
Abstract
Aims:
Incretin signalling is known to prevent the development of arteriosclerosis by relaxation response in endothelial cells via the glucagon-like peptide 1 receptor. It remains unclear, however, whether vascular glucagon-like peptide 1 receptor expression is altered under some conditions. The aim of this study is to examine whether vascular glucagon-like peptide 1 receptor expression is altered by diabetic state as reported in pancreatic β-cells.
Methods:
We used 18-week-old male diabetic db/db mice and control db/m mice. Excised thoracic artery was specifically collected, and vascular endothelial cells were cultured. We compared the glucagon-like peptide 1 receptor expression levels between the db/db and db/m mice.
Results:
Metabolic parameters were significantly worse in db/db mice. The glucagon-like peptide 1 receptor and transcription factor 7-like 2 expression levels in endothelial and smooth muscle cells were significantly lower in db/db mice. Furthermore, siRNA to transcription factor 7-like 2 decreased the transcription factor 7-like 2 levels and such reduction of the transcription factor 7-like 2 resulted in the downregulation of the glucagon-like peptide 1 receptor expressions in cultured vascular endothelial cells.
Conclusion:
The glucagon-like peptide 1 receptor expression level was significantly lower under diabetic condition which was accompanied by the reduction of the transcription factor 7-like 2 expression level. Furthermore, the transcription factor 7-like 2 is a possible regulator of the glucagon-like peptide 1 receptor expression in artery as reported in β-cells.
Keywords
Introduction
The glucagon-like peptide 1 (GLP-1) receptor expression is observed not only in the pancreas but also in artery such as endothelial and smooth muscle cells. 1 GLP-1 signalling in the pancreas facilitates glucose-stimulated insulin secretion, leading to the amelioration of glycaemic control. 2 In endothelial cells, incretin signalling is known to improve vascular relaxation response via endothelial nitric oxide synthase (eNOS) expression and activity. 3 The expressions of plasminogen activator inhibitor-1 (PAI-1) and vascular cell adhesion molecule-1 (VCAM-1) in vascular endothelial cells induced by hyperglycaemia and inflammatory cytokines such as tumour necrosis factor-α (TNF-α) are suppressed by GLP-1 signalling. Moreover, the GLP-1 receptor stimulation is known to increase the expression of eNOS and suppress the intercellular adhesion molecule-1 (ICAM-1) expression of aortic endothelial cells in arteriosclerosis model mice. 4 It is also known that the GLP-1 receptor agonist administration improves flow-mediated dilatation. 5 Taken together, it is likely that GLP-1 signalling in blood vessels improves wall disorder induced by various factors including hyperglycaemia and various inflammatory cytokines. 6 Also in vascular smooth muscle cells, the GLP-1 receptor stimulation is known to prevent the development of atherosclerosis. 7 Finally, the effect was very recently demonstrated in the clinical human study as well. Indeed, it was shown that administration of the GLP-1 receptor agonist to patients with type 2 diabetes led to reduction of cardiovascular-related death and cardiovascular events. 8 Therefore, the role of the GLP-1 receptor in vascular cells is capturing the spotlight now. However, although the GLP-1 receptor expression in pancreatic β-cells is reduced under diabetic conditions9,10 and transcription factor 7-like 2 (TCF7L2) is known to function as a transcription factor for the GLP-1 receptor at least in β-cells, it remains unknown whether vascular GLP-1 receptor expression is altered under some conditions and which factor could influence vascular GLP-1 receptor expression. The aim of this study was to examine whether vascular GLP-1 receptor expression levels could be altered under diabetic conditions and which factor could regulate vascular GLP-1 receptor expression.
Research design and methods
Animals
We used 18-week-old male BKS.Cg-+ Leprdb/+ Lepr db /Jcl (db/db) mice compared to BKS.Cg-m +/+ Lepr db/Jcl (db/m) mice (CLEA, Tokyo, Japan; n = 8 for each group). This study was approved by the Animal Use Committee of Kawasaki Medical School (no. 15–094) and was conducted in compliance with the Animal Use Guidelines of the Kawasaki Medical School.
Vascular endothelial cell extraction from mice
We collected the thoracic artery from 18-week-old mice. And endothelial cells were specifically isolated from its artery using collagenase according to the previously reported method. 11 Vascular endothelial cells were cultured in Endothelial Cell Growth Medium (Lonza Ltd, Basel, Switzerland) for 17 days.
Measurement of biochemical markers
Blood samples were collected from the tail vein. The concentration of plasma insulin (Insulin ELISA kit; Morinaga Institute of Biological Science, Yokohama, Japan) was determined by ELISA. Plasma triglyceride (TG) and plasma nonesterified fatty acids (NEFA) concentration were measured enzymatically using the Triglyceride E-Test Wako and NEFA C-Test Wako (Wako Pure Chemical Industries, Osaka, Japan).
Real-time polymerase chain reaction
Gene expression of various factors in vascular endothelial cells were analysed by real-time reverse transcription polymerase chain reaction (RT-PCR). Primer pairs encoding genes associated with the GLP-1 receptor and factors related to arteriosclerosis were prepared, and real-time RT-PCR with SYBR Green was performed. Each gene expression was semi-quantified by the comparative Ct method with each result in β-actin as a control. Primer sequences were as follows:
GLP-1R (human): (F) CCAAGCCATGGTGACACACA
(R) TTGCAGCCACGAACTCCTT
GLP-1R (mouse): (F) ACTTTCTTTCTCCGCCTTGGT
(R) CCTGGTGCAGTGCAAGTGTCT
TCF7L2 (human): (F) TGTATGAAACCCAGATGTCACCAA
(R) TGGTTCCCACTGCTGCTCA
TCF7L2 (mouse): (F) TGCTGCTGGTGGGTGAAAA
(R) CTCGTCGTTAGCGCCTAGGT
ICAM1(mouse): (F) TCGGAAGGGAGCCAAGTAACT
(R) CGACGCCGCTCAGAAGAA
GIP-receptor(mouse): (F) GGAGCGCAACGAAGTCAAA
(R) CTGGCCCTACCAAGATGGTTAT
Immunostaining
We performed immunostaining using anti-GLP-1 receptor antibody (NLS1205; NOVUS Biologicals, Minneapolis, MN, USA). The sections in citrate buffer solution were heated for 15 min at 90°C in a microwave oven for antigen retrieval. After washing in phosphate-buffered saline with Tween-20 (PBST), sections were blocked with 5% donkey serum for 60 min. And then, the sections were incubated with anti-GLP-1 receptor antibody alone or with anti-α-smooth muscle actin antibody (NB300-978; NOVUS) at 25°C for 14 h. After rinsing with PBST, second antibody (Alexa Fluor 488 donkey anti-rabbit IgG only or with Alexa Fluor 594 donkey anti-goat IgG; Invitrogen Corporation, Carlsbad, CA, USA) was added to sections. To perform immunostaining of the intima, isolectin GS-IB4 Alexa Fluor 594 conjugate (Invitrogen Corporation) was added to the artery sections. After rinsing with PBST, 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) was added for 5 min at 25°C. TCF7L2 staining was performed using a previously reported method. And according to the previously established method, 12 anti-TCF7L2 antibody (Santa Cruz Biotechnology) solely or with anti-α-smooth muscle actin antibody (NOVUS) was used as a primary antibody. Second antibody (Alexa Fluor 488 donkey anti-goat IgG only or with Alexa Fluor 594 donkey anti-rabbit IgG; Invitrogen Corporation) was added to sections. And then, in case of an immunostaining of the intima of the artery, isolectin GS-IB4 Alexa Fluor 594 conjugate (Invitrogen Corporation) was added to the sections. After rinsing with PBST, DAPI (Sigma-Aldrich) was added for 5 min at 25°C. Images were quantified with ImageJ. 12
siTCF7L2 to cultured vascular endothelial cells
We exposed cultured vascular endothelial cells to small interfering RNA (siRNA) directed to TCF7L2 (siTCF7L2) or scrambled control siRNA and cultured for 24 h. And real-time RT-PCR with SYBR Green was performed.
siTCF7L2 to human aortic smooth muscle cells
We purchased HASMC from KURABO and cultured for 2 days. After then, we exposed human aortic smooth muscle cells (HASMC) to siRNA directed to TCF7L2 (siTCF7L2) or scrambled control siRNA and cultured for 24 h. And real-time RT-PCR with SYBR Green was performed.
Statistical analysis
All data are shown as mean ± standard error (SE). The comparison between two groups was performed using the t-test method. And p < 0.05 was regarded as significant. Statistical analysis was performed using JMP9 (SAS, Cary, NC, USA).
Results
Metabolic parameters
Between non-diabetic db/m mice and obese diabetic db/db mice, there were significant differences in body weight (31.4 ± 0.7 g, 37.8 ± 1.5 g), fasting blood glucose (FBG; 67.0 ± 47 mg/dL, 287.2 ± 6.6 mg/dL), insulin (0.34 ± 0.02 ng/mL, 1.35 ± 0.10 ng/mL), TG (118.9 ± 3.0 mg/dL, 188.9 ± 13.1 mg/dL) and free fatty acid (1.08 ± 0.05 mEq/L, 1.45 ± 0.03 mEq/L).
GLP-1 receptor and transcription factor TCF7L2 expression evaluated by immunostaining
To confirm the GLP-1 receptor protein expression in artery, we performed immunostaining with an antibody specific for the GLP-1 receptor. Since GLP-1 receptor antibodies are not regarded as reliable in general, we examined the quality of the GLP-1 receptor antibody (NLS1205; NOVUS Biologicals) using several kinds of negative controls. When the GLP-1 receptor antibody was added to thoracic artery, the internal membrane and the vascular smooth muscle were stained, but they were not stained at all without addition of the GLP-1 receptor antibody (Figure 1(a)). In addition, it is generally considered that the GLP-1 receptor expression is not observed in the liver in non-diabetic animals. Actually, in immunostaining, the GLP-1 receptor was not detected in the liver of non-diabetic mice regardless of with or without the GLP-1 receptor antibody (Figure 1(b)). From these results, we thought that this antibody is reliable. As shown in Figure 1(c), upper eight panels, the GLP-1 receptor expression was significantly lower in endothelial cells in diabetic db/db mice compared to non-diabetic db/m mice. The GLP-1 receptor expression was also significantly lower in smooth muscle cells in diabetic db/db mice compared to non-diabetic db/m mice (Figure 1(c), lower eight panels and Figure 1(e), left panel). Similarly, the expression of the TCF7L2 in endothelial cells and smooth muscle cells was clearly lower in db/db mice compared to db/m mice (Figure 1(d) and (e), right panel).
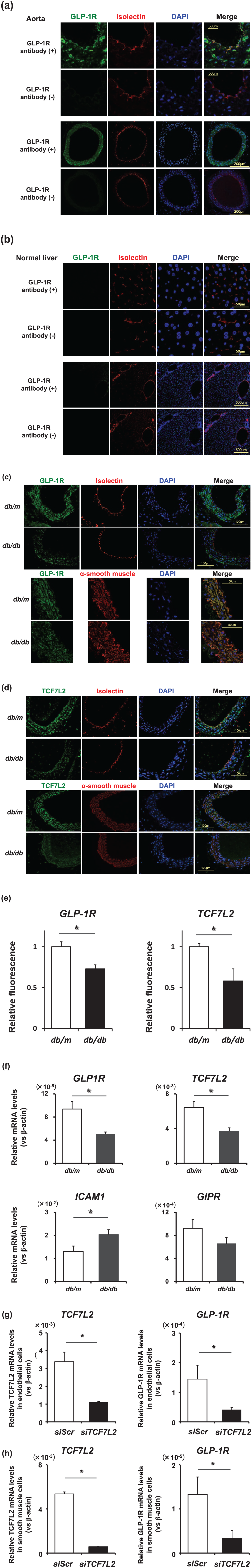
(a) Triple staining for the GLP-1 receptor in green, intima (isolectin) in red, and DAPI in blue in artery sections with or without the GLP-1 receptor antibody. Upper 8 panels: high power fields. Lower 8 panels: low power field (scale bar = 50 μm). (b) Triple staining for the GLP-1 receptor in green, intima (isolectin) in red, and DAPI in blue in liver sections with or without the GLP-1 receptor antibody. Upper 8 panels: high power fields. Lower 8 panels: low power field (scale bar = 200 μm). (c) Upper 8 panels: triple staining for the GLP-1 receptor in green, intima (isolectin) in red, and DAPI in blue in artery sections (scale bar = 100 μm). Lower 9 panels: triple staining for the GLP-1 receptor in green, α-smooth muscle cells in red, and DAPI in blue in artery sections (scale bar = 50 μm). (d) Upper 8 panels: triple staining for the TCF7L2 in green, intima (isolectin) in red, and DAPI in blue in artery sections (scale bar = 100 μm). Lower 9 panels: triple staining for the TCF7L2 in green, α-smooth muscle cells in red, and DAPI in blue in artery sections (scale bar = 100 μm). (e) The GLP-1 receptor and TCF7L2 expression in smooth muscle cells was quantitated using ImageJ. (f) Expression levels of genes related to arteriosclerosis in intima (GLP-1 receptor, TCF7L2, ICAM1, GIP receptor) in non-diabetic db/m mice (white) and diabetic db/db mice (black). (g and h) The TCF7L2 and GLP-1 receptor expression levels in vascular endothelial cells and smooth muscle cells after the treatment with scrambled control (siScr) or TCF7L2 siRNA (siTCF7L2). Data are presented as mean ± SE (*p < 0.05).
GLP-1 receptor and TCF7L2 gene expression
To strengthen our working hypothesis that the GLP-1 receptor and TCF7L2 expression is downregulated under diabetic conditions, we quantitatively evaluated the GLP-1 receptor and TCF7L2 expression by real-time RT-PCR. We compared various gene expression levels between db/db mice and db/m mice in the harvested endothelial cells. As shown in Figure 1(f), the GLP-1 receptor gene expression in the endothelial cells was significantly lower in db/db mice compared to that in db/m mice, which was compatible with the results in immunostaining. The TCF7L2 gene expression in the endothelial cells were also significantly lower in db/db mice compared to that in db/m mice. In contrast, ICAM1 gene expression was significantly higher in db/db mice (Figure 1(f)). There was no statistically significant difference in the GIP receptor expression between db/db mice and db/m mice. Taken together, both the GLP-1 receptor and transcription factor TCF7L2 messenger RNA (mRNA) levels were downregulated in vascular cells under diabetic conditions.
TCF7L2 knock down study
TCF7L2 is known to function as a regulator of the GLP-1 receptor expression at least in pancreatic β-cells. In view of this, we hypothesized that TCF7L2 regulates the GLP-1 receptor expression in endothelial cells as well as in β-cells. We exposed cultured vascular endothelial cells to siRNA directed to TCF7L2 (siTCF7L2) or scrambled control siRNA and cultured them for 24 h. As shown in Figure 1(f) (upper panels), siTCF7L2 significantly decreased the TCF7L2 mRNA levels (p < 0.0001) compared with control. As shown in Figure 1(g), such reduction of the TCF7L2 expression resulted in the significant downregulation of the GLP-1 receptor gene expression (p < 0.05). Furthermore, we exposed aortic smooth muscle cells to siRNA directed to the TCF7L2 (siTCF7L2) or scrambled control siRNA and cultured them for 24 h. As shown in Figure 1(h), siTCF7L2 significantly decreased the TCF7L2 mRNA levels (p < 0.0001) which resulted in the significant downregulation of the GLP-1 receptor gene expression (p < 0.05) in aortic smooth muscle cells as observed in endothelial cells. These data suggest that transcription factor TCF7L2 is, at least in part, involved in the regulation of the GLP-1 receptor expression in vascular cells.
Discussion
The incidence of ischaemic heart disease in subjects with type 2 diabetes is twice to four times higher, and mortality rate is twice higher compared to subjects without diabetes. 13 We also recently reported that maintaining strict glycaemic control without hypoglycaemia was extremely important to prevent ischaemic heart disease. 14 Therefore, it is an urgent need to construct the strategy for preventing ischaemic heart disease. Furthermore, after appearance of the report showing that administration of a GLP-1 receptor agonist in subjects with type 2 diabetes led to a reduction of cardiovascular-related death and cardiovascular events, 8 there is growing interest on the role of the GLP-1 receptor in vascular cells. Therefore, we think that reduction of GLP-1 receptor expression in vascular cells, at least in part, leads to the development of arteriosclerosis and the onset of cardiovascular events.
In this study, we could show that GLP-1 receptor expression in vascular endothelial and smooth muscle cells was downregulated in obese diabetic mice. Considered from our present data, such reduction of the GLP-1 receptor expression in vascular endothelial cells in diabetes model mice could be involved in high frequency of macroangiopathy in subjects with type 2 diabetes. It has been shown so far that the GLP-1 receptor stimulation in vascular endothelial and smooth muscle cells acts against arteriosclerosis. But there was no report showing which condition and/or factors could affect the GLP-1 receptor expression in vascular cells. This study is the first report showing that the GLP-1 receptor expression is lower in vascular cells in diabetic db/db mice compared with control non-diabetic db/m mice and that the GLP-1 receptor expression is, at least in part, regulated by TCF7L2 in vascular cells. It has been shown that in pancreatic β-cells, GLP-1 receptor expression is reduced by hyperglycaemia. Considered from the data in this study, hyperglycaemia is involved in the decreased expression of the GLP-1 receptor in vascular cells, although further study would be necessary to conclude. There are several possibilities concerning the mechanism how chronic hyperglycaemia leads to a reduction of GLP-1 receptor and TCF7L2 expression. It is well known that chronic hyperglycaemia increases oxidative stress and/or endoplasmic reticulum (ER) stress and activates various stress signalling pathways.15,16 Therefore, we assume that such stress and/or subsequent activation of stress signalling pathways are, at least in part, involved in the decrease of the GLP-1 receptor and TCF7L2 expression. There are some limitations in this study. It is very important to evaluate the functional mechanism. However, it was difficult to assess the downstream targets of the GLP-1 receptor in this time because major downstream factors such as pAKT and phosphorylated adenosine monophosphate–activated protein kinase (pAMPK) should be evaluated by western blotting but not by RT-PCR. The amount of endothelial cells obtained from mice artery was too small to perform western blotting. Due to such experimental limitation, we failed to perform such evaluation.
Our findings of this study were that the GLP-1 receptor was downregulated in diabetic state not only in β-cells but also in aorta and that the GLP-1 receptor expression was regulated by the TCF7L2 in vascular cells as observed in β-cells. We think that increasing TCF7L2 and/or GLP-1 receptor expression in vascular cells might be a promising approach to prevent the progression of atherosclerosis. Therefore, future studies are greatly needed. We believe that the data obtained in this study would provide useful information and have great significance in vascular biology research area as well as from the clinical point of view.
Footnotes
Acknowledgements
The authors greatly thank Saeko Moriuchi and Shiori Ikeda for excellent technical assistance. Abstract of this report was presented at the 59th annual meeting of the Japan Diabetes Society (Kyoto) and the 52nd annual meeting of the European Association for the Study of Diabetes (Munich).
Author’s contribution
T.Kim. and H.K. designed the experiments. T.Kim., A.O., Y.N., M.S., S.O., and K.Ko. conducted the experiments. T.Kim. and H.H. analysed the data. T.Kim., A.O., M.S., S.O., H.H., K.Ko., T.Kino., Y.N., S.N., T.M., K.Ka. and H.K. contributed to discussion. T.Kim. wrote the manuscript. H.K. reviewed and edited the manuscript.
Declaration of conflicting interests
H.K. has received honoraria for lectures and received scholarship grants from Sanofi, Novo Nordisk, Lilly, Boehringer Ingelheim, MSD, Takeda, Ono Pharmaceutical, Daiichi Sankyo, Sumitomo Dainippon Pharma, Mitsubishi Tanabe Pharma, Pfizer, Kissei Pharmaceutical, AstraZeneca, Astellas, Novartis, Kowa, Chugai and Taisho Pharmaceutical Co., Ltd. K.K. has been an advisor to, received honoraria for lectures from, and received scholarship grants from Novo Nordisk Pharma, Sanwa Kagaku Kenkyusho, Takeda, Taisho Pharmaceutical Co., Ltd, MSD, Kowa, Sumitomo Dainippon Pharma, Novartis, Mitsubishi Tanabe Pharma, AstraZeneca, Nippon Boehringer Ingelheim Co., Ltd, Chugai, Daiichi Sankyo, and Sanofi.
Funding
This work was supported by a Grant-in-Aid from the Japan Society for the Promotion of Science (No. 16K09770 to H.K.) and Research Project Grants from the Kawasaki Medical School (No. 27-4 to T.K. and No. 27-14 to H.K.).