
Abstract
Background:
Glucagon-like peptide-1 receptor agonists may have a role in modulation of cardiac fibrosis. Our study aimed to determine the effect of the glucagon-like peptide-1 receptor agonist liraglutide in obesity, hypertension and age-induced murine models of cardiac fibrosis and identify associated molecular mechanisms.
Methods:
C57Bl/6J mice on a high-fat diet and C57Bl/6J mice on a normal chow diet treated with angiotensin II were used to induce obesity and hypertension-mediated cardiac fibrosis, respectively. C57Bl/6J mice 20 months old were used to study age-induced cardiac fibrosis. Liraglutide treatment of 30 µg/kg/day–300 µg/kg s.c. twice daily was administered for 4 weeks.
Results:
Liraglutide treatment attenuated obesity, hypertension and age-induced increases in interstitial cardiac fibrosis and expression of inflammatory and oxidative stress markers.
Conclusions:
These observations identify a potential role for liraglutide in the prevention of cardiac fibrosis and identify molecular mechanisms associated with these effects.
Keywords
Introduction
Cardiac fibrosis develops as a consequence of excessive extracellular matrix (ECM) deposition and remodelling resulting from pathological processes, including ischemia, inflammation and neurohormonal activation and is associated with the disruption of normal myocardial structure. 1 Increased fibrosis, through accumulation principally of collagen in the cardiac interstitial and perivascular space, leads to reduction of myocardial and arterial compliance and is a major determinant of increased ventricular stiffness, which together with impaired relaxation forms the foundation for development of diastolic cardiac dysfunction.1–3
Collagen deposition at a site of injury is postulated to be mediated by fibroblasts derived from either fibrocytes from the blood circulation or from epithelia via epithelial–mesenchymal transition (EMT).4,5 Fibroblasts implicated in cardiac fibrosis have a myofibroblast phenotype distinguished by α-smooth muscle actin (α-SMA) expression and resulting in increased secretion of collagen type I and III.6,7 Differentiation of the fibroblast to a myofibroblast phenotype is postulated to be as a consequence of excess fibrogenic pro-inflammatory cytokines frequently mediated by macrophage infiltration and oxidative stress.8,9 Remodelling of excess ECM once deposited in the fibrotic process can occur as a result of expression of matrix metalloproteinases (MMPs) and their inhibitors [tissue inhibitors of matrix metalloproteinases (TIMPs)] with these changes leading to stiffening of the ECM, functional alterations that cause an increase in mechanical stress, cardiac hypertrophy and ultimately cardiac failure.6,7,9,10
Pre-clinical and clinical studies have identified cardioprotective effects of glucagon-like peptide-1 (GLP-1) and glucagon-like peptide-1 receptor (GLP-1R) agonists suggesting that these agents may have a role in modulation of the cardiac fibrosis phenotype.11–14 A single study has recently investigated the effects of liraglutide on cardiac fibrosis in a murine model of obesity. 15 One week, low dose, weight neutral, liraglutide treatment was found to significantly reverse high-fat diet (HFD)-induced increases in perivascular cardiac fibrosis and increased cardiac levels of tumour necrosis factor-α, nuclear translocation of NFκB and expression of procollagen 1A1. 15 Similarly, use of the dipeptidyl peptidase inhibitor-IV (DPP-IV) inhibitor MK0626 was recently demonstrated to attenuate interstitial and perivascular cardiac fibrosis and improved cardiac function, via mechanisms that included a reduction in oxidative stress, in a murine model of obesity-induced cardiac fibrosis. 16 These recent studies highlight the potential for GLP-1R stimulation to attenuate cardiac fibrosis in a non-diabetic setting and posit a possible role for this class of agents in the broader management of cardiac disease.
Our studies investigated the effect of the GLP-1R agonist liraglutide on interstitial cardiac fibrosis in the setting of several disease states including hypertension, obesity and ageing together with determination of the molecular mechanisms associated with these effects.
Materials and methods
In vivo studies
Male C57BL/6J mice weighing approximately 30 g were obtained from Monash Animal Research Laboratories (ARL). Mice were housed in the Pharmacology Animal House, Monash University, in standard mouse cages (with approximately four mice per cage) at 21 C ± 1–5C, with a 12-h light/dark cycle. All mice had access to food and water ad libitum. All treatments and experimental procedures were approved by the School of Biomedical Sciences (SOBS) Animal Ethics Committee, Monash University (Ethics number: SOBSB/PHAR/2013/118).
Obesity model
C57Bl/6J mice 5 months old maintained on a HFD (22% fat and 0.15% cholesterol (Specialty Feeds, Western Australia) from 5 weeks of age were utilised to evoke obesity-induced cardiac fibrosis.
Hypertension model
C57Bl/6J mice 12–16 weeks old maintained on a normal chow diet were treated chronically with angiotensin II (Ang II; 800 ng/kg/day) via osmotic mini-pumps in the final 4 weeks of the study to evoke hypertension-induced cardiac fibrosis.
Ageing model
C57Bl/6J mice 20 months old maintained on a normal chow diet were utilised as a model of age-induced cardiac fibrosis.
Treatment regimens
Treatment regimens for both obesity- and age-induced cardiac fibrosis models consisted of liraglutide 300 µg/kg s.c., twice daily in the final 4 weeks of each experiment. Hypertension treatment groups included saline, Ang II (800 ng/kg/day), Ang II + liraglutide [30 µg/kg/day (non-weight reducing dose)] or Ang II + liraglutide [300 µg/kg/day (weight reducing dose)] via separate mini-osmotic pumps for 4 weeks.
Biometric assessment
Baseline systolic blood pressure (SBP) readings were taken using tail-cuff plethysmography (MC4000 Blood Pressure Analysis System, Hatters Instrument Inc.). Five consecutive cycles of SBP were recorded in each run and the average SBP was then taken from the five runs. During the 4-week treatment period, SBP was monitored and recorded fortnightly commencing before treatment (week 0), middle (week 2) and end of treatment (week 4) using non-invasive tail-cuff plethysmography apparatus. Baseline and end of treatment (4 weeks) body weight (BW) was measured in grams (g).
Surgical procedures
For surgical procedures, mice were anaesthetised using isoflurane inhalation (~1% to 3%). A small area at the back of the neck was shaved and a small incision between the scapulae was made, followed by insertion of mini-osmotic pumps for subcutaneous drug administration. The incision area was stitched using 6/0 DY silk (Dynek Pty Ltd), antibiotic powder applied (Cicatrin, Pfizer) followed by an intramuscular injection of the analgesic cartrophen (0.1 mL of a 1.5 mg/mL stock solution, Biopharm Australia). At the end of treatment, mice were anaesthetised using isoflurane inhalation and euthanised.
Tissue dissection
Organs, fat and blood were collected, with heart and aorta being dissected appropriately as described below. The heart was initially weighed with both the right and left atria intact. The atria were removed and the heart was re-weighed to give ventricular weight (VW). As a gross indicator of cardiac hypertrophy, the VW in milligrams (mg) was compared to BW in grams to give a ratio of VW:BW (mg/g). All organs were then snap frozen in liquid nitrogen and stored at −80°C if not being used for experiments on the day of euthanasia.
The whole heart and aorta were removed and placed in ice-cold Krebs-bicarbonate buffer (pH 7.4) comprising (mM) NaCl 118, KCl 4.7, KH2PO4 1.2, MgSO4·7H2O 1.2, CaCl2 2.5, NaHCO3 25 and glucose 11.7. Fat and connective tissue deposits were removed from both the heart and aorta. The aorta was dissected into abdominal and thoracic sections. Three aortic rings were taken from the abdominal aorta for vascular reactivity assessment using isolated organ baths while the remaining abdominal aorta was embedded in OCT and the thoracic aorta was snap frozen in liquid N2 and stored at −80°C. The heart was then sectioned into two with the upper portion of the heart placed in a cryomould containing Optimum Cutting Temperature compound (OCT), frozen and stored at −80°C and the lower portion was frozen in liquid N2.
Assessment of endothelial vascular reactivity
Three aortic rings (~3 mm/ring) from each mouse were suspended between two 100 µm stainless steel wires connected to an isometric force transducer (FT-03, Grass Instruments). Each mouse had one aortic ring randomly allocated as a time control and two rings for cumulative acetylcholine (ACh)-dose response curves to assess stimulated nitric oxide (NO) release. Aortic rings were immersed in organ baths containing Krebs solution as previously described 17 and were maintained at 37°C with bubbled carbogen (95% O2, 5% CO2). Tissues were gradually stretched to their optimum tension of 0.5 g over a 60-min period. After 15 min equilibration at 0.5 g, a maximum contraction was provoked using U46619 (1.0 µM). When the response plateaued, tissues were washed with Krebs solution a number of times until the tissue returned to baseline tension. All tissues were pre-contracted to 70%–80% maximal contraction of U44619. After the response plateaued, an ACh concentration response curve was performed (1 × 10−9 to 1 × 10−4M) to assess endothelium-dependent relaxation of the aorta. Lastly, 1 × 10−5M of sodium nitroprusside (SNP) was added to each organ bath in order to determine the integrity of the vascular smooth muscle of each preparation. Investigators were blinded to the aortic ring treatment groups for vascular reactivity studies.
Cardiac section preparation and assessment of cardiac fibrosis
Cryostat (Cryocut 1800) was used to generate 10 µm cross-sections of cardiac tissue. Picrosirius red stain (PRS) was used to determine collagen expression levels in transverse cardiac sections. The optimal concentration of PRS used was 0.05%. Collagen staining intensity was analysed under bright field light microscopy. Six different fields of view from the ventricle were randomly selected and photographed with consistent exposure settings for each section under 20× magnification. The percentage of positive interstitial collagen staining per total field of view was quantified using ImageJ 1.45s software (Java, NIH), and averaged as the final percentage collagen content in a particular animal.
Immunohistochemical analysis of cellular and molecular components of cardiac fibrosis
Immunohistochemical analysis of fibroblast and myofibroblast expression was assessed in frozen 10 µm transverse cardiac sections. After optimisation of primary antibody concentrations were completed, experiments were carried out using the primary and secondary antibody dilutions as shown in Table 1.
Primary antibodies and their respective secondary antibody used in immunofluorescence studies.
Immunofluorescent sections were observed under 20× magnification (Olympus, BX51) with six images obtained from the ventricles. The percentage of immunofluorescence was quantified as per total field of view using ImageJ 1.45s software (Java, NIH), and averaged out from a total of six views as the final percentage of marker expression in a particular animal.
Haematoxylin and eosin (H&E) staining protocol to measure cardiomyocyte area
Cardiomyocyte size was assessed using standard haematoxylin and eosin (H&E) staining techniques in 10-µm-thick transverse cardiac sections. H&E stained sections were observed under 20× magnification (Olympus, BX51) and images acquired. The size of the cardiomyocytes was quantified by tracing and measuring the area of 50–80 cardiomyocytes per field of view using ImageJ 1.45s software (Java, NIH), and averaged out from a total of six views as the final cardiomyocyte area in a particular animal.
In vitro studies
In vitro studies utilised the murine macrophage cell line RAW264.7. RAW264.7 cells were cultured to confluence in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% foetal calf serum (FCS). RAW264.7 cells were treated with liraglutide (100 nM) with or without lipopolysaccharide (LPS; 100 ng/mL) in serum-free DMEM for 48 h.
Real-time quantitative polymerase chain reaction
Total RNA was extracted from RAW264.7 cells using Trizol reagent (Invitrogen). An aliquot (2 µL) of total RNA was reverse transcribed in 20 µl reaction mixture at 50°C for 1 h. cDNA was amplified by real-time polymerase chain reaction (PCR) using specific primers:
Mouse Monocyte Chemoattractant Protein (MCP)-1 primers: forward: 5′-CAC-TGT-CAG-CCT-TGG-AGC-TCA-3′, reverse: 5′-AAG- AGG-TCC-CCC-AGA-GTC-TCC-3′. Mouse IL-10 primers: forward: 5′-TCA-GGA-TGC-GGC-TGA-GGC-GCT-G-3′, reverse: 5′-GCT-AAG-AGT-CCC-TGG-ATC-AGA-TT-3′. Mouse NFκB-1 primer: forward: 5′-GCA-AGC-ACT-GTG-AGG-ACG-GGG-T-3′, reverse: GGT-CTG-CTG-CAC-GGC-TGC-CTG-G-3′. Mouse β-actin primers: forward: 5′-GGC-TGT-ATT-CCC-CTC-CAT-CG-3′, reverse: 5′-CCA-GTT-GGT-AAC-AAT-GCC-ATG-T-3′.
Each PCR run also included wells of no template control (NTC). A melting point dissociation curve generated by the instrument was used to confirm that only a single product was present. The fluorescence data were quantitated using the threshold cycle (CT) value. Data were normalised to ß-actin and presented as the mean fold change compared with untreated samples.
Results
Effect of liraglutide on biometric parameters in models of cardiac fibrosis
Systolic blood pressure
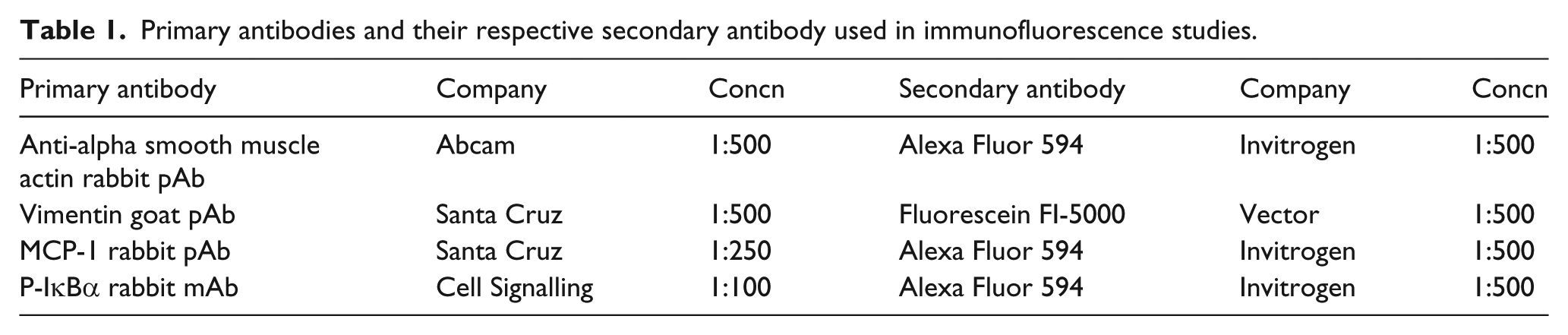
SBP was measured in each mouse prior to treatment, at weeks 2 and 4 (end of treatment). There was no difference in SBP between groups prior to treatment. As expected, there was a significant increase in SBP with Ang II infusion (Figure 1(a-i)) and this was significantly attenuated at both doses of liraglutide (Figure 1(a-i)). Liraglutide treatment also attenuated obesity (HFD) induced increases in SBP with no significant effect in the aged model of cardiac fibrosis (Figure 1(a-ii and iii)).
Effect of liraglutide on biometric parameters in models of cardiac fibrosis. (a) SBP: (i) Mean data for SBP at weeks 0, 2 and 4 (SBP; mmHg) [n = 7–9; *p < 0.05, liraglutide (30 or 300 µg/kg/day) versus saline; **p < 0.01, liraglutide versus Ang II; ****p < 0.0001, Ang II versus saline; two-way ANOVA]. (ii) Mean SBP in 5-month-old HFD-fed C57Bl/6J mice pre- and post-4-week liraglutide treatment 300 µg/kg s.c., bd. (*p < 0.05, two-way ANOVA; n = 4). (iii) Mean SBP in 20-month-old aged normal chow-fed C57Bl/6J mice pre- and post-4-week liraglutide treatment 300 µg/kg s.c., bd. n = 4 = 6. (b) Weight loss: (i) Mean BW (g) at week 0 (pre-treatment), weeks 2 and 4 (end of treatment) in saline, Ang II and Ang II + liraglutide (30 or 300 µg/kg/day), n = 8–10. (ii) Mean BW pre- and post-liraglutide treatment 300 µg/kg s.c., bd in 5-month-old HFD-fed C57Bl/6J mice. (*p < 0.05, HFD pre- versus post-treatment, **p < 0.01, HFD + liraglutide pre- versus post-treatment; two-way ANOVA; n = 4). (iii) Mean BW in 20-month-old aged normal chow-fed C57Bl/6J mice pre- and post-4-week liraglutide treatment 300 µg/kg s.c., bd.***p < 0.001, pre- versus post-liraglutide treatment; two-way ANOVA; n = 4–6). (c) Blood glucose levels: (i) Blood glucose levels in response to glucose load (2 g/kg i.p) over 120 min in saline, Ang II and Ang II + liraglutide 30 or 300 µg/kg/day treated mice. Glucose tolerance test prior to liraglutide treatment (week 0) and after 4 weeks of treatment (n = 8–10; two-way ANOVA). (ii) Fasting blood glucose levels in 5-month old normal diet (ND), HFD-fed C57Bl/6J mice (HFD), HFD C57Bl/6J mice administered 4-week liraglutide treatment 300 µg/kg s.c., bd (HFD + Lirag) and aged 20-month C57Bl/6J mice administered 4-week liraglutide treatment 300 µg/kg s.c., bd (aged + Lirag) **p < 0.01, ND versus HFD-fed mice, *p < 0.05, HFD versus HFD + liraglutide, aged + Lirag; one-way ANOVA, n = 4–7.
Body weight
A trend towards weight loss was observed with high-dose liraglutide treatment compared to saline in the Ang II hypertension model (Figure 1(b-i)). HFD-induced weight gain was significantly attenuated with liraglutide treatment (Figure 1(b-ii)) and significant weight loss compared to control was observed in aged mice (Figure 1(b-iii)).
Blood glucose
In Ang II–infused mice, glucose tolerance test demonstrated no difference in blood glucose levels prior to and after 4 weeks of treatment in all groups (Figure 1(c-i)), consistent with previous studies. 18 In the HFD-induced obesity model, fasting hyperglycaemia was observed compared with mice on a normal chow diet. HFD-mediated induction of hyperglycaemia was significantly attenuated by liraglutide treatment (Figure 1(c-ii)). While aged mice treated with liraglutide had significantly lower fasting blood glucose levels compared to HFD-fed mice (Figure 1(c-ii)), no significant difference in glucose levels was observed between aged normal chow-fed mice untreated or treated with liraglutide, consistent with previous reports using GLP-1R agonists in aged C57/Bl6 mice. 19
Effect of liraglutide on vascular reactivity in models of cardiac fibrosis
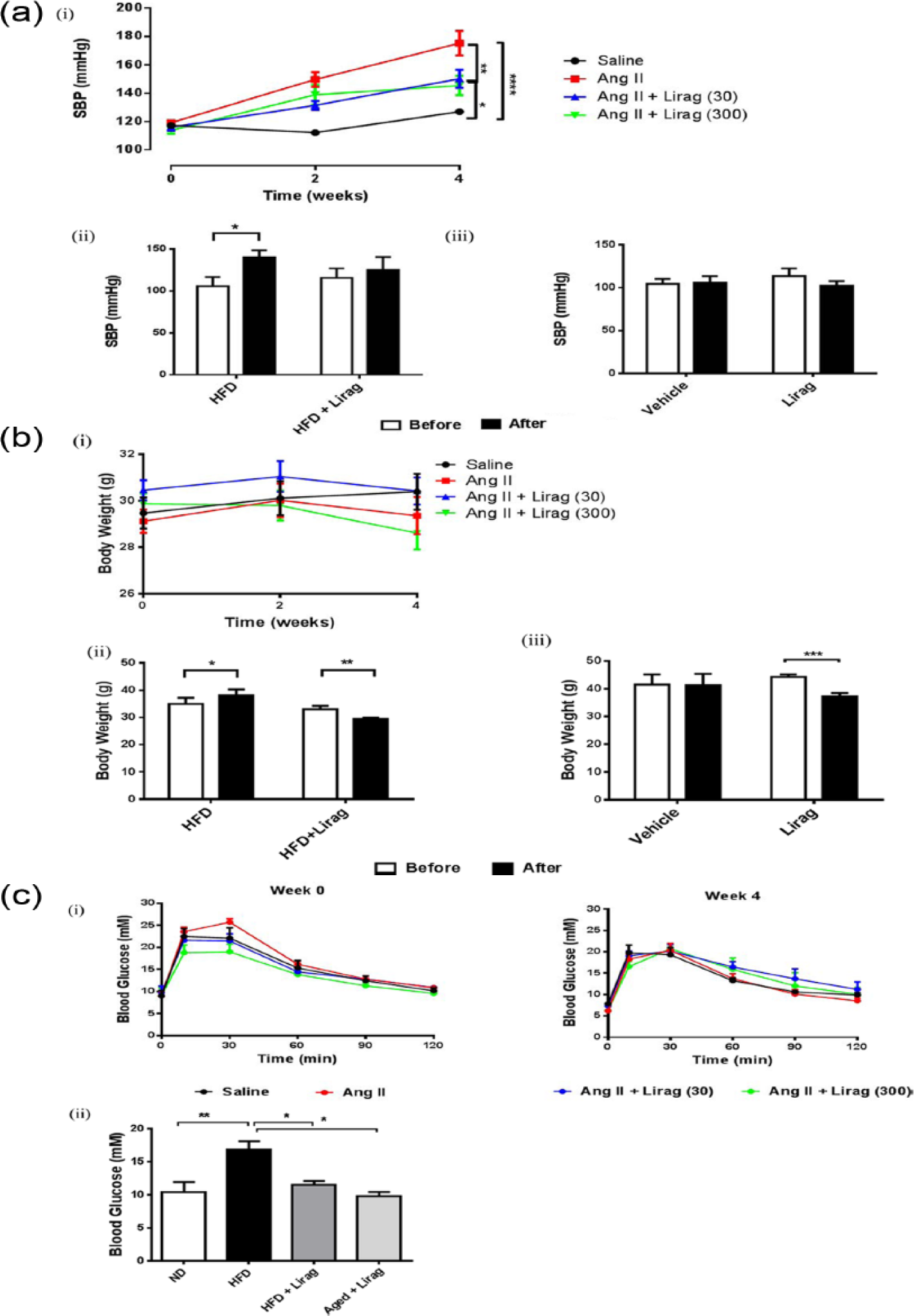
Treatment with either 30 or 300 µg/kg/day doses of liraglutide abolished Ang II-induced endothelial dysfunction (Figure 2(a-i)). No effect of either Ang II alone or in combination with either dose of liraglutide on response to the endothelium-independent vasodilator, SNP confirmed the endothelial dependence of this effect (Figure 2(a-ii)). A significant age-dependent reduction in endothelial-dependent vasorelaxation was observed in aortas taken from 20-month old C57Bl/6J mice compared to effects seen in young mice (Figure 2(b)). Four-week liraglutide treatment of aged mice reversed age-induced endothelial dysfunction so that there was no longer any difference in endothelial function compared to aorta from young mice (Figure 2(b)). We found no significant effect of either HFD alone or in combination with liraglutide treatment on endothelial function when compared to vasorelaxation seen in aorta taken from mice fed a normal diet (Figure 2(c)).
Effect of liraglutide on vascular reactivity in models of cardiac fibrosis. (a): (i) Concentration response curves to endothelium-dependent vasodilator ACh in abdominal aortic rings (n = 5–9; **p < 0.001, Sham, Ang II + Lirag 30 µg/kg/day, Ang II + liraglutide 300 µg/kg/day versus Ang II; two-way RM ANOVA). (ii) Mean data for relaxation in response to SNP (n = 5–8; one-way ANOVA. (b): Concentration response curves to endothelium-dependent vasodilator ACh in abdominal aortic rings in young 5-month-old mice and 20-month-old aged C57Bl/6J mice treated with either vehicle or liraglutide 300 µg/kg s.c., bd, n = 4–8. *p < 0.05, young versus aged controls; p = 0.0692, aged control versus aged + liraglutide-treated mice; two-way RM ANOVA. (c): Concentration response curves to endothelium-dependent vasodilator ACh in abdominal aortic rings from mice fed a normal diet (ND) and mice fed a HFD ± liraglutide 300 µg/kg s.c., bd, n = 4–6.
Effect of liraglutide on cardiac hypertrophy in a model of cardiac fibrosis
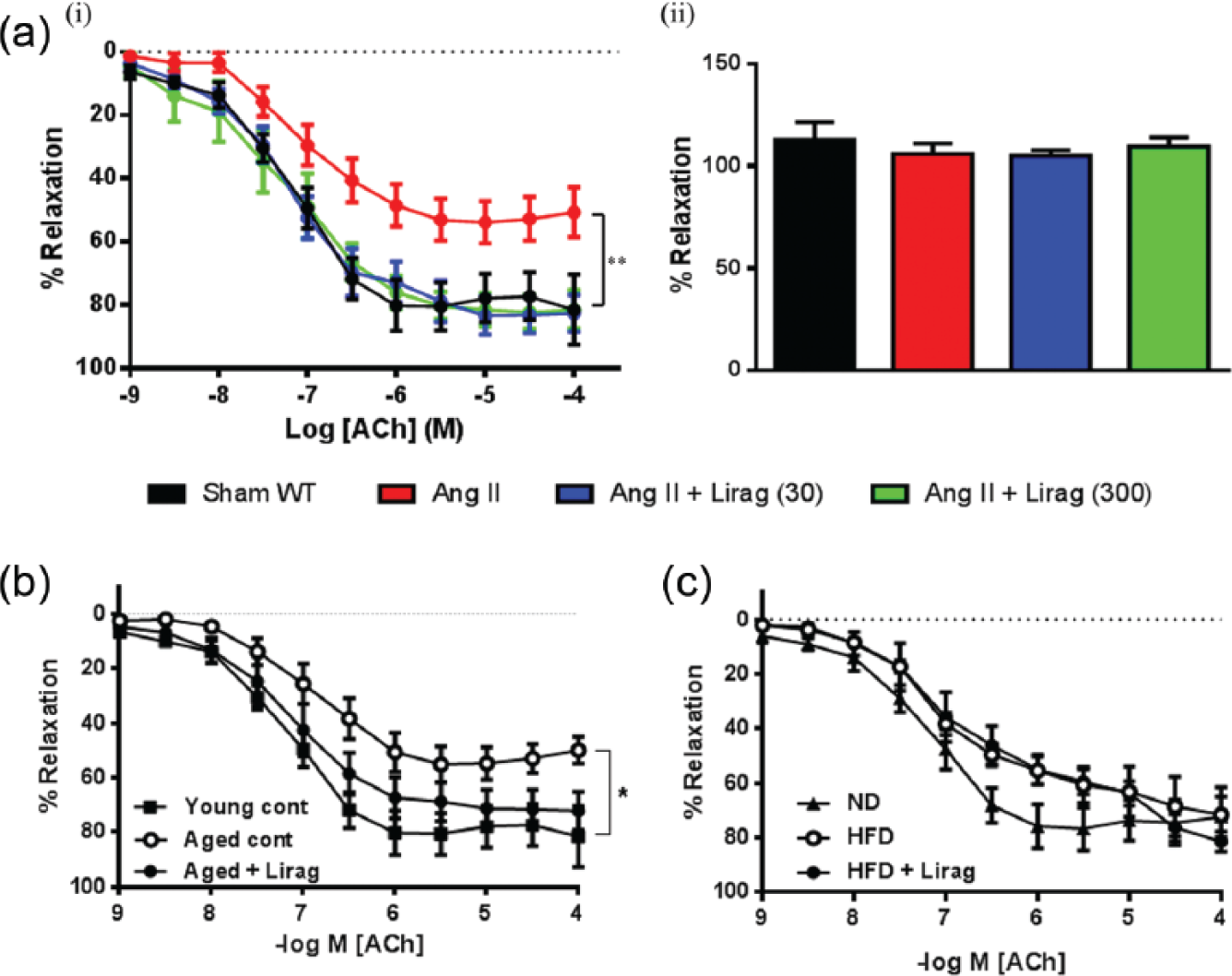
As a gross measure of cardiac hypertrophy, VW to BW ratio was assessed. Ang II-treatment significantly increased VW:BW ratio compared to saline-treated WT mice (Figure 3(a)). Co-treatment of Ang II–infused mice with liraglutide did not significantly attenuate this effect (Figure 3(a)). As a more accurate assessment of cardiac hypertrophy, the cardiomyocyte cross-sectional area was determined with Ang II treatment significantly increasing cardiomyocyte cross-sectional area (Figure 3(b) and (c)). Liraglutide treatment at low and high dose was able to significantly attenuate the Ang II-induced increase in cardiomyocyte area (Figure 3(b) and (c)).
Effect of liraglutide on cardiac hypertrophy in models of cardiac fibrosis. (a) Ventricular weight: body weight (VW:BW) ratio (n = 7–9; *p < 0.05, Ang II versus sham; one-way ANOVA). (b) Cardiomyocyte cross-sectional area (µm2) (n = 7–9; ****p < 0.0001, Ang II versus sham; ##p < 0.01, Ang II + liraglutide 30 µg/kg/day versus Ang II; ###p < 0.001, Ang II + liraglutide 300 µg/kg/day versus Ang II; one-way ANOVA). (c) Representative H&E stained cardiac sections. Black horizontal bar = 200 µm.
Effect of liraglutide on cardiac fibrosis
Chronic Ang II infusion increased collagen deposition in comparison with saline-treated controls (Figure 4(a)). Ang II-induced increase in cardiac collagen deposition was abolished when mice were co-treated with either dose of liraglutide (Figure 4(a)). Aged 20-month-old C57Bl/6J mice maintained on a normal chow diet and 5-month-old C57Bl/6J mice maintained on a HFD demonstrated a significant increase in cardiac fibrosis compared with 5-month-old normal chow-fed mice (Figure 4(b)). Liraglutide treatment for 4 weeks significantly attenuated the development of cardiac fibrosis in both models such that interstitial collagen levels were similar to those found in 5-month-old C57Bl/6J mice maintained on a normal diet (Figure 4(b)).
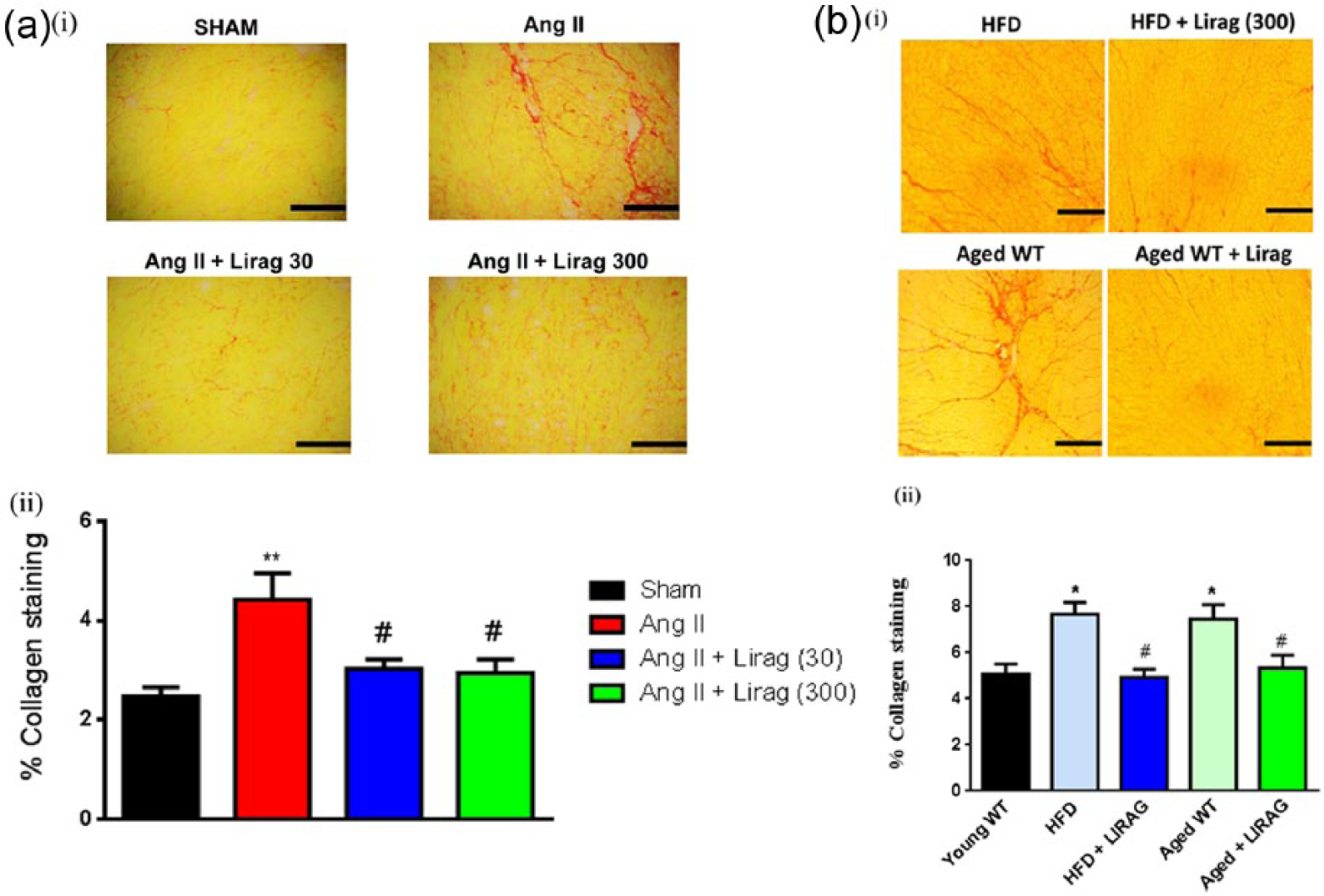
Effect of liraglutide on cardiac fibrosis. (a): (i) Representative sections under bright field microscopy of picrosirius red staining for interstitial collagen distribution (20× magnification) and (ii) quantification of collagen expressed as percentage of collagen staining in cardiac sections (n = 7–10; **p < 0.01, Ang II versus sham; #p < 0.05, Ang II + liraglutide 30 or 300 µg/kg/day versus Ang II; one-way ANOVA). Black horizontal bar = 200 µm. (b): (i) Representative sections under bright field microscopy of picrosirius red staining for interstitial collagen distribution (20× magnification). HFD = cardiac sections of HFD-induced cardiac fibrosis in 5-month-old C57Bl/6J mice, HFD + Lirag (300) = cardiac sections of HFD-induced cardiac fibrosis in 5-month-old C57Bl/6J mice treated with 4 weeks liraglutide 300 µg/kg s.c., bd, aged WT = 20-month-old aged normal chow-fed C57Bl/6J mice, aged + Lirag = 20-month-old aged normal chow-fed C57Bl/6J mice treated with 4 weeks liraglutide 300 µg/kg s.c., bd. Young WT = 5-month-old C57Bl/6J mice maintained on a normal chow diet,*p < 0.01, young WT versus HFD and aged WT, n = 4–6. #p < 0.01, HFD and aged WT versus, HFD + Lirag 300 and aged WT + Lirag, one-way ANOVA, n = 4–6. Black horizontal bar = 200 µm.
Effect of liraglutide on fibroblast-myofibroblast phenotype in models of cardiac fibrosis
Fibroblast expression using vimentin and myofibroblast expression using α-SMA was evaluated in cardiac sections. A significant increase in both vimentin and α-SMA was identified in the Ang II hypertension model of cardiac fibrosis together with trends to increase the expression of fibroblasts and myofibroblasts in both the HFD obesity and 20-month-old ageing models of cardiac fibrosis (Figure 5(a) and (b)). Liraglutide treatment (30 and 300 µg/kg/day) in the Ang II hypertension model of cardiac fibrosis significantly reduced the increase in cardiac expression of both fibroblast and myofibroblast cell types (Figure 5(a)). A reduction in both fibroblast and myofibroblast cell types with liraglutide treatment was also identified in both the HFD obesity and 20-month-old ageing models of cardiac fibrosis to levels similar to that expressed in young WT mice (Figure 5(b)).
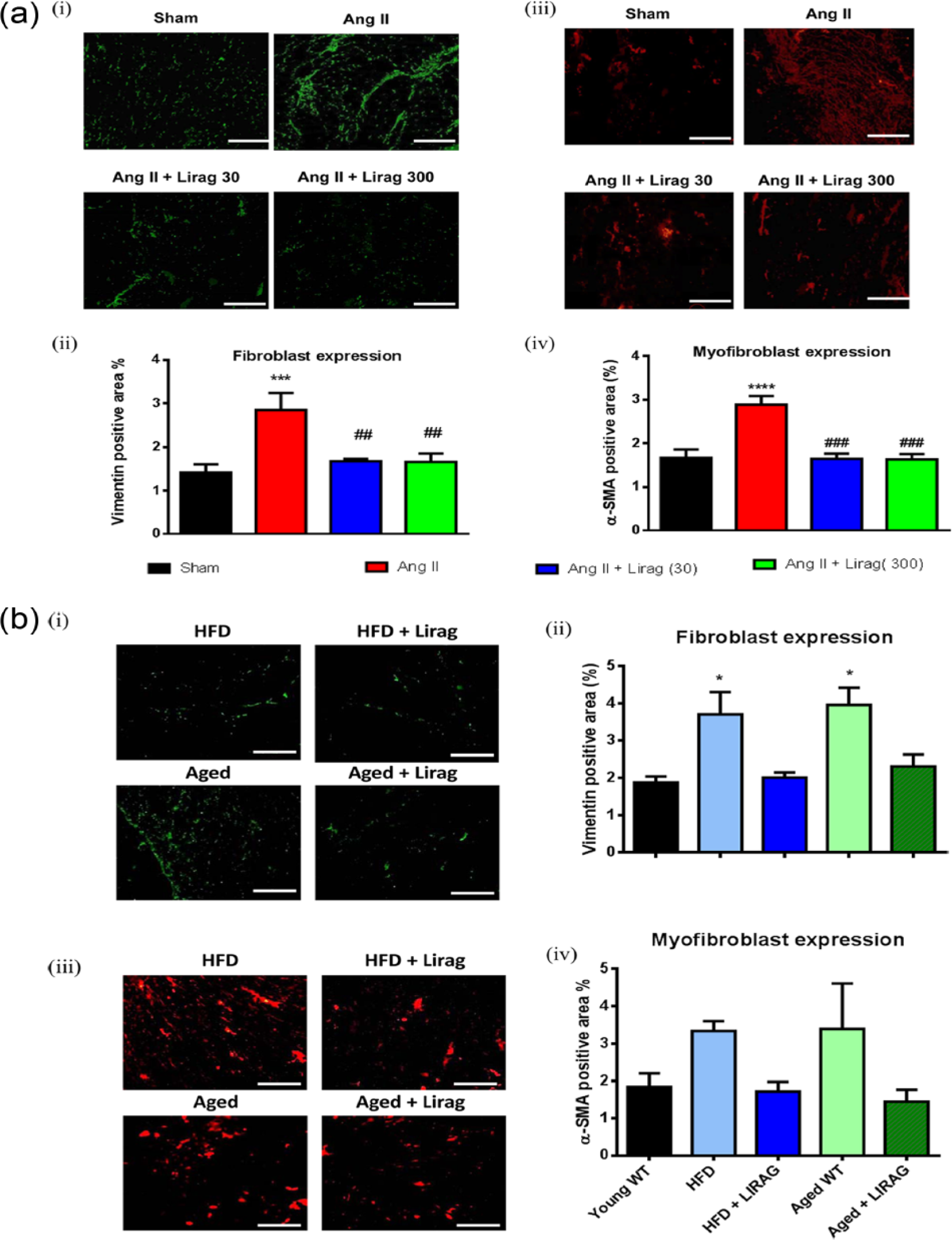
Effect of liraglutide on fibroblast-myofibroblast phenotype in models of cardiac fibrosis. (a): (i) Representative sections of immunofluorescence staining for vimentin in cardiac sections (20× magnification) and (ii) quantification of vimentin expressed as percentage of positive stained area in cardiac sections (n = 7–10; ***p < 0.001, Ang II versus sham; ##p < 0.01, Ang II + liraglutide 30 or 300 µg/kg/day versus Ang II; one-way RM ANOVA). (iii) Representative sections of immunofluorescence staining for α-SMA in cardiac sections (20× magnification) and (iv) quantification of α-SMA expressed as percentage of positive stained area in cardiac sections (n = 7–10; ****p < 0.0001, Ang II versus sham; ###p < 0.001, Ang II + liraglutide 30 or 300 µg/kg/day versus Ang II; one-way RM ANOVA). White horizontal bar = 200 µm. (b): (i) Representative cardiac sections of immunofluorescence staining for vimentin in HFD, HFD + liraglutide 300 µg/kg s.c., bd (HFD + lirag), aged 20-month-old normal chow-fed C57Bl/6J mice (aged WT) and aged 20-month-old normal chow-fed C57Bl/6J mice + liraglutide 300 µg/kg s.c., bd (aged + lirag) (20× magnification). (ii) Quantification of vimentin expressed as percentage of positive stained area in cardiac sections. (iii) Representative cardiac sections of immunofluorescence staining for α-SMA (20× magnification) (iv) Quantification of α-SMA expressed as percentage of positive stained area in cardiac sections in *p < 0.05, young WT (5-month-old C57Bl/6J mice on normal chow diet) versus HFD-fed or aged WT; one-way ANOVA, n = 4–6. White horizontal bar = 200 µm.
Effect of liraglutide on NFκB expression and activity in models of cardiac fibrosis
Activation of NFκB transcription is directly correlated with the phosphorylation of the inhibitory unit I-kBα. p-IkBα expression was significantly increased in cardiac sections from Ang II–infused hypertensive mice compared with controls (Figure 6(a)). Treatment with liraglutide prevented Ang II-induced NFκB activation with the effect being significant at higher doses of liraglutide (Figure 6(a)). In vitro assessment of NFκB mRNA expression in the murine macrophage cell line RAW264.7 demonstrated significant attenuation of LPS-mediated induction of NFκB mRNA expression when treated with liraglutide for 48 h (Figure 6(b)).
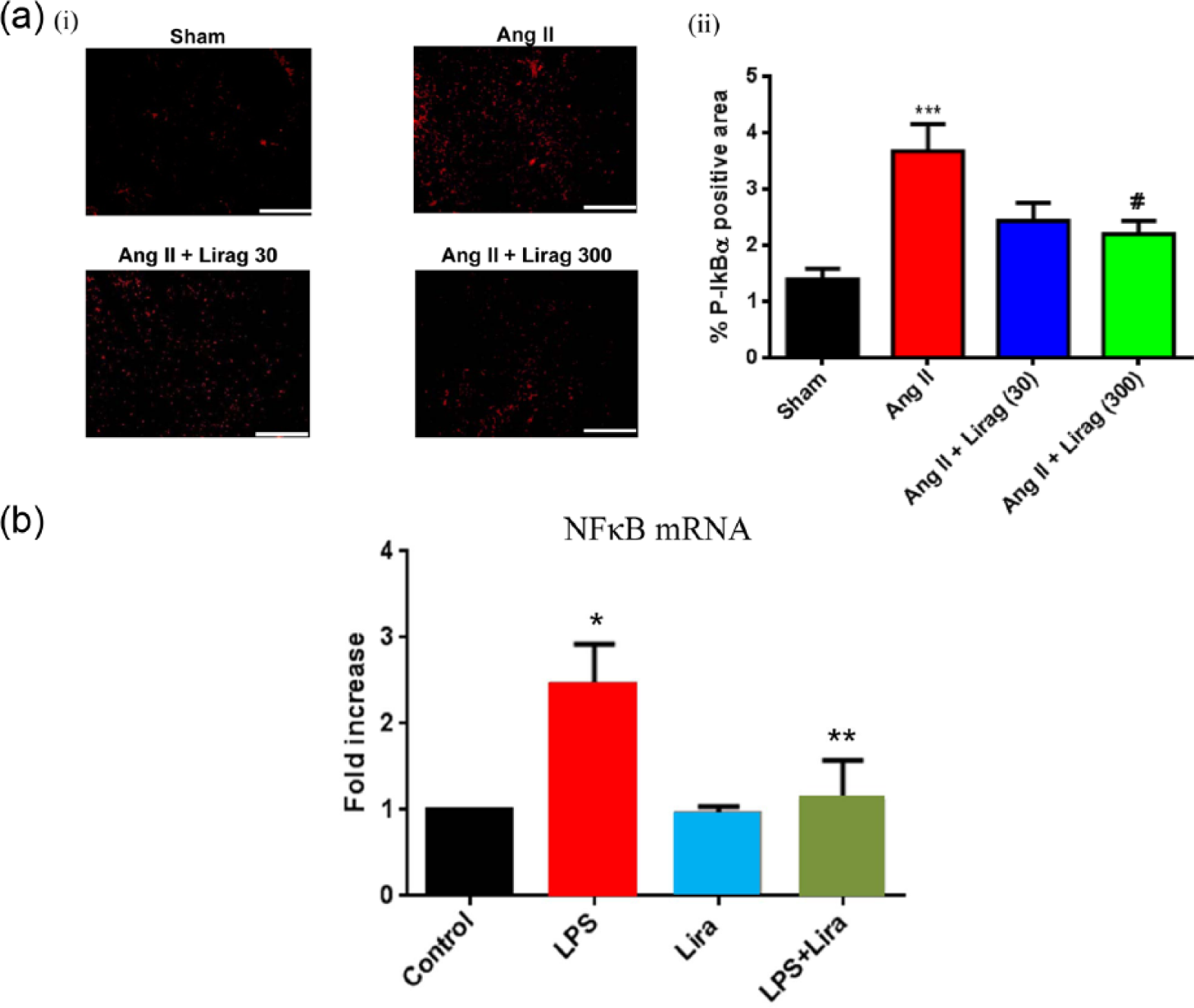
Effect of liraglutide on NFκB expression and activity in models of cardiac fibrosis. (a): (i) Representative cardiac sections of immunofluorescence staining for p-IκBα, a marker of NFκB activation (20× magnification) and (ii) quantification of p-IκBα expressed as percentage of positive stained area in cardiac sections (n = 4–5; ***p < 0.001, Ang II versus sham; #p < 0.05, Ang II + liraglutide 300 µg/kg/day versus Ang II; one-way ANOVA). White horizontal bar = 200 µm. (b): NFκB mRNA expression in RAW246.7 cells. Control = untreated, LPS = 100 ng/mL LPS, Lira = 100 nM liraglutide (*p < 0.05, control versus LPS, **p < 0.05, LPS versus LPS + liraglutide; one-way ANOVA, n = 3).
Effect of liraglutide on pro- and anti-inflammatory chemokine expression in models of cardiac fibrosis
NFκB-mediated induction of pro-inflammatory chemokines is well recognised in the pathogenesis of cardiac fibrosis. 20 Expression of the pro-inflammatory chemokine MCP-1 was significantly increased in the Ang II-mediated hypertensive model of cardiac fibrosis (Figure 7(a)). Ang II-mediated induction of MCP-1 expression was significantly attenuated with liraglutide treatment (Figure 7(a)). In vitro assessment of MCP-1 mRNA expression in the murine macrophage cell line RAW264.7 demonstrated significant attenuation of LPS-mediated induction of MCP-1 mRNA expression when treated with liraglutide for 48 h (Figure 7(b-i)). Interestingly, liraglutide treatment simultaneously and significantly induced expression of the anti-inflammatory chemokine IL-10 in the murine macrophage cell line RAW264.7 (Figure 7(b-ii)).
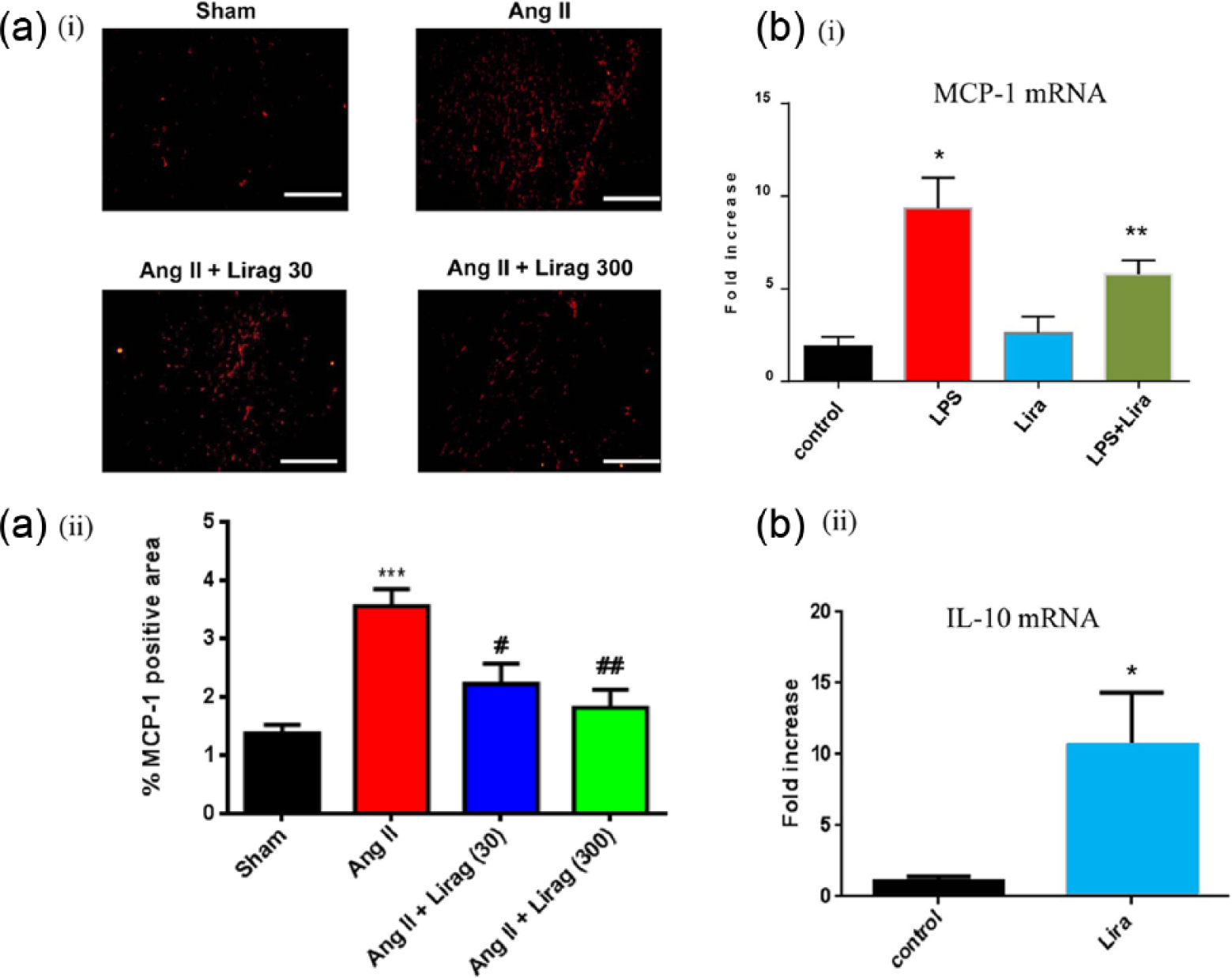
Effect of liraglutide on pro- and anti-inflammatory chemokine expression in models of cardiac fibrosis. (a): (i) Representative sections of immunofluorescence staining for MCP-1 in cardiac section (20× magnification) and (ii) quantification of MCP-1 expressed as percentage of positive stained area in heart sections (n = 4–5; ***p < 0.001, Ang II versus sham; #p < 0.05, Ang II + liraglutide 30 µg/kg/day versus Ang II; ##p < 0.01, Ang II + liraglutide 300 µg/kg/day versus Ang II; one-way RM ANOVA). White horizontal bar = 200 µm. (b): (i) MCP-1 and (ii) IL-10 mRNA expression in RAW246.7 cells. Control = untreated, LPS = 100 ng/mL LPS, Lira = 100 nM liraglutide. (*p < 0.05, control versus LPS, lira, **p < 0.05, LPS versus LPS + liraglutide, n = 3).
Effect of liraglutide on superoxide production in models of cardiac fibrosis
Superoxide production was significantly increased in the Ang II hypertension model of cardiac fibrosis and this was dose dependently reduced with liraglutide treatment. Specificity of the assay for superoxide was confirmed by quenching of the superoxide signal using superoxide dismutase (PEG-SOD) (Figure 8).
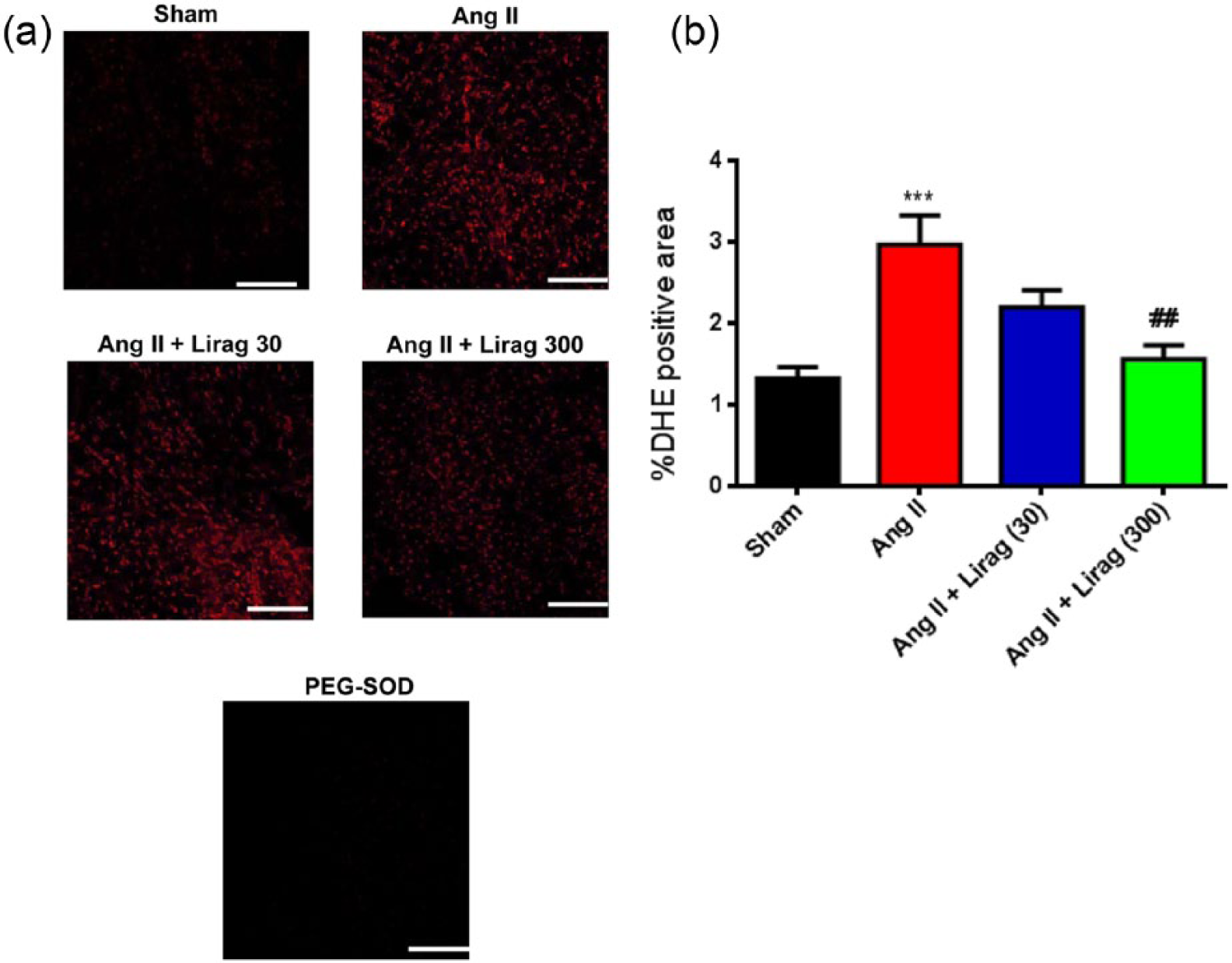
Effect of liraglutide on superoxide production in models of cardiac fibrosis: (a) Representative images of cardiac sections with red stain indicative of superoxide. PEG-SOD = superoxide dismutase. White horizontal bar = 200 µm. (b) Quantification of superoxide expressed as percentage of DHE positive stained area ***p < 0.001, Ang II versus sham; ##p < 0.01, Ang II + liraglutide 300 µg/kg/day versus Ang II; one-way RM ANOVA, n = 5–7.
Discussion
Cardiac fibrosis is associated with several cardiac disease states and is principally mediated via inflammatory mechanisms. 1 Unchecked cardiac fibrosis results in increased myocardial stiffness, a precursor to cardiac failure, a condition associated with considerable morbidity and mortality in both diabetic and non-diabetic patient populations.1–3,10
Given previous clinical studies demonstrating improvements in cardiac function with GLP-1 and GLP-1R agonist treatment together with pre-clinical observations of attenuation of cardiac fibrosis in the setting of GLP-1R agonist and DPP-IV treatment,11–16 we were interested in characterising the potential therapeutic effects of the GLP-1R agonist liraglutide in several murine models of cardiac fibrosis and delineating the pathophysiological and molecular mechanisms associated with these effects.
Our observations have identified attenuation of cardiac fibrosis in hypertensive, obesity and age-related murine models of cardiac fibrosis with these findings demonstrated using weight loss associated doses of liraglutide (300 µg/kg s.c., twice daily) utilised in previous in vivo studies21,22 together with weight neutral, normoglycemic, low-dose liraglutide (30 µg/kg/day) treatment in the Ang II model of hypertensive cardiac fibrosis suggesting that they are at least in part independent of liraglutide-mediated effects on BW and plasma glucose levels. These observations correlate with attenuation of cardiac fibroblast and myofibroblast expression, together with inhibition of cardiomyocyte hypertrophy and suggest an effect of liraglutide on the secretory fibroblast phenotype previously identified to be regulated by both Ang II and transforming growth factor-β1 (TGF) signalling events.10,23 The molecular mechanisms responsible for potential liraglutide-mediated modulation of Ang II and/or TGF-β1 signalling in the setting of cardiac fibrosis are unknown; however, recent observations suggest a role for the orphan nuclear receptor NR4A1 in modulation of TGF-β signalling in the setting of fibrosis, 24 a molecule previously demonstrated to be regulated by liraglutide treatment. 25
Improvements in vascular reactivity in both hypertensive and ageing models of cardiac fibrosis suggest a potential therapeutic association between these processes. Previous demonstration of improvements in vascular reactivity correlating with improvements in cardiac function and attenuation of cardiac failure/fibrosis lend support to this contention. 26 While the molecular mechanisms responsible for putative therapeutic effects of improvements in vascular reactivity on cardiac fibrosis are unknown reduction in oxidative stress and the concomitant attenuation of expression of inflammatory cytokines from vascular endothelial cells is thought to contribute to this effect. 27
In an attempt to further delineate the molecular mechanisms responsible for the attenuation of cardiac fibrosis in our murine models evaluation both in vitro and in vivo of liraglutide treatment on several inflammatory and oxidative stress markers was performed. In vivo and in vitro attenuation of NFκB activity and expression with liraglutide treatment (Figure 6(a) and (b)) supports previous studies identifying liraglutide-mediated in vitro suppression of NFκB expression in HUVEC 28 together with recent in vivo identification of attenuation of NFκB translocation and activity in murine models of fibrosis.15,29 These observations suggest a potentially broad anti-inflammatory effect of liraglutide, consistent with reports in relation to effects of other GLP-1R agonists, 30 in the setting of cardiac fibrosis.
Recent studies demonstrating an absence of GLP-1R expression in ventricular cardiomyocytes31,32 raise concerns regarding potential cellular mechanisms responsible for the posited cardioprotective effects of liraglutide; 11 however, clear demonstration of GLP-1R expression in macrophages 33 and inhibitory effects of GLP-1 on macrophage tissue infiltration and inflammation in in vivo studies 34 lends support to the concept of a cell type specific therapeutic anti-inflammatory effect of liraglutide in the myocardium or points to an as yet unidentified mechanism for these effects, potentially involving vascular endothelial or smooth muscle cell GLP-1R expression 35 as has been postulated in recent cardiomyocyte-targeted GLP-1R knockout studies. 36
Potential downstream inflammatory mediators of NFκB signalling were also investigated to further dissect the molecular mechanisms associated with the effects of liraglutide in our models of cardiac fibrosis. MCP-1 expression, a critical target of pathological inflammatory stimuli, was attenuated both in vitro and in vivo with liraglutide treatment (Figure 7(a)) and attests to the anti-inflammatory activity of this molecule. Intriguingly in vitro identification of concurrent upregulation of expression of the anti-inflammatory cytokine IL-10 in murine macrophages further reinforces the global anti-inflammatory activity of liraglutide in this cell type (Figure 7(b)) and supports previous studies demonstrating GLP-1-dependent polarisation of macrophages towards the M2 anti-inflammatory phenotype. 33
The role of oxidative stress as an independent mediator of tissue inflammation is widely acknowledged, particularly in cardiovascular tissue. 37 In order to characterise the effects of liraglutide on cardiac oxidative stress, evaluation of cardiac superoxide production was undertaken. Liraglutide treatment significantly attenuated cardiac superoxide production (Figure 8) identifying an additional component of the potential molecular mechanism responsible for attenuation of cardiac fibrosis by liraglutide in our murine models and supporting several pre-clinical studies.38–41
Our study is the first to evaluate the potential beneficial effects of a GLP-1R agonist in an ageing model of cardiac fibrosis with restitution of fibrosis to levels demonstrated in ‘young’ 5-month-old mice in aged mice treated with liraglutide (Figure 4(b)). The unique characteristics associated with age-induced cardiac fibrosis include direct senescence-associated fibrogenic actions and impaired responses to cardiac damage including a reduced capacity to degrade collagen. 42 In addition, activation of several distinct molecular pathways facilitating production of reactive oxygen species, chemokine-mediated recruitment of mononuclear cells and fibroblast progenitors and utilising TGF-β activation, endothelin-1 and Ang II signalling cascades mediate interstitial and perivascular fibrosis in the senescent heart.42,43 The therapeutic effect of liraglutide in attenuating age-induced cardiac fibrosis is likely to be mediated via modulation of several aspects of the molecular pathogenesis of this condition including potential effects on reactive oxygen species and chemokine production (Figures 6–8), as demonstrated in our Ang II hypertension model of cardiac fibrosis, and conceivably involving, as previously mentioned, modulation of Nr4A1 expression.
Extra-pancreatic effects of DPP-IV inhibitors and GLP-1R agonists, in relation to potential influences on the cardiovascular system generally and cardiac fibrosis specifically, are a source of considerable research interest and significance given the predisposition of type 2 diabetic patients to accelerated cardiovascular disease. While large-scale clinical studies, possibly subject to uncontrolled variables, have yet to confirm the potentially beneficial cardiovascular effects of these agents, 44 pre-clinical studies continue to suggest a cardioprotective/anti-fibrotic role for this class of agents, potentially mediated via glucose lowering, 45 influences on hormones regulating glucose homeostasis 46 and direct effects on the myocardium.
Our previous pre-clinical in vivo observations in murine models of advanced macrovascular disease have identified liraglutide-mediated attenuation of atherosclerosis and enhanced plaque stabilisation.21,22 Our current studies identify the potential for the GLP-1R agonist liraglutide to attenuate cardiac fibrosis in multiple pathological settings identified in both diabetic and non-diabetic states and elucidates the molecular mechanisms associated with these effects. These observations suggest evaluation of this agent to determine the in vivo functional sequel of attenuation of cardiac fibrosis together with the downstream effects of this condition including cardiac failure in diabetic and non-diabetic settings may be warranted.
Footnotes
Declaration of conflicting interests
The study was, in-part, funded by a scientific research grant from Novo Nordisk A/S Denmark.