
Abstract
Breast cancer is the most common malignancy in women worldwide, and major challenges in its treatment include drug resistance and metastasis. Three-dimensional cell culture systems have received widespread attention in drug discovery studies but existing models have limitations, that warrant the development of a simple and repeatable three-dimensional culture model for high-throughput screening. In this study, we designed a simple, reproducible, and highly efficient microencapsulated device to co-culture MCF-7 cells and HUVECs in microcapsules to establish an in vitro vascularized micro-tumor model for chemotherapeutic drug screening. First, to construct a three-dimensional micro-tumor model, cell encapsulation devices were created using three different sizes of flat-mouthed needles. Immunohistochemistry and immunofluorescence assays were conducted to determine vascular lumen formation. Cell proliferation was detected using the Cell Counting Kit-8 assay. Finally, to observe the drug reactions between the models, anticancer drugs (doxorubicin or paclitaxel) were added 12 h after the two-dimensional cultured cells were plated or 7 days after cell growth in the core-shell microcapsules. Vascularized micro-tumors were obtained after 14 days of three-dimensional culture. The proliferation rate in the three-dimensional cultured cells was slower than that of two-dimensional cultured cells. Three-dimensional cultured cells were more resistant to anticancer drugs than two-dimensional cultured cells. This novel sample encapsulation device formed core-shell microcapsules and can be used to successfully construct 3D vascularized micro-tumors in vitro. The three-dimensional culture model may provide a platform for drug screening and is valuable for studying tumor development and angiogenesis.
Introduction
Breast cancer (BC) is the most common type of cancer in women, accounting for approximately 31% of all new diagnoses. 1 In drug discovery, the lack of efficacy and safety are the main reasons for the failure of the clinical development phase. 2 Owing to the high investment and low success rate in drug discovery, finding new methods to improve the success rate of drug discovery is necessary. Two-dimensional cell cultures are the most commonly used in vitro methods for pre-clinical drug discovery but they fail to mimic the natural tumor microenvironment (TME) in vivo.3,4 Three-dimensional (3D) cell cultures are superior to two-dimensional (2D) cell cultures in studying cell-cell and cell-matrix interactions. 5 Moreover, 3D tumor models can more accurately simulate the biological characteristics of solid tumors, such as cellular heterogeneity, gene expression, and drug resistance. 4
The TME is a complex ecosystem full of heterogeneity, including cancer cells, stromal cells, blood vessels, nerve fibers, extracellular matrix (ECM), and related acellular components. 6 Stromal cells (including endothelial cells) may affect the sensitivity of cancer cells to anticancer drugs by modifying ECM and paracrine factors. 7 However, most 3D tumor models currently lack a vascular system. Owing to limitations with diffusion, living cells only exist within 200 μm on the surface of 3D tumor models, including spheroids. 8
Cell microencapsulation is a relatively new 3D culture technique that immobilizes well-functioning and living cells in a suitable matrix. 9 The appropriate matrix should mimic the ECM, be biocompatible, and support cell survival. Alginate, collagen, fibrin, gelatin, and hyaluronic acid are widely used natural polymer biomaterials because of their similarity to ECM components and can be used for cytotoxicity testing of various anticancer drugs in 3D cell models. 10 Sodium alginate is the most commonly used culture material, including (1,4) -linked β-d-mannose (M block) and α-l-glucose acid blocks (G block), which can be crosslinked with divalent cations (such as Ca2+) to form hydrogels.11,12 Collagen, the main component of ECM protein, has low antigenicity, low inflammatory reactivity, and biocompatibility, providing mechanical support for antagonistic forces.13,14 Several methods are available for encapsulating cells into microcapsules, including electrostatic spraying, photolithography, emulsification, and droplet microfluidics. However, traditional cell encapsulation technology requires professional equipment and special processes, and its manufacturing complexity and high cost hinder the wide application of drug testing.15,16 In this study, we designed a simple, reproducible, and highly efficient microencapsulated device to co-culture MCF-7 cells and HUVECs in microcapsules to establish an in vitro vascularized micro-tumor model for chemotherapeutic drug screening.
Materials and Methods
Cell Source and Culture
MCF-7 cells (breast cancer cell line) and HUVEC were donated by Professor Wu's and Professor Zhao's laboratories, respectively. The cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco, NY, USA) containing 10% fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin solution (Beyotime Biotechnology, Shanghai, China) and placed in a 5% CO2 humidified incubator at 37 °C. The cells were digested in a culture flask using trypsin and centrifuged at 1200 rpm for 5 min. The supernatant was discarded, and the cells were deposited on ice for later use.
Preparation of Cell Encapsulation Device
The cell encapsulation device comprised three different sizes of flat needles, including purple, pink, and black needles with inner diameters of 510 μm, 600 μm, and 740 μm, respectively, as shown in Figure S1. First, the purple needle was placed inside the pink needle so that the two needles formed concentric circles in cross-sections. Subsequently, the black needle was vertically inserted into the pink needle, and the three needles were bonded using a hot melt adhesive, as shown in Figure 1. Next, the simple device was connected through the ‘cross’ scratch on the 1.5 mL EP tube cover. Finally, the whole device was fixed in a 50 mL centrifugation tube with a hollow foam.

Schematic diagram of the encapsulation device used for forming core-shell microcapsules.
Detection of Viscosity of Sodium Alginate
We measured the viscosity of 1%, 2%, and 3% sodium alginate at 25 °C room temperature with a viscometer (NDJ-5S, Shenzhen Qun Long Instrument Equipment Co., Ltd, Shenzhen, China) to select the appropriate concentration of sodium alginate encapsulated cells, and each group was assessed at least thrice.
Cell Encapsulation and Micro-Tumor Culture
Before the experiment, the cell encapsulation device was soaked in anhydrous ethanol and irradiated with UV light for 30 min, and sterile saline was used to flush the device channels. The device has two microchannels: inlet 1 as the core phase and inlet 2 as the shell phase. We mixed the neutralized rat tail tendon collagen type I (#C8062, Solarbio, Beijing, China) at an initial concentration of 5 mg/mL with MCF-7 cells/HUVECs (total density 2 × 106 cells/mL), added 2% sodium carboxymethyl cellulose to increase the hardness of the ECM, maintained the collagen concentration at 1.5 mg/mL, and injected it into inlet 1. Next, 2% sodium alginate or a mixture containing HUVECs was injected from inlet 2. The device's outlet was connected to a 1.5 mL EP tube containing 0.1 M CaCl2 solution. All solutions were filtered using a 0.22 μm needle filter.
After starting the centrifugation and adjusting the speed, sodium alginate was gelatinized in the CaCl2 solution to form microcapsules. The collected microcapsules were transferred to a complete medium containing EGM-2 cell culture factor (#CC-4176; Lonza, Basel, Switzerland) and cultured in a 5% CO2 incubator at 37 °C for 14 days.
Cell Viability Assay
The cell-containing microcapsules were taken out after 1, 3, 5, and 7 days of culture, and a Calcein AM/PI staining kit (Beyotime Biotechnology) was used to detect cell activity. First, the Calcein AM/PI detection working fluid was prepared following the manufacturer's instructions. The microcapsules were washed with PBS, added to an appropriate amount of the Calcein AM/PI detection working solution, and incubated at 37 °C in the dark for 30 min. Finally, the staining was observed using a fluorescence microscope.
Detection of the Vascular Lumen in Micro-Tumors by Immunofluorescence and Immunohistochemistry (IHC)
The cell-containing microcapsules cells were taken after 14 days of culture, and the shell (sodium alginate) was dissolved with 75 mM sodium citrate to release micro-tumors. After washing thrice with PBS, the collected cell aggregates were transferred to a slide and fixed with 4% paraformaldehyde. After washing thrice with PBS, 0.5% Triton X-100 (Biosharp Life Sciences, Guangzhou, China) was added, followed by incubation at 37 °C for 15 min to increase the permeability of the cell membrane to the antibody. After washing with PBS, 2% BSA prepared with PBS was added and incubated at 37 °C for 1 h. Next, CD31 antibody with a dilution of 1:100 (#11265-1-AP; Proteintech, Wuhan, China) and actin antibody at a dilution of 1:200 (#T0021; Affinity Biosciences, OH, USA) were added and incubated overnight at 4 °C in a wet box. The next day, after washing thrice with PBS, fluorescent secondary antibodies with a dilution of 1:500 were added in the dark environment, including goat anti-mouse antibody coupled to DyLight 488 (#A23210; Abbkine Scientific Co., CA, USA) and goat anti-rabbit antibody conjugated to DyLight 594 (#A23420; Abbkine), and incubated at 37 °C in the dark for 1 h. Next, DAPI (Biosharp Life Sciences) was added and incubated at 37 °C for 10 min. After washing thrice with PBS, an anti-fluorescence quenching agent was added dropwise and sealed. Finally, the staining was observed using a confocal microscope.
The cell-containing microcapsules were taken out after two weeks of culture, and the shell (sodium alginate) was dissolved with 75 mM sodium citrate to release micro-tumors. After washing thrice with PBS, the collected cell aggregates were fixed with 4% paraformaldehyde for 24 h. The collected cell aggregates were dehydrated, embedded, and prepared into 4 μm paraffin sections. The paraffin sections were baked in a 65 °C incubator for 2 h, and dewaxed with xylene to water. The 0.01 mol/L sodium citrate buffer was prepared, and the antigenic sites were exposed using high-pressure thermal repair. CD31 antibody (1:800; #11265-1-AP; Proteintech) was dripped and incubated overnight at 4 °C. After washing thrice with PBS the next day, an appropriate amount of MaxVisionTM HRP polymer anti-rabbit IHC reagent (#Kit-5006; MXB Biotechnology, Fuzhou, China) was added, followed by incubation at 37 °C for 15 min. After washing thrice with PBS, a 3,3′-diaminobenzidine staining solution was added. Then, the slices were immersed in hematoxylin for staining, dehydrated, and sealed. Finally, the imaging was observed using an optical microscope.
Cell Proliferation in 2D and 3D Culture
As 3D culture groups, microcapsules containing MCF-7 cells, HUVECs, and a mixture of both (1:1) were inoculated on a 96-well plate (100 /well) and cultured in a complete medium containing the EGM-2 cell culture factor. As 2D culture groups, MCF-7 cells, HUVECs, and a mixture of both (1:1) were inoculated in a 96-well plate (500 cells/well) and cultured in a complete medium containing the EGM-2 cell culture factor. On days 1, 3, 5, and 7 after culture, the CCK-8 reagent (Beyotime Biotechnology) was used to measure the cells’ absorbance at 450 nm.
Sensitivity of 2D- and 3D-Cultured Cells to Chemotherapeutic Drugs
Doxorubicin (DOX; #KGA8184) was purchased from Keygen Biotech, Jiangsu, China. Paclitaxel (PTX; #IP0020) was purchased from Solarbio Life Sciences, Beijing, China. DMSO was added to fully dissolve the drug to prepare a storage solution and stored at −80 °C. Before the experiments, the drug was diluted with fresh medium at the appropriate concentration, with the final DMSO concentration <0.1%.
As 3D culture groups, microcapsules containing cells cultured for one week were inoculated in a 96-well plate (100 /well), including microcapsules containing MCF-7 cells, HUVEC, and a mixture of both (1:1). As 2D culture groups, MCF-7 cells, HUVEC, and a mixture of both (1:1) were inoculated in a 96-well plate (500 cells/well) and cultured for 12 h. Then, the diluted DOX/PTX (0-100 μg/mL) was added and incubated for 24 h. The absorbance of the cells at 450 nm was determined using the CCK-8 reagent. The IC50 of each experimental group was calculated using GraphPad Prism 8.0.
Statistical Analysis
All data are expressed as average ± standard deviation (SD) of the results of at least three independent runs. GraphPad Prism8.0 was used for statistical analysis, and the Student's t-test was used for inter-group comparison. A P-value less than .05 was considered statistically significant.
Results
Preparation of Core-Shell Microcapsules
Before the preparation of core-shell cell microcapsules, we prepared monolayer cell microcapsules, as shown in Figure S2. Although the process of monolayer cell encapsulation is simpler than that of core-shell, it is not conducive to cell growth. The formation process of core-shell microcapsules is shown in Figure 1. During the process of cell encapsulation, using hydrogels with appropriate viscosity is essential; the most suitable viscosity is usually 300–3000 mPa·s. A hydrogel solution with viscosity <300 mPa·s will lead to mechanical instability of the structure. Conversely, increasing the viscosity of the hydrogel (≤100 000 mPa·s) will lead to mechanical integrity but requires high pressure to squeeze the aqueous solution of the hydrogel, decreasing cell viability and proliferation. The viscosity of different concentrations of sodium alginate was measured using a viscometer; the results showed that 2% sodium alginate (4050 mPa·s) was more suitable for cell encapsulation (Figure 2a). We collected the formed microcapsules for further observation and statistical analysis and found that the microcapsules were homogeneous and their diameters had an approximately normal distribution, with an average diameter of 444 μm (Figure 2b).

Formation of core-shell microcapsules.
As shown in Figure 2c, we compared the effects of three models on cell activity. The core of Model A was a mixture of type I collagen and 2% sodium carboxymethyl cellulose (3:6.5) containing MCF-7 cells and HUVEC, and the shell was 2% sodium alginate. The core of model B was a mixture of type I collagen, 2% sodium carboxymethyl cellulose, and 2% sodium alginate (3:5.5:1) containing MCF-7 cells and HUVEC, and the shell was 2% sodium alginate. The core of model C was a mixture of type I collagen and 2% sodium carboxymethyl cellulose (3∶6.5) containing MCF-7 cells, and the shell was 2% sodium alginate containing HUVEC. During the one-week culture, cell activity was tested by sampling every two days. Cells growing in a 3D microenvironment were surrounded by the matrix and gradually aggregated and maintained high activity (Figure 2d–f). We also found that the cells in model A had the lowest death rate and the greatest ability to aggregate.
Formation of a 3D Vascularized Micro-Tumor
We further observed micro-tumor vascularization in microcapsules produced by each of the two models (A and C) under the conventional static culture in 12-well plates. Since the cells in the microcapsules produced by model B had a high mortality rate during culture, no further experiments were performed. The core of Model A was a mixture of type I collagen and 2% sodium carboxymethyl cellulose containing MCF-7 cells and HUVEC (1:1), and the shell was 2% sodium alginate. The core of model C was a mixture of type I collagen and 2% sodium carboxymethyl cellulose containing MCF-7 cells, and the shell was 2% sodium alginate containing the same cell number of HUVEC as MCF-7 cells. The difference between the two models is whether there is direct cell-cell contact between MCF-7 cells and HUVEC after encapsulation.
The microcapsules produced by the two models were transferred to a complete medium containing the EGM-2 cell culture factor, including cytokines, such as vascular endothelial growth factor (VEGF), human fibroblast growth factor (hFGF), human endothelial growth factor (hEGF), and R3-insulin-like growth factor 1 (R3-IGF-1), to promote vasculogenesis and angiogenesis and then collected after two weeks of culture. CD31 expression in micro-tumor tissues was detected using immunohistochemistry and immunofluorescence. We observed that the microcapsules generated by both models had evident lumens formed by HUVEC (Figure 3a and b).

The formation of the vascular lumen in micro-tumors.
To study cell proliferation in the 3D-engineered system, we selected model A with low mortality, high aggregation, and angiogenesis for further experiments. As shown in Figure 4b–d, we observed that cell proliferation in the 3D culture system was generally slower than that in the 2D culture system.

Cell proliferation in 2D and 3D cultures.
In Vitro Drug Response
We compared the sensitivity of 2D- and 3D-cultured cells to chemotherapeutic drugs (DOX/PTX). As shown in Figure 5a–f, 2D-cultured cells were more sensitive to DOX and PTX. The average inhibitory concentration to achieve 50% cell killing (IC50) of free DOX was 10.27 μg/mL and 23.57 μg/mL in a mixture of MCF-7 cells and HUVEC in 2D and 3D cultures, respectively (a 1.3-fold increase). The IC50 of free DOX in the 3D culture (28.78 μg/mL) of MCF-7 cells was 7.89 times that of the 2D culture (3.647 μg/mL), while the IC50 of free DOX in the 3D culture (128.7 μg/mL) of HUVEC was seven times that of the 2D culture (18.37 μg/mL). In the 2D and 3D cultures, the IC50 of free PTX in the MCF-7 cells and HUVEC mixture was 39.51 μg/mL and 61.74 μg/mL, respectively (a 1.5-fold increase). The IC50 value of free PTX in the 3D culture (90.04 μg/mL) of MCF-7 cells was 3.34 times that in the 2D culture (26.91 μg/mL), while the IC50 of free PTX in the 3D culture (148.1 μg/mL) of HUVEC was 4.3 times that in the 2D culture (30.44 μg/mL).
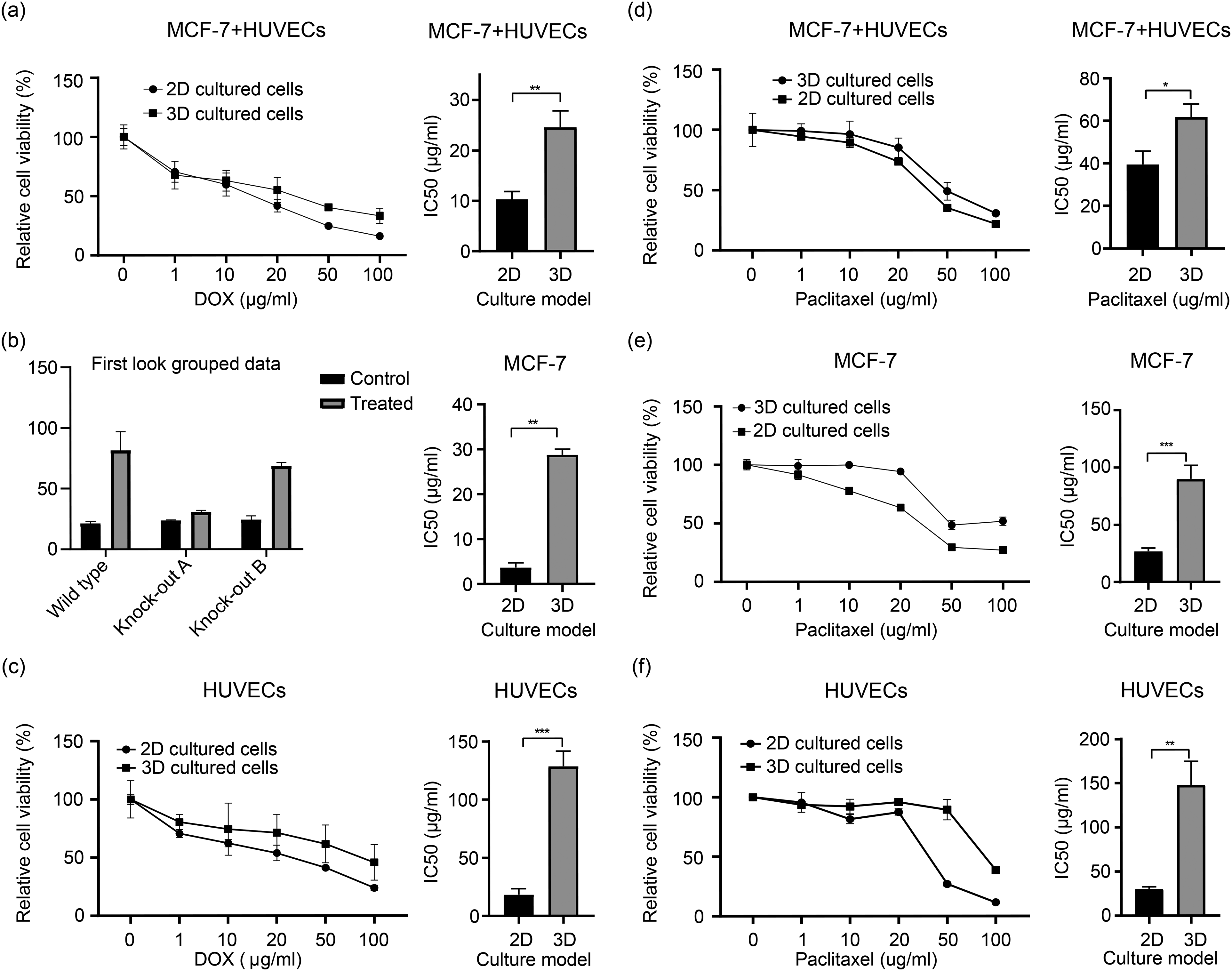
The sensitivity of 2D- and 3D-cultured cells to chemotherapeutic drugs.
Discussion
Three-dimensional cell cultures have gained increasing attention in drug screening studies. However, many available 3D culture techniques cannot be widely used because they are time-consuming, expensive, and show poor repeatability. We designed a simple, reproducible, and efficient cell encapsulation device by which MCF-7 cells and HUVEC were encapsulated in semi-permeable hydrogels. Although the biocompatibility of sodium alginate has been widely studied, the effect of alginate composition on its biocompatibility is controversial.
The residual impurities in sodium alginate are the main causes of poor biocompatibility, foreign body reaction, and inflammatory response. 17 Purified sodium alginate can reduce the host's potential immune response. 18 This is consistent with our experimental results. We found that cells encapsulated in hydrogels containing sodium alginate appeared to have higher mortality and poorer cohesion. Therefore, we used collagen with low antigenicity as the core and added sodium carboxymethyl cellulose to increase the viscosity of the solution. This method achieves good biocompatibility and a suitable porous structure for efficient cell attachment with good cell activity, proliferation, and ECM remodeling.
Angiogenesis can promote tumor development and progression, while solid tumors require blood vessels to grow.19,20 During angiogenesis, endothelial cells respond to growth signals (basic fibroblast growth factor, VEGF) to proliferate and migrate to form new blood vessels.20,21 Although several growth factors are involved in angiogenesis, VEGF is the most effective angiogenesis stimulator. 22 Cancer cells in 3D micro-tumors can release VEGF to activate the VEGF/VEGFR signaling pathway, playing a crucial role in angiogenesis and tumor growth.7,23 Therefore, we cultured the generated microcapsules in a medium supplemented with endothelial cell growth factors, including VEGF, hFGF-B, R3-IGF-1, and hEGF. After 14 days of culture, CD31 expression was detected by immunofluorescence and immunohistochemistry, and HUVECs formed tubular lumens in the micro-tumor tissues. These results indicated that we successfully constructed in vitro 3D vascularized micro-tumors.
Evidence suggests that the growth rate difference between 2D monolayer and 3D culture systems depends on the cell line and ECM. 24 Luca et al found that the proliferation of colorectal cancer cell lines in a laminin-rich ECM was significantly reduced. 25 Hongisto et al reported that the growth rate of JIMT1 cells cultured in Matrigel was 1.86-fold faster than that of the 2D monolayer culture. Interestingly, the growth rate of the same cells in poly-2-hydroxyethyl methacrylate was 7.2-fold slower than that of the 2D monolayer culture. 26 Our results indicate that the growth rate of cells in the 3D culture (model A) was slower than that in the 2D monolayer culture.
Many studies have found that 2D- and 3D-cultured cells respond differently to anticancer drugs, and 3D-cultured cells have lower sensitivity to anticancer drugs.27,28 Compared with in vivo tumors, in vitro 2D-cultured tumor cells exhibit slower tumor progression and lower drug resistance, leading to lower efficiency of drug screening and testing. 29 Imamura et al found that different BC cell lines have various sensitivities to PTX and DOX. 30 In the 3D culture, dense multicellular spheroids formed by BT-549, BT-474, and T-47D showed stronger resistance, whereas loose multicellular spheroids formed by MCF-7, HCC-1954, and MDA-MB-231 showed similar sensitivities to that of the 2D culture. The present study found that the resistance index of 3D-cultured cells to anticancer drugs (DOX and PTX) was 2.3 to 7.89 times that of 2D-cultured cells. This further indicates that TME significantly affects cell drug resistance. However, the mechanism of drug resistance in this 3D culture model is unclear and requires further exploration and research.
In this study, despite the considerable work undertaken, some limitations warrant further consideration. For example, due to the workload, we only validated doxorubicin and paclitaxel using the micro-tumor model. In future studies, we plan to include more drugs for validation. We exclusively utilized breast cancer cell lines for experimentation. We intend to build upon this foundation by isolating primary tumor cells from patients to construct micro-tumor tissue models. These models will be used for the screening of chemotherapeutic drugs to support personalized clinical treatments.
Conclusion
In summary, we demonstrated the ability of this sample encapsulation device to form core-shell microcapsules and successfully constructed 3D vascularized micro-tumors in vitro. The 3D-cultured cells had a slower proliferation rate and higher drug resistance than the 2D-cultured cells. Therefore, the 3D culture model may be valuable for studying the mechanism of tumor angiogenesis, drug resistance of tumor cells, and screening anticancer drugs.
Supplemental Material
sj-docx-1-tct-10.1177_15330338241286755 - Supplemental material for Establishment of an in Vitro Three-Dimensional Vascularized Micro-Tumor Model and Screening of Chemotherapeutic Drugs
Supplemental material, sj-docx-1-tct-10.1177_15330338241286755 for Establishment of an in Vitro Three-Dimensional Vascularized Micro-Tumor Model and Screening of Chemotherapeutic Drugs by Qian Ye, Juanru Wang, Boyang Wang, Min Zhao, Zhengsheng Wu and Xiaoli Liu in Technology in Cancer Research & Treatment
Footnotes
Abbreviations
Acknowledgments
We thank Professor Zhao Gang (CryoBME & Miocro-Nano-Bio- Systems Engineering Laboratory, University of Science and Technology of China, China) for the generous gift of the HUVE cells, as well as Professor Wu Qiang (Pathology Laboratory, Anhui Medical University, China) for the generous gift of the MCF-7 cells.
Author Contributions
Experiments were designed and carried out by Qian Ye and Juanru Wang. The manuscript was drafted and the data were interpreted by Qian Ye. Statistical analyses were performed by Boyang Wang. Experiments were designed and the manuscript was revised by Xiaoli Liu and Zhengsheng Wu. The equipment and materials were provided by Min Zhao. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
All data generated or analyzed during this study are included in the manuscript. Further inquiries should be directed to the corresponding author.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the BSKY Scientific Research Project of Anhui Medical University, Basic and Clinical Cooperative Research Promotion Program of Anhui Medical University, Applied Medical Research Project of Hefei Health Commission, (grant number XJ201801, 2020xkjT012, Hwk2021yb012).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.