
Abstract
Keywords
Introduction
Primary pulmonary lymphoma (PPL) accounts for <0.5% of primary lung cancers, <1% of lymphomas, and about 3% to 4% of all extranodal lymphomas. 1 PPL was first described by Emanuele Pekelis who reported a case of “peribronchial lymphosarcoma.” 2 PPL, a clonal lymphoid proliferation condition, affects either or both lungs (parenchyma and/or bronchi) in individuals with undetectable extra-pulmonary involvement or bone marrow condition at diagnosis time or during the successive 3 months. 3 PPL can be grouped into non-Hodgkin's lymphoma (NHL) or Hodgkin's lymphoma (HL). 4 Mucosa-associated lymphoid tissue lymphoma (MALT) comprises 60% to 80% of all cases while diffuse large B-cell lymphoma (DLBCL) accounts for 10% to 25%. 5 Other rare types include lymphomatoid granulomatosis, plasmacytoma, small lymphocytic lymphoma (SLL), mantle B-cell lymphoma (MCL), follicular lymphoma (FL), and Burkitt lymphoma. PPL of T-cell origins are extremely rare, anaplastic large cell lymphoma (ALCL) is the main phenotype. 4 Patients with PPL often have nonspecific respiratory manifestations, such as expectoration, cough, bloody sputum, chest tightness, and chest pain, while some patients may have no symptoms. 6 Chest x-ray is first performed, although the results are unspecific (eg, solitary or multiple nodules, mass or pleural effusions). 7 Therefore, computed tomography (CT) is the main examination method of PPL, which plays a crucial role in better defining thoracic alterations in patients. The prognosis of PPL varies with different pathologic types. Currently, PPL is an uncommon entity with nonspecific manifestations, as well as radiographic results, which makes its diagnosis difficult and there is no standard protocol for treatment. Therefore, this study aims to raise awareness among clinicians, to raise awareness of PPL and to share the experiences of treating and managing the patients. Clinical data from 50 individuals with PPL at the First Affiliated Hospital of Nanchang University was collected and analyzed.
Materials and Methods
Study Population
We retrospectively reviewed the medical records of all patients with pathologically confirmed PPL. A total of 83 PPL patients at the First Affiliated Hospital of Nanchang University (China) from January 2009 to December 2019 were collected. Out of these, 33 patients (30 had secondary pulmonary lymphoma while 3 did not have all clinical data) were excluded. Therefore, as per the inclusion and exclusion criteria, 50 patients were enrolled.
Inclusion and Exclusion Criteria
This was a single-center retrospective study based on 2017 revised WHO classification and diagnostic criteria of cancers of lymphoid and hematopoietic tissues. 8 The inclusion criteria consisted of (1) patients who were 18 years old and above; (2) clear lymphoma pathological diagnosis; (3) lung involvement, either bilaterally, or unilaterally with or without the involvement of mediastinal or hilar lymph node; and (4) without evidence of invasion of extranodal, as well as extrathoracic tissues up to 3 months following the primary diagnosis; and (5) no lymphoma history. Patients were excluded on the basis of: (i) aged <18 years old; (ii) the presence of extra-pulmonary lymphoma; and (iii) had other tumors. 9 All patients underwent contrast-enhanced CT scans coupled with histology. The participants’ demographic features, pathologic changes, clinical symptoms, staging, imaging features, IPI (international prognostic index) scores, treatment measures, along with prognosis were documented. Written informed consents were obtained from the patients. The Ethical Committee of the First Affiliated Hospital of Nanchang University approved this study (approval no. 2021-5-085).
Statistical Analysis
SPSS 22.0 was used for statistical analyses. Descriptive analysis was performed using counts and percentages of categorical variables to summarize medical records. Fisher's exact test were using analyzes Enumeration data. Considered to be statistically remarkable was a 2-tailed P value <.05. We defined overall survival (OS) as the time beginning from diagnosis to death or the last follow-up, patients who became progression-free and/or were lost to follow-up were considered censored data; and progression-free survival (PFS) after first-line chemotherapy as the time beginning from the onset of first-line drug therapy to disease progression and death. Log-rank survival analysis was carried out through the Kaplan-Meier approach and Cox regression models for prognostic factors.
Results
General Characteristics
Among the 50 patients with PPL, about 27 were male and 23 female patients. The sex ratio was 1.2:1 (27:23) and the mean age of the patients was 57.6 ± 15.6 years. PPL diagnosis was confirmed via pathology and immunohistochemistry (IHC). The clinical characteristic details of patient were presented in Table 1.
Clinical Characteristics of the Patients.

Abbreviations: DLBCL, diffuse large B-cell lymphoma; HL, Hodgkin's lymphoma; MALT, mucosa-associated lymphoid tissue lymphoma.
Clinical Manifestations
All the 50 patients had different degrees of symptoms, including cough (n = 37, 74%), expectoration (n = 25, 50%), bloody sputum (n = 12, 24%), fever (n = 7, 14%), chest distress (n = 13, 26%), chest pain (n = 12, 24%), night sweats and weight loss (n = 1, 2%). Only 2 patients were admitted to hospital due to upper limb and lower back pain.
Imaging Findings
All 50 patients with PPL underwent a chest CT examination. Solitary mass was reported in 31 cases (62%), 13 cases (26%) had multiple nodules or masses, and 6 cases (12%) had mixed type masses. Unilateral lung involvement was common, 16 cases (32%) were in the left lung (there were 8 patients in each upper and lower lobe of left lung), 21 cases (42%) in the right lung (9 cases in upper lobe and 12 cases in lower right lobe), 8 cases (16%) in the middle lung lobe, and 5 cases (10%) in bilateral lungs. Results for air bronchogram (n = 5, 10%) and pleural effusion (n = 7, 14%) were also obtained (Figure 1).

Representative CT images of the patient lungs. (A) In the middle and upper lobe of the right lung, there is a patch under the anterior pleura, which shows the air bearing sign of the bronchus. (B) Diffuse nodules and mass shadows in both lungs. (C) Soft tissue mass shadow is seen in the middle and lower lobe of the right lung, measuring approximately 4.5 × 6.1 cm. (E) The density of the right upper lobe of the lung was increased. (F) The space-occupying the lesion in the lower left lung, measuring approximately 6 × 5 cm in size, accompanied by pleura stretch.
Diagnosis Methods and Pathology
The main sources of specimens from the 50 patients diagnosed with PPL were fine needle aspiration (FNA) and diagnostic thoracotomy when FNA is difficult to obtain specimens, including video-assisted thoracoscopy, thoracotomy (lobectomy or biopsy, pleural biopsy), EBUS-FNA and CT-guided percutaneous needle lung biopsies (Table 2).
Diagnostic Methods in the 50 Patients Diagnosed with Primary Pulmonary Lymphoma.

All patients underwent pathologic evaluation. We had 2 HL cases and 48 NHL cases. In the phenotype of NHL, MALT (n = 21, including 2 cases had diffuse large B-cell transformation and 1 case with plasmacyte differentiation), DLBCL (n = 12), SLL (n = 2), MCL (n = 2), FL (n = 1), 8 cases of B-cell lymphoma lacking further categorization, as well as T-cell lymphoma (n = 2) (Table 1).
The histologic features of HL show the lymph nodes have a relatively conserved nodal architecture harboring diffuse invasion of medium- or small-sized lymphocytes with mild irregularities in the nucleus. Small-accumulated clusters or foci of a few giant cells with unevenly lobulated, extremely convoluted Hodgkin's or Reed-Sternberg cell-like nuclei scattered across the expanded paracortex (Figure 2A). IHC staining showed that large mononucleated and multinucleated cells resulted immunoreactive for positive for CD3, as illustrated in Figure 2B, CD15 (weak positive), as shown in Figure 2C, CD30 (Figure 2D), as well as CD79a (Figure 2E), negative for CD20 (Figure 2F).

Primary pulmonary HL. HE staining shows the lymph nodes exhibit a relatively preserved nodal architecture with diffuse infiltration of small- or medium-sized lymphocytes with mild nuclear irregularity. Small aggregated foci or clusters of a few giant cells with irregularly lobulated, highly convoluted Reed-Sternberg or Hodgkin's cell-like nuclei are scattered throughout the expanded paracortex (A) (40 × ). Immunohistochemical staining shows positive for CD3 (B), CD15 (weak positive) (C), CD30 (D), and CD79a (E), negative for CD20 (F) (40 × ).
The most common B-cell lymphoma is MALT. HE staining showed thick lymphoid proliferations with well circumscribed borders (bottom) that extended into the adjacent lung parenchyma following a lymphangitic spread pattern. Lymphoid proliferations expanded the marginal zones of reactive follicles and interfollicular areas (Figure 3A). CD20 and CD79a were strongly positive in IHC staining (Figure 3B and C), implying that they express Bcl-2. Moreover, neoplastic monocytoid cells are Bcl-2 + , while reactive monocytoid lymphocytes are Bcl-2−. In contrast, neoplastic cells are CD3 + (Figure 3D), EBER + (Figure 3E). Ki-67 labeling indices in tumor cells were found to be low (<10%) (Figure 3F).
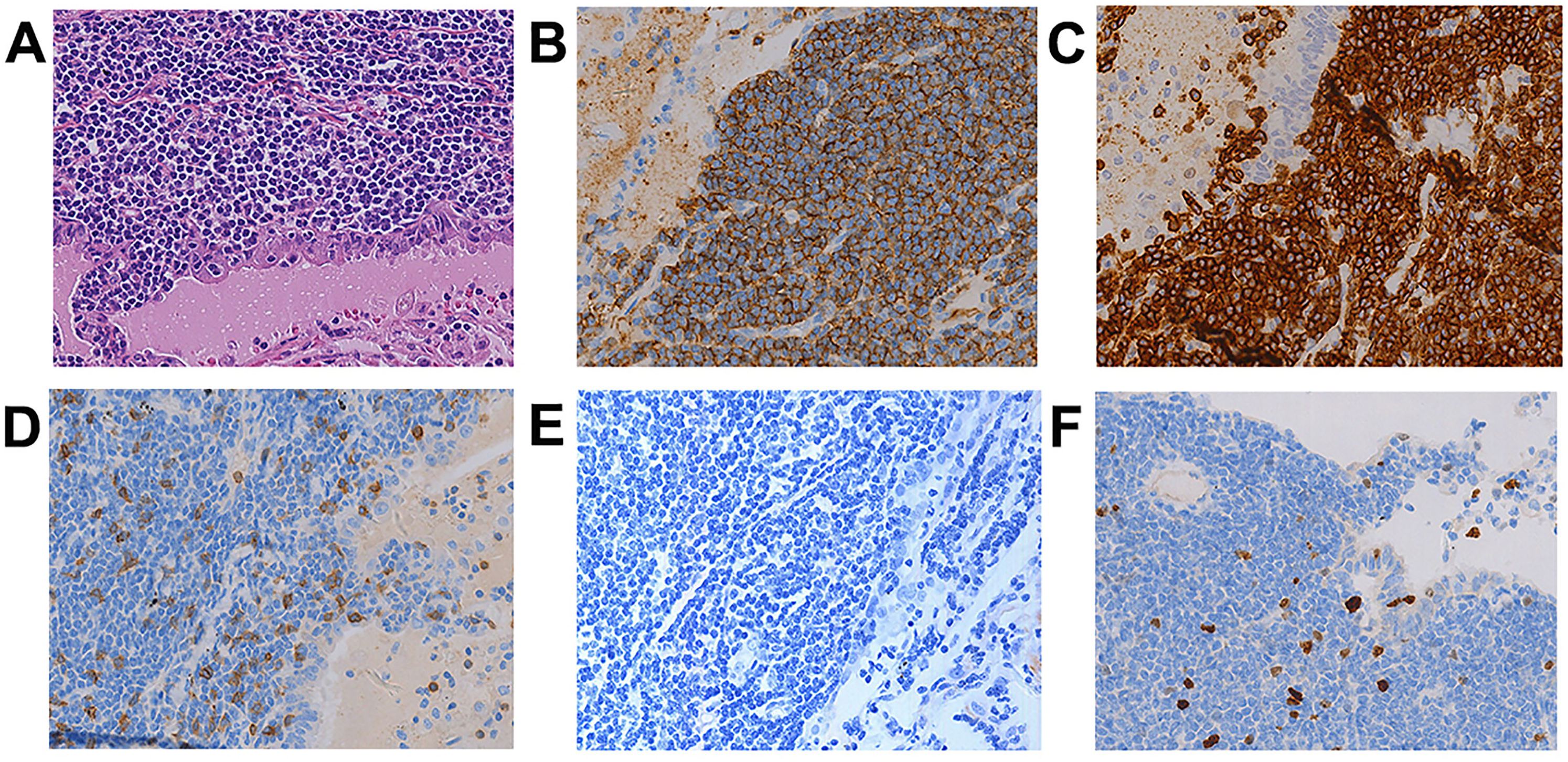
Primary pulmonary MALT. HE staining shows a dense lymphoid proliferation with relatively well circumscribed border (bottom) extending into adjacent lung parenchyma in a lymphangitic pattern of spread (A) (40 × ). Immunohistochemical staining shows diffuse positive for CD20 (B) and CD79a (C) (40 × ), negative for CD3 (D), EBER (E), and low Ki-67 (<10%) (F) (40 × ).
DLBCL is the second major category of PPL. Histologically, confluent sheets of atypical lymphoid cells that were ≥2 times larger than “mature” lymphocytes were observed. The cells exhibited a immunoblastic or centroblastic morphologies (Figure 4A). IHC staining was strongly positive for CD79a as well as CD20 (Figure 4B, C). Few or rare scattered CD3 + T cells in the background were present (Figure 4E). IHC staining was negative for EBER (Figure 4F). Ki-67 labeling indices were high (60% to 90%) (Figure 4D). In the presence of extensive necrotic tumors, CD20 may be important.

Primary pulmonary DLBCL. HE staining shows confluents sheets of large atypical lymphoid cells, at least 2 times larger than a “mature” lymphocyte, with coarse nuclear chromatin, prominent nucleoli, and a rim of amphophilic to slightly eosinophilic cytoplasm (A) (40 × ). Immunohistochemical staining shows diffuse positive for CD20 (B), CD79a (C) and high Ki-67 (60%-90%) (D), negative for CD3 (E) and EBER (F). (40 × ).
Small B-cell lymphomas are rare. They have been documented as case series of primary non-Hodgkin lung lymphomas. The main pathological types of small B-cell lymphomas include SLL, MCL, and FL. Histologically, upon imaging, small B-cell lymphomas appear as either single or multiple masses or as bilateral reticulonodular infiltrates and exhibit a lymphangitic spread via lung parenchyma. IHC is the main method to identify different subtypes. IHC staining showed that neoplastic cells were CD20 + . CD5 positive in SLL and MCL, but negative in FL. CD23 was positive in SLL, but negative in MCL. CyclinD1 was positive in MCL, but negative in SLL (Figure 5A-L).

Primary pulmonary small B-cell lymphomas. HE staining shows SLL consists of a monotonous population of small lymphocytes with round nucleus and coarsely granular chromatin (A) (40 × ), immunohistochemical staining shows positive for CD20 (B) and CD5 (C), negative for cyclinD1 (D) (40 × ). FL is composed of centrocytes and only a few centroblasts, <5/HPF. Lymphoid tissue hyperplasia, follicles are formed in different sizes, tumor boundary is not clear and infiltrating growth to lung tissue (E) (40 × ). Immunohistochemical staining shows positive for CD20 (F) and Bcl-2 (G) and CD23 (follicular dendritic reticulum was positive) (H) (40 × ). MCL consists of small- to medium-sized lymphoid elements with irregular nuclear contours, somewhat dispersed chromatin, nuclei are round to ovoid and moderately hyperchromatic (I) (40 × ). Immunohistochemical staining shows positive for CD20 (J) and CD5 (K) and CyclinD1 (L) (40 × ).
The primary T-cell lymphomas of the lungs are exceedingly sparse, only a few cases are documented in available literature. Among all lymphomas of the T-cells, primary ALCL is a frequent subtype with primary pulmonary T-cell lymphoma being second. Herein, just 2 cases of primary pulmonary T-cell lymphomas; both patients were peripheral T-cell lymphoma (PTL). Microscopically, PTL was necrotic in the center, with dense peripheral cells that were medium-sized, irregular nuclei as well as homogeneous chromatin (Figure 6A). Through IHC, the expression of T-cell biomarker varies in PTL. The malignant cells are usually positive for CD3, CD4, as well as Ki-67 (65% + ), but negative for CD20 and CD8 (Figure 6B-D).

Primary pulmonary lymphoma of peripheral T-cell. HE staining shows the tumor was necrotic in the center, with dense peripheral cells, medium-sized cells, irregular nuclei and homogeneous chromatin (A) (40 × ). Immunohistochemical staining shows positive for CD3 (B) and CD4 (C), Ki-67 (65% + ) (D), negative for CD8 (E) and CD20 (F) (40 × ).
Staging
As per the pasquale extranodal lymphomas staging 6 : (i) Stage I E: lung involvement only (might be bilateral); (ii) Stage II 1E: lung along with hilar lymph node involvement; (iii) Stage II 2E: lung coupled with mediastinal lymph node involvement; (iv) Stage II 2EW: lung along with diaphragm or neighboring chest wall involvement; (v) Stage III: lung, as well as lymph node below the diaphragm involvement; (vi) Stage IV: 1 or more extra lymphatic tissue or organ involvement.
Herein, according to Ferraro's staging of extranodal lymphoma based on Ann Arbor's lymphoma staging method, the specific stages of all cases are as follows, stage I E (10 cases, 20%), stage II 1E (8 cases, 16%), stage II 2E (17 cases, 34%), (12 cases, 24%) of stage II 2EW, as well as stage III (3 cases, 6%) (Table 1). No stage IV patients.
Treatment and Follow-up
Three (6%) patients were treated with surgery, 25 (50%) patients were treated using chemotherapy, 2 (4%) patients received chemotherapy + radiotherapy, and 9 (18%) patients received surgery + chemotherapy + radiotherapy. The treatment options of 11 (22%) patients were not clear (Table 1).
About 45 patients received outpatient or telephone follow-up, we lost 5 patients in the follow-up. One hundred twenty three months was the longest follow-up duration and 40 patients were survived. The 3-year OS along with PFS rates were 57.8% and 53.3%, respectively, and the 5-year OS, as well as PFS were 46.7% and 44.4%, respectively. Median survival and the median PFS times were 52 months and 45 months, respectively. The OS and PFS curves are illustrated in Figure 7.

Overall survival (OS) curve and progression-free survival (PFS) curve.
Prognostic Factors
As shown in Table 3, the factors that contributed to OS were used for univariate analyses. Age was remarkably linked to a poor 5-year OS (P < .05), but aggressive lymphomas, sex, treatment, stage, histology, lactate dehydrogenase (LDH) levels not remarkably linked with it (P > .05). Age was found to be an independent predictor for 5-year survival (hazard ratio, 8.900; P = .038), illustrating poor prognosis for patients who were aged ≥60 compared to those aged <60 (P < .05) (Table 3).
Univariate and Multivariate Analysis of Prognostic Factors.

Discussion
PPL is a clonal lymphoid proliferation that affects the lung's bronchi or parenchyma. PPL is rare and is responsible for 0.4% of all cancerous lymphomas. 10 The study cohort's median age at diagnosis was 57.6 ± 15.6 years, congruent with previous researches, which documented that PPL incidence commonly peaks in 6th and 7th decades of life. 11 PPL incidence has been shown to be relatively higher in males in contrast with females, 12 nevertheless, some studies opine this disease is relatively frequent in female. 13 Herein, there are 27 male and 23 female patients, the sex ratio was 1.2:1.
PPL with a low incidence often occurred with nondistinct clinical manifestations, with high diagnosis in the clinic. PPL clinical features are nondistinct, with about 36% of PPL patients exhibiting nonspecific clinical features at diagnosis, 3 congruent with our data. Herein, more than 50% of the patients present with a cough as the first manifestation, and other symptoms include fever, chest tightness, chest pain, and other common nonspecific respiratory symptoms.
The most frequent CT results in PPL consist of the existence of masses and consolidations, along with focal/diffuse ground-glass opacities, as well as peribronchovascular and perilymphatic lesions’ spread. 14 Fluoro-18-fluorodeoxyglucose positron emission tomography/CT (18F-FDG PET/CT) is another vital imaging tool, which has been widely used in staging, resetting, and follow-up of various tumors including HL and NHL.15–17 Among low-grade NHL, mass or nodular, as well as solitary lobe pneumonia are more frequent; however, multiple lobe lesions tend to present in the high-grade NHL. Besides, associated features were CT angiogram sign, air bronchogram, and pleural effusion. Air bronchogram constitutes a remarkable manifestation of PPL, especially in MALT lymphoma and manifests in 50% of individuals with MALT. 7 Here, we established that solitary mass, pneumonia, multiple nodules, or their consolidations were the main imaging manifestations of primary pulmonary NHL. Five cases had air bronchogram in this study and 4 of the cases were MALT. These results were consistent with those reported in the literature. D. Yao, et al studied imaging data of 19 Primary pulmonary NHL patients, which exhibits consisting of a fuzzy shadow at the lung mass edge exhibiting air bronchogram, lung mass shadow's long-term stability, and a manifestation that is pneumonia-like lacking infectious clinical or lab manifestations. In their study, 68.4% patients were initially mis-diagnosed with pneumonia, lung cancer, as well as tuberculosis. 18 In addition, it has been reported that about 10% of the patients are accompanied by pleural effusion.19–21 In the current study, 7 cases (14%) showed pleural effusion and results are consistent with those reported in the literature. Previous researches 22 report that primary pulmonary NHL might be accompanied by bronchiectasis, which can disappear after treatment, and can be distinguished from bronchiectasis caused by lung cancer. However, only 1 patient with bronchiectasis was reported in our study.
Pathological examination remains the gold standard for PPL diagnosis. However, it is still a challenge to distinctly and pathologically diagnose some patients. Herein, the major way to obtain pathological tissue is through EBUS-FNA and CT-guided percutaneous needle lung biopsies, it is safer, as well as less invasive in contrast with conventional invasive procedure. When it is impossible to obtain enough pathological diagnosis samples, invasive procedure should be considered. Invasive procedure for diagnostic includes lobectomy, VATS, and conventional surgery. Every approach of specimen collection presents with its own advantages along with disadvantages, and hence requires profound consideration before use. In this study, 12 patients underwent a surgical operation to obtain lesion tissue; 4 patients underwent vats, and 8 patients underwent thoracotomy. Besides, 27 cases were diagnosed with PPL by EBUS-FNA and 11 cases via CT-guided per-cutaneous needle lung biopsies.
IHC analysis has an indispensable role in validating the diagnosis. IHC can further be used to clarify the diagnosis and pathological classification. The 2017 WHO classification categorizes cancerous lymphomas into 5 classes 8 : mature T/NK-cell lymphoma, precursor lymphoid hematopoietic system tumors, mature B-cell lymphoma, histocytes and dendritic cell neoplasms (HDCN), and HL. Primary pulmonary HL is very scarce and 2 cases were reported in this study. Pathological examination showed reactive nonneoplastic inflammatory cells and R-S cells, which is consistent with a previous report. 23 Primary pulmonary NHL accounts for most of PPL. A frequent kind of PPL is derived with MALT along with DLBCL, which exhibits >95% of cases. The rest of the cases consist of rarer entities, for example, FL and SLL. 4 We confirmed the previous epidemiological data: 42% of PPL were MALT, 24% were DLBCL, 4% were T-cell lymphoma and SLL 4%, MCL 4%, FL 1%, and B-cell lymphoma lacking further classification about 16%.
The most frequent pathological type of primary pulmonary NHL is MALT, as well as DLBCL, which is reported to account for 90%, 24 the pathological features of these 2 types were described emphatically in the result part of the article. The pathological morphology of MALT lymphoma is very similar to that of benign lymphoproliferative disease of the lung, and follicular bronchiolitis, lymphocytic interstitial pneumonia, and lymphocytic inflammatory pseudotumor need to be differentiated. 25 DLBCL constitutes the second most frequent PPL subclass, defined as a connection of sheets of huge aberrant cells, which are at least 2-fold larger relative to a “mature” lymphocytes whose nuclei are equivalent in size to a macrophage nuclei. Primary pulmonary DLBCL can independently develop, however, they might occur as a result of transformations from low-grade B-cell lymphomas, for instance MALT. The concrete HE staining and immunohistochemical pathological features were also consistent with those reported in the previous literature. 26
Currently, the standard treatment modality for PPL is still debated. 3 At present, many studies believe that surgical therapy for stages IE-II1E might enable diagnostic precision and might additionally eliminate lesions completely with satisfactory treatment effects, particularly for low-grade MALT lymphoma.13,27–29 They believe that surgical therapy or local radiotherapy should be considered if the imaging results reveal that the lesion is restricted, with no distant infiltration, complete surgical removal of lesions might be useful to patients. Nonetheless, they found most PPLs since the lesions were not restricted to one region; with the involvement of lymph nodes or neighboring tissues. Surgical treatment was not effective at removing the lesions. In this situation, adjuvant chemotherapy and subsequent radical surgical abscission might not offer additional survival benefits, 6 chemotherapy or radiotherapy should be recommended. In the case of MALT lymphoma, a regimen of CHOP therapy has attained noble results. Moreover, Rituximab has achieved good results in MALT lymphoma patients, particularly in CD20 + patients. New treatment strategies are being evaluated in clinical trials. For instance, Noy et al. have documented the long-term clinical safety as well as efficacies of ibrutinib, a Bruton's tyrosine kinase inhibitor, in a refractory/relapsed MALT lymphoma cohort previously treated with rituximab, alone or in combination. 30 PI3K inhibitors, opanlisib and umbrasilib, have shown exceptional activities in MALT lymphoma. 31 For infiltrative DLBCL, a regimen of R-CHOP achieved good results, with a complete rate of remission of 70% to 80%. Herein, just 3 cases of early stage patients only received surgical treatment, and the survival time was more than 5 years. The 9 individuals with MALT lymphomas, who were treated with surgery and subsequently by chemotherapy along with radiotherapy, had high OS in contrast with patients under other treatments, nonetheless, this difference was not remarkable. The remaining 27 patients were treated CHOP or rituximab-CHOP combined with radiotherapy. Two HL patients treated with ABVD regimen (doxorubicin, dacarbazine, bleomycin, as well as vinblastine) and a partial remission was achieved. Three years after chemotherapy, two patients were still alive and in excellent health condition.
In our study, the survival times of individuals with primary pulmonary NHL vary because of heterogeneous groups of patients. Generally, the median period to mortality was 7 years, with the 3-year OS and 5-year OS of 86% and 57% to 75%. The patients who had indolent lymphomas, such as MALT lymphomas, their 5-year survival rates were 68% and 10-year were 53%. 32 On the contrary, severe lymphomas which appeared to be more diffuse, as well as destructive, including DLBCL, the 5-year survival rates were between 0% and 65%. 24 T-cell lymphoma in PPL is very sparse, and only seen in case reports, the pathological features are vascular centered, invasive and destructive, with dismal prognosis and short survival time.33–35 Herein, PPL was more frequent in older people, with the age older than 60 years being remarkably linked to dismal prognosis, similar to previous studies.27,36 However, OS was not significantly associated with aggressive lymphomas sex, treatment, stage, histology, and LDH levels, but this did not mean that its OS had no definite effect on prognosis. Due to the relatively small sample size, there is a need for further studies to clarify these findings.
Admittedly, our study had numerous limitations. Firstly, given the characteristic limitations of retrospective studies, as well as single center studies, bias involved with selection of participants and limited subject numbers constitute inevitable drawbacks. Secondly, the data analyzed in this study were obtained from the past 13 years, some follow-up data were missing. Hence, we did not evaluate the association of the clinico-pathological features. Thirdly, participant number in every subgroup of non-MALT lymphoma group was small. Therefore, subgroup analyses should be conducted and summarized once more data is collected.
Conclusions
PPL is a rare, with atypical clinical manifestations and is often misdiagnosed. IHC is currently the standard used in pathologic evaluation of PPL. CT is an effective diagnostic method, but pathological biopsy is still needed to confirm the diagnosis. Chemoradiotherapy is an effective treatment for PPL, but the standard effective treatment still remains controversial. MALT prognosis is better when compared to that of other PPL types. However, the sample size of current research is generally small and our research size is limited too, so the prognostic factors of PPL need to be further studied.
Footnotes
Abbreviations
Ethical Approval
Our study was approved by the Ethics Committee of the First Affiliated Hospital of NanChang University (approval no. 2021-5-085). All patients provided written informed consent prior to enrollment in the study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Science and technology plan of Jiangxi Provincial Health Commission, No. 20203121.