum levels were not correlated with age. Correlations between serum and CSF BDNF levels were also examined by age category. There was a negative correlation among subjects <45 years (r = −.307, P = .029, n = 51), but this was not significant for subjects >45 years (r = −.113, P = .590, n = 25).
BDNF CSF and Serum Level Associations With Demographic and Injury Variables. a
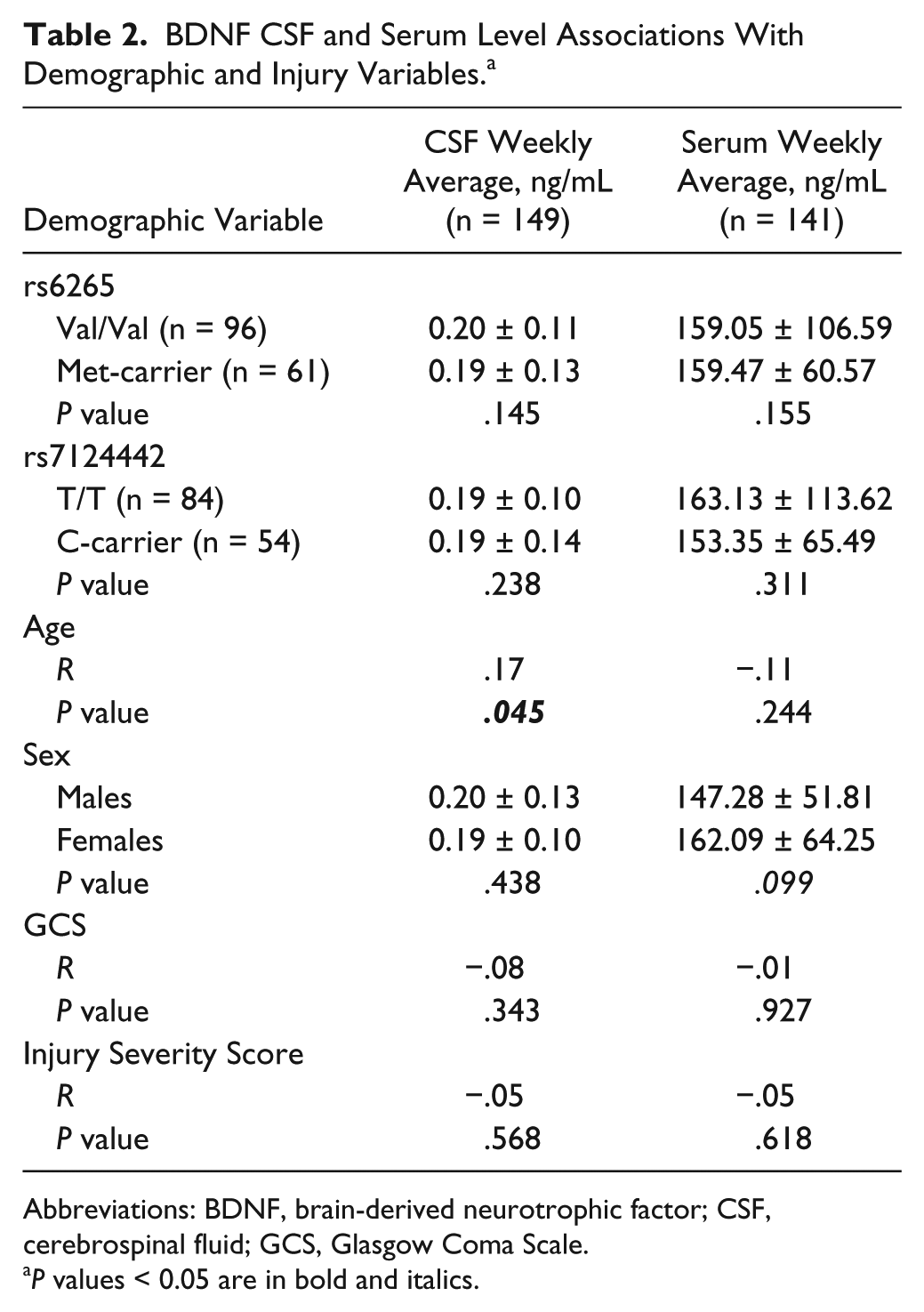
Abbreviations: BDNF, brain-derived neurotrophic factor; CSF, cerebrospinal fluid; GCS, Glasgow Coma Scale.
P values < 0.05 are in bold and italics.
CSF and Serum BDNF Associations With Mortality
Consistent with previous work, 10 CSF and serum BDNF were examined for associations within 2 mortality time epochs and the total survival period. Mortality associations are presented in Table 3. Age and GCS were associated with mortality during both time epochs and across the total survival period. Average CSF BDNF levels were higher in those who died within the postacute time epoch. Serum BDNF levels tended to be associated with acute mortality; participants who died 0 to 7 days postinjury had lower serum levels (130.75 ± 17.26 ng/mL) versus survivors (159.47 ± 60.57 ng/mL).
Demographic Associations With Mortality Time Epochs. a
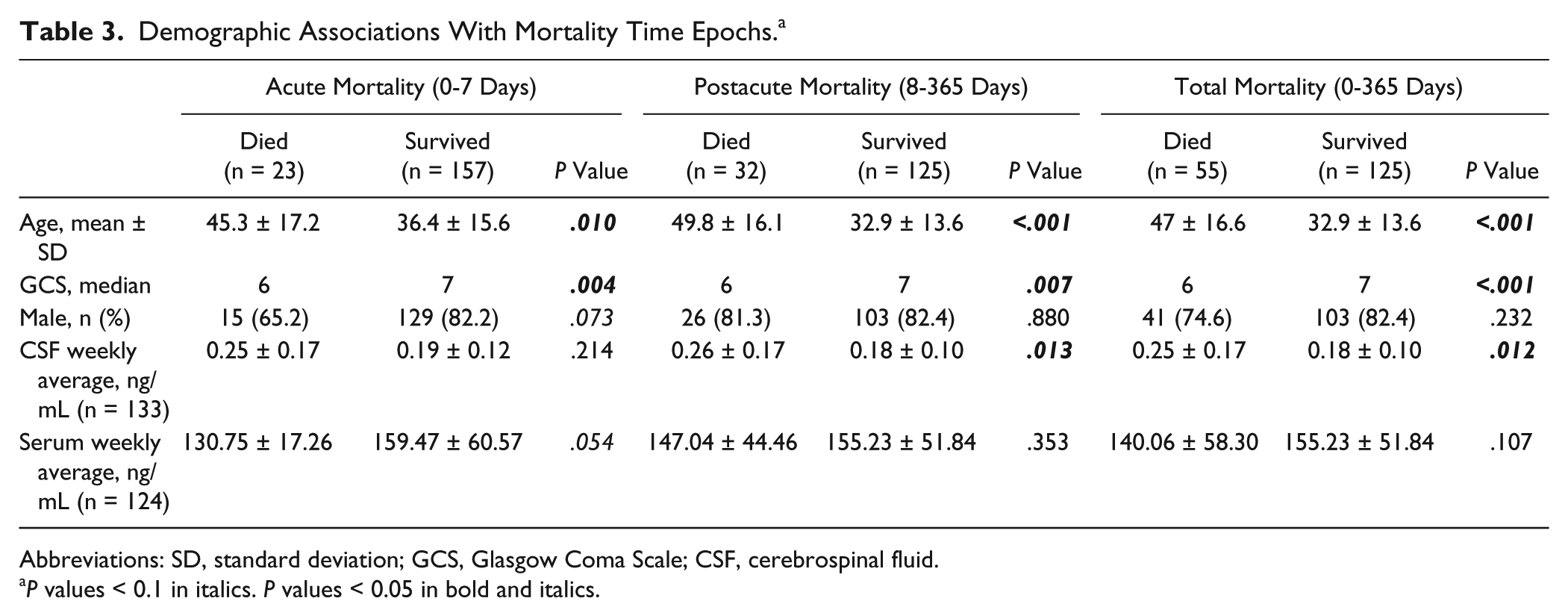
Abbreviations: SD, standard deviation; GCS, Glasgow Coma Scale; CSF, cerebrospinal fluid.
P values < 0.1 in italics. P values < 0.05 in bold and italics.
Associations between CSF BDNF levels and TUD were examined across the first year (0-365; Table 4A). In this model, average CSF BDNF levels predicted TUD (P = .042, hazard ratio [HR] = 10.973) even when including age * GRS interaction (P = .0561, HR = 0.968) and GCS (P = .004, HR = 0.728) as covariates. Importantly, when CSF BDNF was excluded from this model, age * GRS was significant (P = .034, HR = 0.971), where similar to our previous report, 10 older individuals were at higher risk for mortality if they carried fewer of the hypothesized risk (ie, low BDNF secretion) alleles.
Modeling Time Until Death Across Recovery (0-365 Days). a
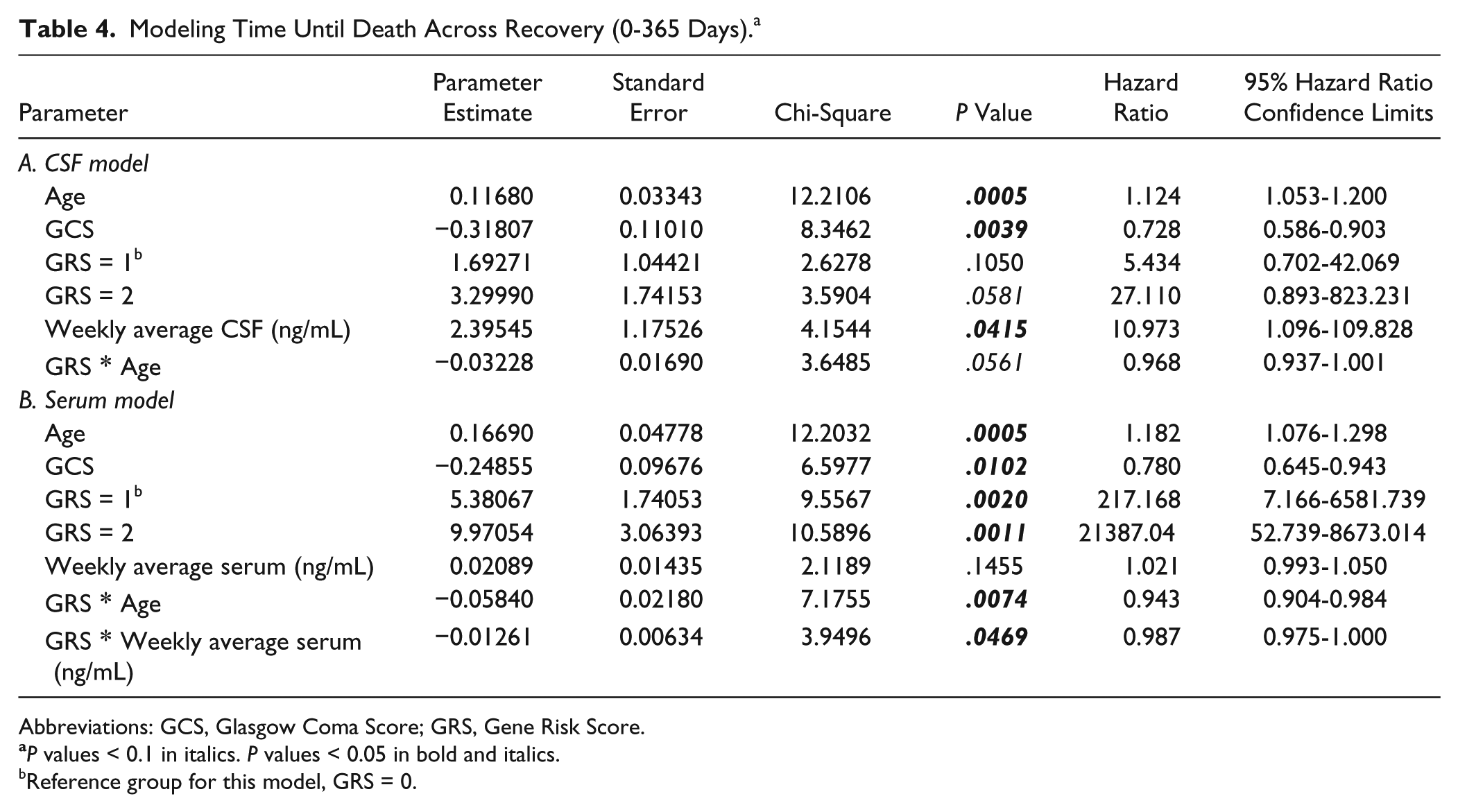
Abbreviations: GCS, Glasgow Coma Score; GRS, Gene Risk Score.
P values < 0.1 in italics. P values < 0.05 in bold and italics.
Reference group for this model, GRS = 0.
Associations between average serum BDNF and TUD were examined across the first year of recovery (days 0-365). There was a significant serum BDNF * GRS interaction (P = .047, HR = 0.987), even when age * GRS (P = .007, HR = 0.943) and GCS (P = .010, HR = 0.780) were included as covariates (Table 4B).
CSF and Serum BDNF Associations With GOS
In a secondary analysis, we evaluated CSF and serum BDNF associations with global outcome (GOS) to determine if BDNF levels were indicative of mortality or graded across a range of outcomes. Average CSF BDNF levels differed at 6 months between subjects who died versus those who survived (with both favorable and unfavorable outcome groups; Figure 2A). At 12 months, those with unfavorable outcome had CSF levels that was compared to those with favorable outcomes and those that died, but were not significantly different from either group. However, CSF BDNF levels were higher in those who died by 12 months versus those with favorable outcome. Serum BDNF was not associated with 6-month GOS scores. Serum BDNF tended to be lower in subjects who died by 12 months (140.06 ± 8.99 ng/mL) versus those with favorable outcomes (167.97 ± 8.23 ng/mL, P = .051; Figure 2B).
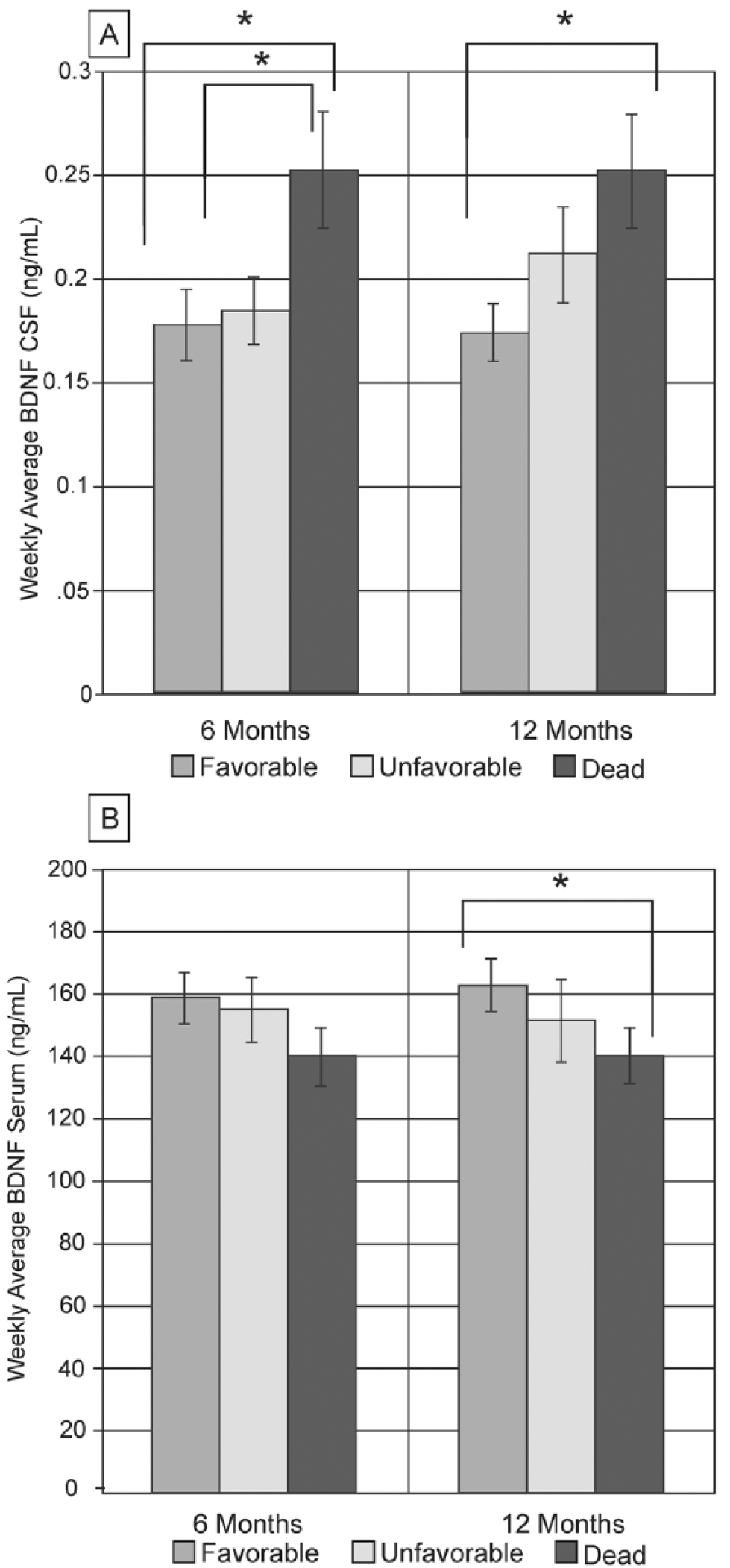
Glasgow Outcome Scale (GOS) was subdivided as favorable (4-5) and unfavorable (2-3) for analyses with brain-derived neurotrophic factor (BDNF) levels. (A) At 6 months, weekly averages of cerebrospinal fluid (CSF) were significantly higher in subjects who died compared to subjects who survived (both favorable and unfavorable outcomes, P < .05). At 12 months, GOS levels were significantly different between subjects who died and subjects with favorable outcomes (P < .05). (B) Weekly serum levels tended to be lower in subjects who died by 12 months compared to subjects who had favorable outcomes (P = .051), but there was no significant association at 6 months.
Discussion
This study investigated BDNF as a dynamic and genetically modifiable biomarker for mortality and global outcome following TBI. Compared to controls, serum BDNF levels were reduced and CSF BDNF levels were modestly increased 1 week post-TBI. Additionally, relationships between age, BDNF genetic variation, and BDNF levels post-TBI showed 2 important interactions in mortality predictions, gene * age and gene * serum. This work suggests BDNF is a biomarker for mortality post-TBI, possibly due to BDNF’s role in neuronal survival/apoptotic signaling as well as autonomic nervous system reactivity.
Clinical TBI biomarker studies evaluating BDNF are limited, and to our knowledge, none have investigated BDNF levels and TBI outcome, especially in the context of genetic variation. One study in pediatric TBI showed increases in CSF BDNF levels immediately following injury (2 hours postinjury) that remained higher than controls throughout the first 24 hours. 49 Kalish and Phillips 39 found serum BDNF was correlated with injury severity post-TBI, such that subjects with mild injuries had the highest levels. Yet BDNF levels have never been examined as a marker for TBI-related mortality.
BDNF is an intriguing biomarker for TBI-related mortality and outcome. There is evidence of BDNF upregulation immediately following experimental TBI,29,50 findings that were thought to aid neuroprotection, given BDNF’s role in neuronal survival.51,52 Yet subsequent studies demonstrated that acute infusions of BDNF following injury do not attenuate tissue loss or improve motor or cognitive recovery. 13 Additionally, BDNF may be relevant in TBI due to its relationship to autonomic nervous system functioning.14,15 HPA axis dysregulation is common following TBI,22,53 and thus variation in plasticity and HPA axis reactivity associated with BDNF signaling 19 could affect mortality and/or outcomes postinjury.
Additional support for BDNF as a biomarker comes from our previous research, which demonstrated important BDNF gene variation interactions with age that influenced mortality predictions post-TBI. 10 We have previously suggested age-specific risk profiles could be due to a known balance shift in BDNF receptor ratios with aging, from pro-survival to pro-apoptotic.26-28 Injury can also shift receptor balance. Experimental TBI induces transient increases in hippocampal TrkB.FL followed by delayed increases in TrkB.T (truncated, inactive form) with regionally specific p75NTR increases up to 8 weeks post-TBI. 29 Relevant to acute TBI mortality, TrkB.FL/Trk.T expression ratios may support cell survival during excitotoxic injury.54,55 With a shift in BDNF receptor ratios occurring with injury and aging, our work indicates that understanding the effects of both is likely necessary to generate sensitive TBI-related mortality prediction models. Importantly, this shifting receptor profiles suggest BDNF could be both beneficial and detrimental following neurological injury depending on receptor balances.
Our current study examined how BDNF levels in CSF and serum predicted mortality and outcome post-TBI, especially in concert with BDNF genetic variation and age. Interestingly, elevated CSF levels were associated with increased mortality risk. We suggest mortality risk associated with higher BDNF signaling could be due to the age or injury-related changes in BDNF target receptor expression that favor apoptotic (p75) pathways, effectively making BDNF exposure detrimental, especially early after injury.
In uninjured populations, serum BDNF levels appear to reflect brain and CSF BDNF levels where BDNF is released into the blood from brain.56,57 However, in our study, there tended to be a negative correlation between CSF and serum that differs from healthy populations 37 and rodent models. 36 There may be multiple reasons for this negative correlation between compartments. In addition to the brain, BDNF is also synthesized and secreted from vascular endothelial cells and may have peripheral actions. 58 BDNF is also stored and released from platelets, 59 especially in response to injured tissue. 60 The negative correlation between CSF and serum following TBI could be due to blood–brain barrier (BBB) disruption, with platelets dumping BDNF and serum BDNF transit into the CNS acutely after TBI. BDNF transit into the CNS could also effectively represent an unsuccessful compensatory mechanism after TBI. Also, serum BDNF reductions may be attributable to increased autonomic function, as HPA axis activation can diminish BDNF levels. 61 Lower serum BDNF associations with mortality may again reflect transit or dumping of BDNF into the CNS and/or reflect autonomic changes in BDNF levels related to HPA axis reactivity. Thus, higher CSF BDNF and lower serum BDNF associations with mortality may represent target receptor expression profiles, BBB disruption, and compensatory BDNF flow regulation.
When examining BDNF levels (either serum or CSF) associations with mortality, the BDNF-GRS * age interaction remained significant or a trend over the 1-year monitoring period. In this current study, BDNF levels predicted mortality in addition to age * GRS interactions, suggesting BDNF levels contribute additional information about mortality beyond age * GRS. However, the magnitude and significance of the age * GRS interaction was reduced when accounting for BDNF levels in this study, findings that indicate that the age * GRS interaction may capture some element of aging effects on BDNF secretion in the context of TBI, in addition to aging influences on target receptor milieu associated with normal aging. Notably, BDNF serum levels are reduced in older individuals in studies without TBI,62,63 although an age * BDNF levels interaction with mortality was not observed in our study.
Our final mortality model includes GRS * age and GRS * serum BDNF interactions. While additional research is needed to understand the mechanisms of these interactions, we suggest BDNF profiles immediately postinjury could reflect both neuronal survival/apoptotic signaling as well as autonomic nervous system effects on outcome. Given findings regarding BDNF variation in autonomic function,19,20 and aging effects on autonomic function/reactivity, 64 GRS * age interactions may also reflect HPA axis reactivity. Yet the GRS * serum interaction likely reflects BDNF gene control over BDNF secretion.30,31 There may be the possibility for a 3-variable interaction of GRS * age * serum, due to age-related increases in BBB permeability,65,66 however this study is underpowered for exploring a 3-variable interaction. CSF BDNF levels did not interact with GRS in mortality. Regardless of GRS, high CSF BDNF levels were associated with mortality, suggesting other factors like BBB transport and neuronal BDNF production affect CSF BDNF contributions to mortality.
We also examined CSF and serum relationships to 6- and 12-month GOS. GOS includes mortality as an outcome, and thus CSF relationships with GOS were driven by mortality status. Serum levels showed no relationship to 6-month GOS, but there was some capacity for mortality prediction with 12-month GOS. Similar to CSF analyses, the primary significant comparison at 12 months was between those who died versus those with favorable outcomes. This finding suggests acute BDNF is more reflective of mortality status, and this biomarker may be lacking when discriminating global outcome among survivors. However, future BDNF evaluation in the context of other survivor-specific outcome measures should be explored.
BDNF biomeasurements have many important caveats to consider, such as processing time, storage temperature, and interassay variation.67-70 To combat these issues, we used freshly frozen samples, reduced processing time, and stored samples at −80°C. Long storage times at −80°C could affect BDNF serum measurements, but importantly, TBI and control samples were subject to similar storage times and conditions. Additionally, the BDNF ELISA used here does not differentiate between pro-BDNF and mature BDNF. Future studies may investigate differences in pro-BDNF versus mature BDNF. Serum BDNF levels can also be affected by circadian rhythms. 71 However, it is not clear if/how BDNF levels vary daily in the context of acute TBI as studies report a loss of normal circadian rhythms acutely after TBI. 6 As BDNF levels are associated with depression, participants depressed at the time of injury may have lower serum BDNF initially. Examining preinjury function and premorbidity may better delineate these issues. One study suggests plasma BDNF levels predict mortality in intensive care unit patients without TBI. While associated mechanisms are unclear, 33 with altered metabolic homeostasis and autonomic dysfunction immediately following TBI, there may be vascular BDNF actions that influence systemic contributors to mortality. Additionally, the cause of death for individuals is not clear as the Social Security Database does not provide cause of death. Larger studies may generate meaningful information and categorization strategies comparing cause of death and BDNF pathology.
While there are important limitations to consider, this study suggests that BDNF pathology is likely an important new target in relationship to individual variation in mortality predictions post-TBI. With the increasingly higher incidence of TBI in elderly populations, the implications of altered BDNF signaling in the context of advanced age could inform geriatric TBI care. Additionally, experimental TBI studies in aging/aged animals could confirm mechanisms that link age and injury alterations on BDNF target receptors to mortality post-TBI. Incorporating this work into a Rehabilomics40,41 framework for further study with neurological outcomes and autonomic function after TBI may allow for more individualized rehabilitation and medical interventions with regard to BDNF pathology, genetics, and age. Still future studies are needed to examine how BDNF pathology evolves over time, especially in regard to outcomes like depression 72 and cognitive functioning 73 where higher BDNF levels may contribute to positive outcomes. Additionally, this work may have applications in other acquired brain injuries like stroke, where BDNF signaling is altered under ischemic conditions.54,55 Understanding BDNF signaling in relation to other secondary injury phenomena like HPA axis dysfunction, age, and/or target receptor regulation in neurological injury may inform new and more tailored TBI treatments.
Footnotes
Acknowledgements
The authors would like to thank the University of Pittsburgh Medical Center Trauma Registry for some elements of data collected.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National Institute on Disability and Rehabilitation Research (Grant Number: NIDRR H133A120087, Wagner); National Institute of Nursing Research (Grant Number: R01NR008424, Conley; R01NR013342, Conley); US Department of Defense (Grant Number: W81XWH-07-1-0701, Wagner); National Institute of Child Health and Human Development (Grant Number: R01HD048162, Wagner).