
Abstract
In epidermal growth factor receptor (EGFR) mutant non-small cell lung cancer (NSCLC), acquired resistance to EGFR tyrosine kinase inhibitors (TKI) leads to disease progression. Strategies to overcome the resistance are required in treatment for advanced lung cancer. In this study, we investigated the therapeutic effect of afatinib and HangAmDan-B1 (HAD-B1) co-administration in gefitinib-resistant NSCLC using HCC827-GR, NSCLC cell line with gefitinib resistance, and the HCC827-GR cell implanted mouse model. HAD-B1 consists of 4 herbs, Panax notoginseng Radix, Cordyceps militaris, Panax ginseng C. A. Mey, and Boswellia carteri Birdwood, and has been reported to be effective in patients with advanced lung cancer in clinical practice. Our findings demonstrated that HAD-B1 combined with afatinib markedly inhibited cell proliferation and induced apoptosis compared to afatinib monotherapy and HAD-B1 monotherapy. Inhibition of HCC827-GR cell proliferation by HAD-B1 occurred through MET amplification and reduced phosphorylation, and the synergistic effect of afatinib and HAD-B1 induced cell cycle arrest and apoptosis in HCC827-GR cells via the downregulation of ERK and mTOR signaling pathways. In hematology and biochemistry tests, HAD-B1 alleviated the toxicity of tumor. In conclusion, HAD-B1 combined with afatinib would be a promising therapeutic strategy for NSCLC with EGFR-TKI resistance.
Introduction
According to a United States cancer observatory study in 2021, lung cancer is still the most common cause of cancer-related death in the world. 1 In Korea, lung cancer has been the leading cause of cancer death in both sexes, demonstrating the highest proportions of patients diagnosed at the distant metastatic stage, approximately 50%. 2 Lung cancer is categorized into non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) by using the size and the morphology of the cancer cells. 3 NSCLC patients account for almost 75% of all lung cancer patients, and most of them have epidermal growth factor receptor (EGFR) mutations. 4 Therefore, the EGFR tyrosine kinase inhibitor (TKI) is the most common clinical therapeutic. 5
Treatment with EGFR-TKIs, such as first-generation EGFR-TKIs (gefitinib and erlotinib) or second-generation EGFR-TKI (afatinib), has become the standard treatment for progressive NSCLC with EGFR mutations demonstrating an improved progression-free survival (PFS) compared to standard chemotherapy.6-9 The first-generation targeted therapy inhibits EGFR by competitive binding with ATP and second-generation TKIs such as afatinib have the characteristic of irreversibly inhibiting all 4 ErbB receptors including EGFR, which could be favorable compared to first-generation TKIs.10-13 However, target drugs induce the T790M mutation on EGFR, and this mutated cancer gradually acquires EGFR-TKI resistance, followed by cancer relapse. 14 The mechanism of resistance to EGFR-TKIs has not been clarified yet; thus, these limitations remain a challenge in the treatment of advanced NSCLC. A third-generation EGFR-TKI (Osimertinib) targeting T790M EGFR-mutated NSCLC was developed, but it does not resolve the problems caused by every resistance mechanism. Therefore, a viable treatment option would be to identify supplementary drugs to overcome the resistance when used in combination with EGFR-TKIs.
HangAmDan-B1 (HAD-B1) is a blended extract of Korean natural products that is composed of Panax notoginseng Radix, Cordyceps militaris, Panax ginseng C. A. Mey, and Boswellia carteri Birdwood. HAD-B1 is a modified form of HangAmDan-B (HAD-B), which was previously developed at the East West Cancer Center (EWCC; Daejeon Korean Medicine Hospital, Daejeon University, Daejeon, Korea) as an anti-cancer herbal medicine.15-17 In our previous clinical study, HAD-B showed improved survival outcomes for lung cancer patients.18-20 Moreover, HAD-B1, which is based on HAD-B, demonstrated an anti-cancer effect on A549 cells with cisplatin resistance. 21 In this study, we investigated the synergetic effect of HAD-B1 and afatinib on an HCC827-GR, NSCLC cell line with gefitinib resistance that might have been caused by drug-resistant mutations in the first and the second generation EGFR-TKIs such as T790M.22,23
Materials and Methods
Generation of Gefitinib-Resistant HCC827 Cells In Vitro
Gefitinib-resistant HCC827 cells were developed by exposing HCC827 cells to increasing concentrations of gefitinib (LC laboratories, MA, USA), starting with 1 µM, and increasing by 100 µM when the cells resumed near normal growth kinetics. Parental cells were maintained concomitantly without the drug. After 4 months, the treated cells, designated HCC827-GR, were able to grow in the presence of 100-nM gefitinib and could be maintained at that concentration. The experiments were performed after the cells had grown in the presence of the drug at that concentration for 6 months.
Preparation of Afatinib and HAD-B1
Afatinib was purchased from LC laboratories (Woburn, MA, USA). HAD-B1 is a blended herbal extract composed of Panax notoginseng Radix, Cordyceps militaris, Panax ginseng C. A. Mey, and Boswellia carteri Birdwood (Table 1). It was manufactured by Daehan Biopharm (Gyeonggi, Republic of Korea) and provided by the EWCC (Daejeon Korean Medicine Hospital, Daejeon University, Korea) in August 2019. In this study, HAD-B1 suspended in dimethyl sulfoxide was centrifuged, and only the supernatant was used.
Ingredients of HangAmDan-B1 (HAD-B1).

A previous study identified 6 critical components of HAD-B1 by using high performance liquid chromatography (HPLC). 24 Each HAD-B1 tablet contained 0.320 mg of cordycepin, 1.110 mg of notoginsenoside R1, 0.610 mg of ginsenoside Rg1, 1.270 mg of ginsenoside Rb1, 0.031 mg of α-boswellic acid, and 0.058 mg of β-boswellic acid.
Cell Culture
HCC827 and HCC827-GR cell lines were cultured in Roswell Park Memorial Institute 1640 (RPMI 1640, Welgene Inc. Daejeon, Republic of Korea) containing 10% fetal bovine serum (FBS, Welgene Inc.). The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Cell Viability Assay
Cell viability was determined using the CCK-8 assay. HCC827-GR cells (5 × 103 cells/well) were seeded on 96-well plates and incubated at 37°C in a CO2 incubator. The cells were treated with afatinib (5 µM–50 µM) or HAD-B1 (5 µg/m-5 mg/mL), respectively, to determine the Combination Index (CI) for 72 hours. Twenty microliters of CCK-8 (Donginbiotech Co., Ltd. Seoul, Republic of Korea) were then added to each well, after which the wells were heated at 37°C in a CO2 incubator for 2 hours. Next, the absorbance of the colored solution was measured at a wavelength of 450 nm by using a microplate reader (Spectramax ID3, Molecular Device, USA).
Apoptosis and Cell Cycle Analysis
HCC827-GR cells were treated with HAD-B1 and afatinib, after which cell viability and apoptosis were determined using a Muse Annexin V and Dead Cell kit (Luminex, Austin, TX, USA) in accordance with the manufacturer’s protocol. Cell cycle analyses were performed using a Muse Cell Cycle kit (Luminex, Austin, TX, USA). Apoptosis and the cell cycle were analyzed using a Muse® Cell Analyzer (EMD Millipore).
Western Blot Analysis
HCC827-GR cells were treated with afatinib and/or HAD-B1. The cells were harvested, and their lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The gel was transferred to a polyvinylidene difluoride membrane for Western blot analyses. Membranes were stained with antibodies against p-MET (Tyr1234/1235), MET, p-EGFR (Tyr1068), EGFR, p-ERK1/2 (Thr202/Tyr204), ERK1/2, p-S6 (Ser235/236), S6, and GAPDH (Cell Signaling Technology, Beverly, MA, USA) and were then treated with secondary anti-mouse or anti-rabbit peroxidase-conjugated antibodies. The membrane imaging was performed using an iBrignt CL1500 system (ThermoFisher Scientific, Waltham, MA, USA) with enhanced chemiluminescence (EZ-Western Kit; DoGen Bio, Seoul, Republic of Korea).
In Vivo Tumor Growth Assay in a Xenograft Animal Model of Human HCC827-GR Lung Cancer Cells
HCC827-GR cells were injected subcutaneously to generate a xenograft model in mice. Then the mice were randomly divided into 4 groups (n = 6): the normal group (non-treatment), the negative control group (10 mL/kg saline), the afatinib group (10 mg/kg), the HAD-B1 group (400 mg/kg), and the combined group (afatinib with 10 mg/kg plus HAD-B1 with 400 mg/kg). Once the tumor size had reached 100 mm 3 , HAD-B1 and afatinib were administered orally once daily for 4 weeks, during which time the tumor size and the body weight were checked daily. Hematology and blood chemistry were performed using a hematology analyzer (Ac. T diff™, BECKMAN COULTER) and a biochemical analyzer (A7020, Hitachi). All care and handling of the animals were performed according to the Guide for the Care and Use of Laboratory Animals (HTRC-16-37(1)).
Statistical Analysis
All data are expressed as the means ± standard deviations, and statistical comparisons were performed using the student’s t-test. Statistical analyses were performed using Microsoft® Excel® Office 365 (Microsoft Corporation, Redmond, WA). P < .05 was considered to indicate a statistically significant difference.
The combination index (CI) is widely used to quantify drug synergism based on the Chou-Talalay multiple drug effect equation. The CI values were determined for each concentration of afatinib, HAD-B1, and their combination in cell proliferation assays using CompuSyn (ComboSyn, Inc. Paramus, NJ; Table 1). CI < 1 indicates synergism, CI = 1.0 indicates an additive effect, and CI > 1.0 indicates antagonism.
Results
Establishment of a Gefitinib-Resistant HCC827 Cell Line
We exposed HCC827 cells to increasing gefitinib concentration for 6 months in order to establish a gefitinib-resistant (GR) HCC827 cell line. That cell line, HCC827-GR, showed a half maximal inhibition concentration (IC50) of 66.12 µM, which was higher than that of the parental cells (0.0074 µM; Figure 1A). Both HCC827 and HCC827-GR cells inhibited phosphorylation of EGFR when treated with gefitinib. However, unlike HCC827 cells, the HCC827-GR cells increased the expression of MET and maintained their phosphorylation regardless of gefitinib treatment (Figure 1B). These results suggested that resistance to gefitinib had been successfully generated in HCC827-GR cells and that the resistance was mediated by MET amplification.
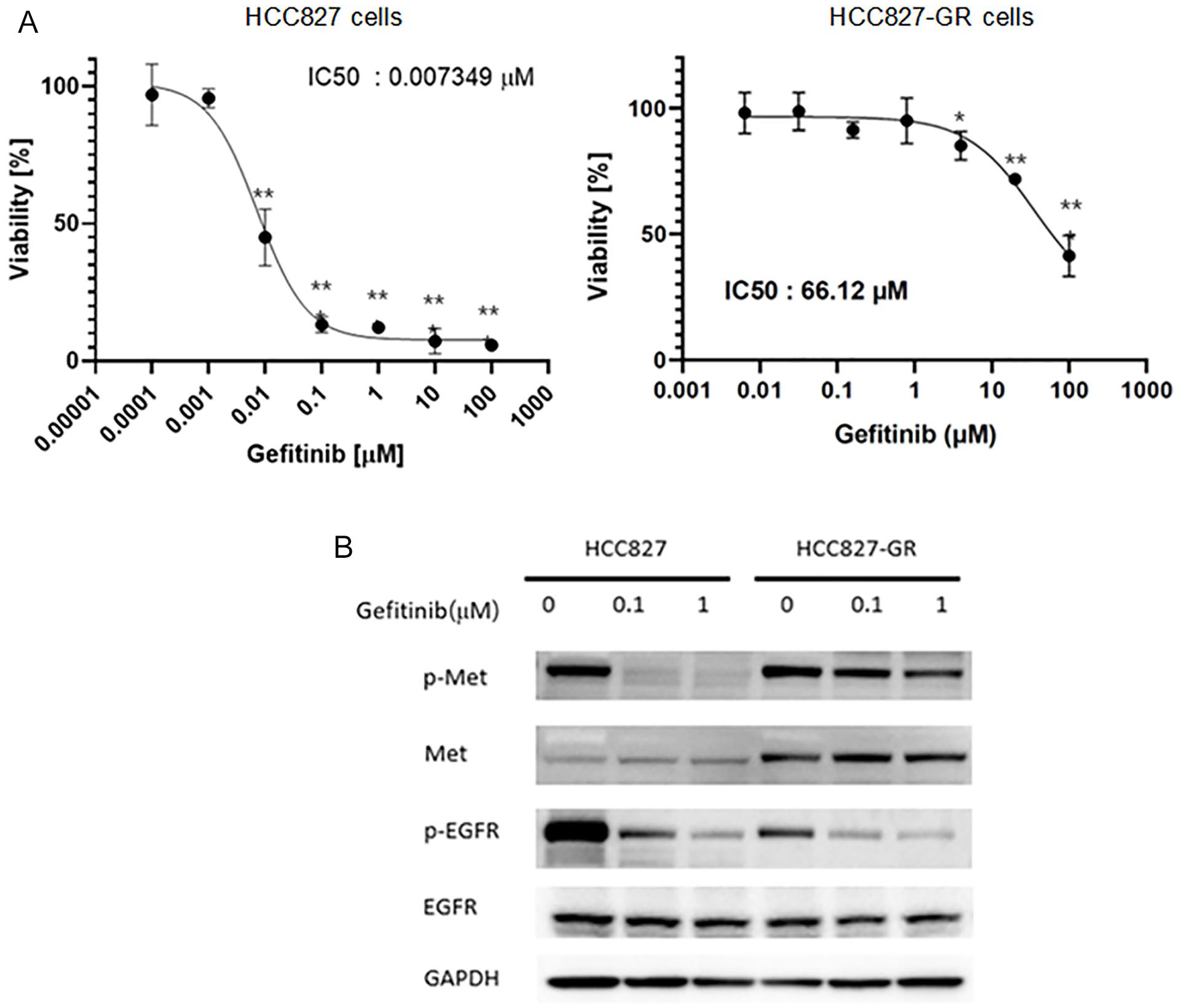
The HCC827-GR cells are resistant to gefitinib in vitro and show MET amplification. (A) Cell viability assay of HCC827 and HCC827-GR cells treated with increasing concentrations of gefitinib for 72 hours. (B) Western blot analysis of cell lysates from HCC827 and HCC827-GR cells treated with gefitinib for 24 hours. Data shown are the means ± standard deviations (SDs), and statistical analyses were performed using the student’s t-test (n = 3; *P < .05, **P < .01, and ***P < .001, vs control).
A Combination of Afatinib and HAD-B1 Decreases Cell Viability of HCC827-GR
On the basis of previous studies that had shown HAD-B1 to have an anti-cancer effect, we assessed the effects of a combination of afatinib and HAD-B1 on growth inhibition in HCC827-GR cells. 21 We performed a CCK-8 cell viability assay and used the method of non-constant ratio drug combination proposed by Chou and Talalay to determine the synergistic, additive, or antagonistic effects of afatinib and HAD-B1. The half maximal inhibition (IC50) of HCC827-GR cell viability due to the combined treatment was observed at an afatinib concentration of 0.1 µM to be 0.135 mg/mL (Figure 2B) whereas the monotherapy groups were higher (afatinib 12.87 µM and HAD-B1 0.190 mg/mL, respectively; Figure 2A). The CI values based on the data from the cell viability assay demonstrated that the combined treatment of HAD-B1 with afatinib had a synergistic effect (Table 2). These results indicate that HAD-B1 has a synergistic effect, even in NSCLC cells with resistance to EGFR-TKI, in suppressing cell viability when used in combination with afatinib.

Growth inhibitory effect of afatinib and HAD-B1 on HCC827-GR cell lines. The HCC827-GR cells were treated with increasing concentrations of (A) afatinib alone and HAD-B1 alone or (B) a combination of the 2 drugs (afatinib 0.1 μM) (B) for 72 hours. (C) The synergy between the drugs was determined using the method of Chou and Talalay. The dashed horizontal line at 1 indicates the line of the additive effect. Data shown are the means ± SDs, and statistical analyses were performed using the student’s t-test (n = 3; *P < .05, **P < .01, and ***P < .001, vs control).
Combination Index (CI) Analysis of HangAmDan-B1 (HAD-B1) Combined with Afatinib at a Non-Constant Ratio in HCC827-GR Cells a .

Data are values from Figure 1.
CI = 1.00, additive; CI < 1.00, synergistic; CI > 1.00, antagonistic.
Induction of Apoptosis and Cell Cycle Change in HCC827-GR Cells by Afatinib and/or HAD-B1
In previous our study, we confirmed that HAD-B1 regulated the cell cycle and induced apoptosis in H1975, an EGFR-L858R/T790M double mutation NSCLC cell line. 25 Therefore, we confirmed that apoptosis induction and cell cycle changes had taken place in HCC827-GR cells due to HAD-B1 treatment. In comparison to a treatment with HAD-B1 or with afatinib, the combined treatment showed a further induction of apoptosis in the HCC827-GR cells: control: early Apop. 1.66%, late Apop. 10.20%: HAD-B1: 27.56%, 7.00%; afatinib: 19.34%, 13.56%; HAD-B1 and afatinib: 30.66%, 50.10%; Figure 3). HCC827-GR cells were found to be arrested in the G2/M phase when treated with afatinib, the S phase when treated with HAD-B1, and the G2/M phase when treated with both (Figure 4).

Effect of afatinib and HangAmDan-B1 (HAD-B1) on apoptosis. The HCC827-GR cells were treated with afatinib and/or HAD-B1 for 48 hours. (A) Apoptosis was assessed by using annexin staining and a Mini Flow Cytometry Muse Cell Analyzer. (B) Apoptosis distributions obtained from the histogram analysis shown in (A).
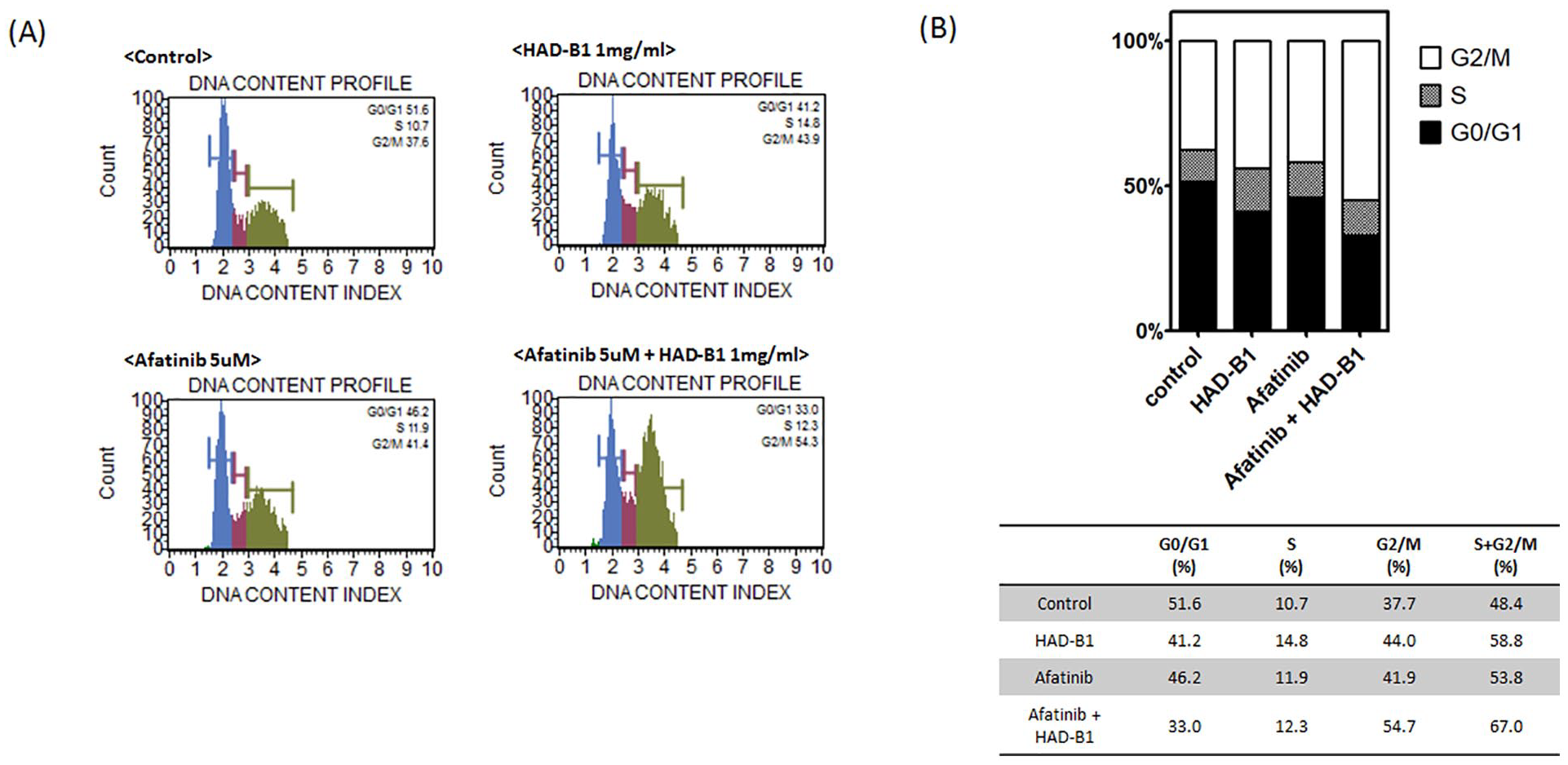
Effect of afatinib and HangAmDan-B1 (HAD-B1) on the cell cycle. The HCC827-GR cells were treated with afatinib and/or HAD-B1 for 48 hours. (A) Cell cycle distribution was analyzed by using a Muse cell cycle kit (Merck Millipore, Billerica, MA). (B) Cell cycle distributions obtained from the histogram analysis shown in (A).
Inhibition of EGFR Phosphorylation and MET Amplification Leads to Down-Regulation of ERK Phosphorylation and S6 Phosphorylation in HCC827-GR Cells
To investigate the mechanism of the anti-cancer effect of HAD-B1 on HCC827-GR cells, we performed western blot analyses of endogenous cell signaling proteins in HCC827-GR cells treated with afatinib or with HAD-B1. Although HAD-B1 did not affect EGFR phosphorylation, which was significantly reduced by afatinib, it did reduce the expression of MET in a dose-dependent manner (Figure 5A). Furthermore, when afatinib and HAD-B1 were used together, phosphorylation of ERK1/2 and phosphorylation of S6 in the mTOR pathways were synergistically reduced (Figure 5B). These results demonstrate that the inhibitory effect of HAD-B1 on HCC827-GR cell proliferation results from inhibitions of the ERK and the mTOR signaling pathways due to reductions in both MET amplification and S6 phosphorylation.

Inhibitory effect of afatinib and HangAmDan-B1 (HAD-B1) on MET and ERK signaling. Western blot analyses of cell lysates from HCC827-GR cells treated with afatinib and HAD-B1. (A) The HCC827-GR cells were treated with afatinib (0.1 μM) or HAD-B1 with the indicated doses for 24 hours. (B) The HCC827-GR cells were treated with afatinib (0.1 μM) and/or HAD-B1 (1 mg/mL) for 12 hours.
Inhibitory Effect of the Combination Therapy on Gefitinib-Resistant Tumor-Bearing Xenograft Mice
For evaluating the synergistic anti-cancer effect of HAD-B1 with afatinib in vivo, we injected HCC827-GR cells into immunodeficient nude mice. After the tumor volume had grown to approximately 100 mm3, HAD-B1, afatinib, and a combination of the 2 were administered orally once daily for 4 weeks. After the treatments, the mean tumor growth was noticeably reduced in the combined treatment group compared to the tumor growths in the other groups (negative control: 927.32 mm 3 , afatinib only: 525.29 mm 3 , HAD-B1 only: 737.31 mm 3 , combination: 277.49 mm 3 ; Figure 6A and B). Moreover, the combination treatment significantly reduced tumor weight (Figure 6C). Toxicity was assessed by measuring body weight, food intake, complete blood cell count, and biochemical blood chemistry. There was no significant difference between each group in body weight and food intake (Figure 7). The hematology and the biochemistry results revealed that HAD-B1 did not cause any toxicity effect but also showed significant improvement in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels compared to the negative control group (Table 3). Taken together, these data suggest that HAD-B1 can improve, without any side effects, the anti-cancer effect of afatinib in the treatment of solid tumors derived from HCC827-GR cells.
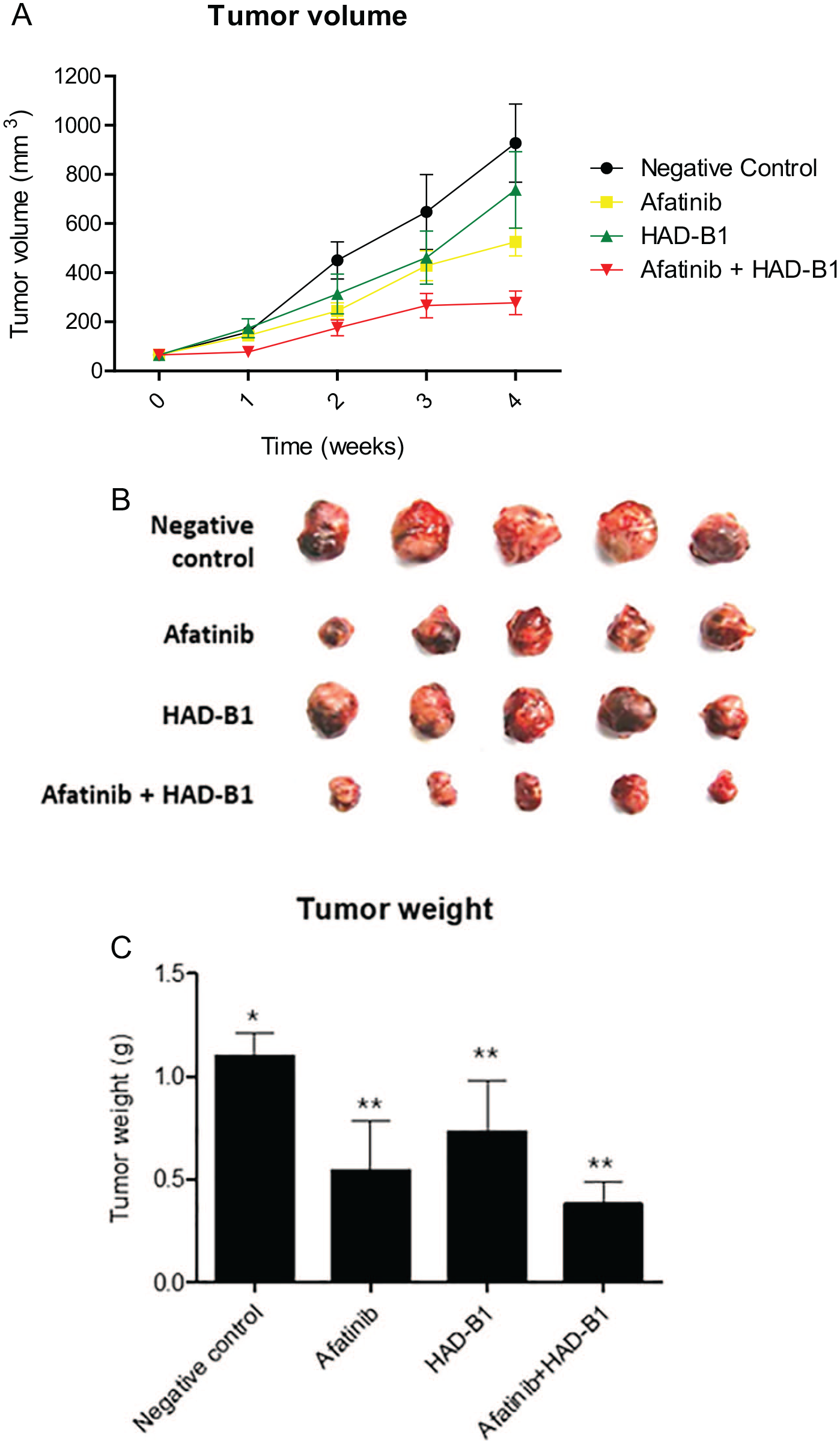
Inhibitory effect of HangAmDan-B1 (HAD-B1) on solid tumor growth in the HCC827-GR lung tumor-cell xenograft model. (A) The growth curves represent the average values for 5 mice in each group. Error bars indicate standard deviations. (B) Pictures from the different tumor groups. (C) Tumor weights of the different groups. (*P < .05, **P < .01, and ***P < .001, vs control).

Toxicity analysis for the HCC827-GR lung tumor-cell xenograft animals treated with afatinib and HangAmDan-B1 (HAD-B1): (A) body weights and (B) and food intakes.
Four Weeks After Inoculation of the Tumor Cells, Blood Was Collected and Examined Using Complete Blood Count and Blood Biochemistry.
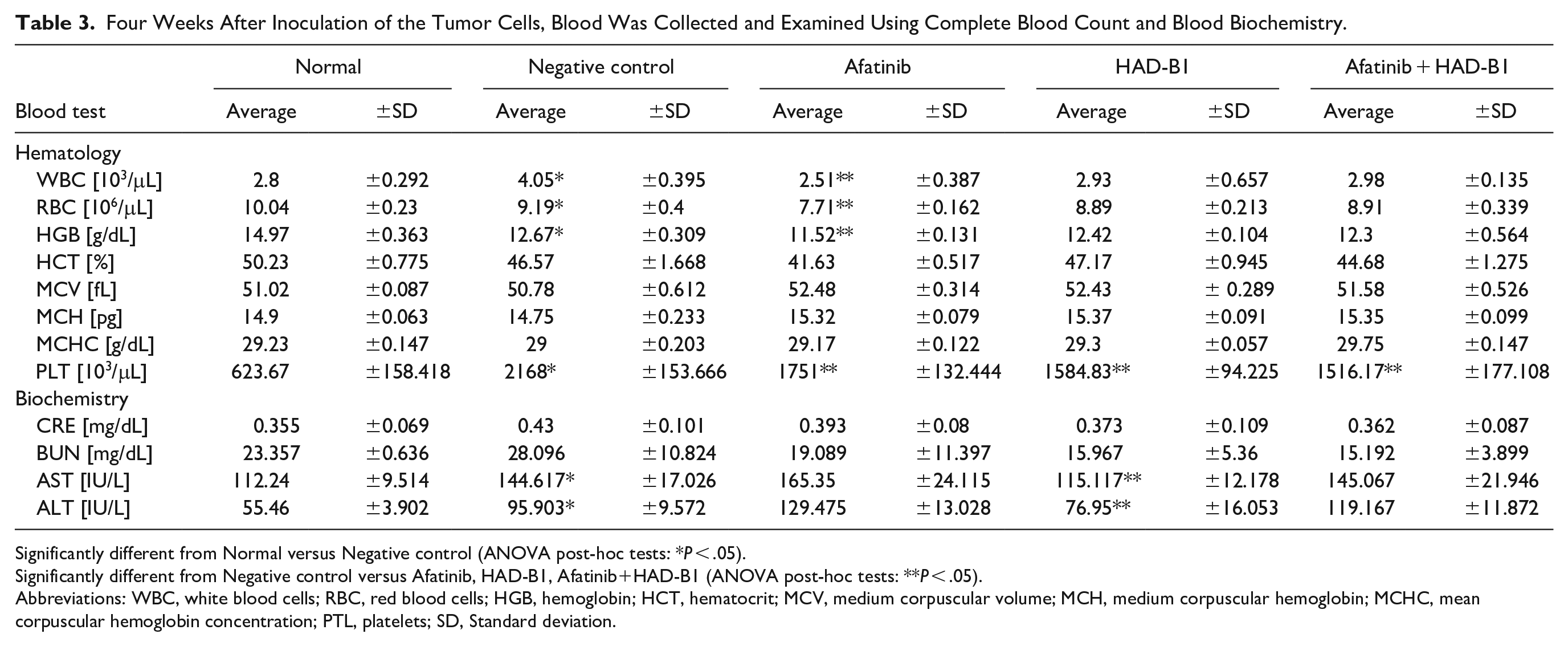
Significantly different from Normal versus Negative control (ANOVA post-hoc tests: *P < .05).
Significantly different from Negative control versus Afatinib, HAD-B1, Afatinib+HAD-B1 (ANOVA post-hoc tests: **P < .05).
Abbreviations: WBC, white blood cells; RBC, red blood cells; HGB, hemoglobin; HCT, hematocrit; MCV, medium corpuscular volume; MCH, medium corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; PTL, platelets; SD, Standard deviation.
Discussion
Afatinib is a second-generation EGFR-TKI, which is active in first-generation resistant tumors, including L858R/T790M double mutations of EGFR, and shows higher response rates and progression-free survival compared to first-generation EGFR-TKI. However, because afatinib causes dose-dependent side effects, drugs that can reduce those side effects and overcome the resistance caused by EGFR-TKI must be developed. HAD-B1 is an anti-cancer drug developed to focus on the treatment of lung cancer based on existing cases and consists of 4 herbs: Panax notoginseng Radix, Cordyceps militaris, Panax ginseng C. A. Mey, and Boswellia carteri Birdwood. The 3-dimensional HPLC analysis of HAD-B1 showed that it contains 6 critical compounds: cordycepin, R1, Rg1, Rb1, α-boswellic acid, and β-boswellic acid. 24 In NSCLC, EGFR-TKI resistance occurs through various mechanisms; thus, HAD-B1, which is composed of complex components, should be able to inhibit that resistance more effectively than a single-component drug. In fact, in HCC827-GR, a NSCLC cell line that is resistant to EGFR-TKI, the HAD-B1 combined treatment group showed synergistically inhibited cell proliferation as compared to the group treated with afatinib alone (Figure 2). In addition, the combination treatment of afatinib and HAD-B1 induced cell cycle arrest and apoptosis more effectively (Figures 3 and 4).
ERK1/2 is a member of the mitogen-activated protein kinase (MAPK) family and functions as a serine/threonine protein kinase. Phosphorylated ERK1/2 can regulate gene expression, cell proliferation, cell differentiation, and cell migration and is known to inhibit apoptosis. In contrast, inhibition of ERK1/2 promotes apoptosis. In the mTOR pathway, the mTOR phosphorylates S6K and the S6K phosphorylates S6 to promote increased gene transcription, cell growth, and cell proliferation. Because the HCC827-GR cells amplify MET compared to the parent HCC827 cells, even if EGFR activation is suppressed by afatinib treatment, the phosphorylation of ERK and S6 still remain through the MET signaling pathway. However, the phosphorylation of ERK and S6 has been confirmed to be reduced when MET is suppressed simultaneously due to HAD-B1 treatment (Figure 5). Afatinib inhibits activation of EGFR, and HAD-B1 reduces amplification of MET; thus, the 2 drugs when used together, should have a synergistic anticancer effect (Figure 8). In the tumor growth experiment using the HCC827-GR tumor xenograft model, we confirmed the synergistic effect arising from treatment with HAD-B1 and afatinib together (Figure 6). No significant side effects occurred when HAD-B1 was administered; rather, the administration of HAD-B1 relieved the side effects caused by treatment with afatinib (Figure 7).

Molecular mechanism underlying the action of HangAmDan-B1 (HAD-B1) and afatinib. Afatinib is an inhibitor of receptor tyrosine kinases, including EGFR and HER2. The inhibitory effect of HAD-B1 on HCC827-GR cell proliferation occurs through MET amplification and phosphorylation reduction. The synergistic effect of afatinib and HAD-B1 inhibits the ERK and mTOR signaling pathways, resulting in cell cycle arrest and apoptosis of cancer cells.
HAD-B1 has been demonstrated to have a potential anti-proliferative effect in cancer cells, regardless of EGFR-TKI resistance. Furthermore, HAD-B1 combined with afatinib has been shown to suppress cancer cell viability efficiently. In cancer cells, as compared to normal cells, cell division and proliferation are upregulated because of multiple oncogenes. Our previous study discovered that HAD-B1 induces apoptosis in cancer cells by targeting their cell cycle. 25 Thus, HAD-B1 not only has an anticancer effect on its own, but also has a synergistic effect when combined with afatinib.
The types of targeted therapies used in the treatment of NSCLC and the range of therapeutic applications are continuously increasing. At the same time, research on the mechanisms of molecular genetics related to carcinogenesis and the development of therapeutic agents targeting genetic mutations is continuing. Currently, a third-generation EGFR-TKI is being developed and used as first-line therapy for patients with NSCLC; nevertheless, cancer cells have been reported to acquire resistance to that therapy, resulting in severe symptoms and even recurrence. Therefore, HAD-B1, with its distinctive anti-tumor effect, should be very valuable for overcoming EGFR-TKI resistance. Our findings suggest that HAD-B1 may be an excellent therapeutic option for patients with NSCLC who have acquired resistance to TKIs. We expect combination therapy with HAD-B1, a natural product, and afatinib to have a synergistic therapeutic effect in the treatment of patients with EGFR-TKI-resistant NSCLC.
In this study, the roles of important pathways other than the MET and the ERK pathways in the anticancer effect of HAD-B1 on HCC827-GR are unclear, which is a limitation of this study. Thus, the mechanisms underlying the effect of the main active compounds in HAD-B1 need to be explored further and identified.
Conclusion
In conclusion, the combination treatment with HAD-B1 and afatinib demonstrated a synergistic effect against gefitinib-resistant NSCLC by inhibiting the ERK and mTOR signaling pathways via MET amplification reduction, resulting in cell cycle arrest and apoptosis of cancer cells. Therefore, HAD-B1 should be an effective and safe alternative therapeutic strategy for treating NSCLC patients with acquired resistance to EGFR-TKIs.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by a grant from the Ministry of Health & Welfare, Republic of Korea (HI19C1046).