
Abstract
Background:
Insulin exerts vasculoprotective effects on endothelial cells (ECs) and growth-promoting effects on vascular smooth muscle cells (SMCs) in vitro, and suppresses neointimal growth in vivo. Here we determined the role of ECs and SMCs in the effect of insulin on neointimal growth.
Methods:
Mice with transgene CreERT2 under the control of EC-specific Tie2 (Tie2-Cre) or SMC-specific smooth muscle myosin heavy chain promoter/enhancer (SMMHC-Cre) or littermate controls were crossbred with mice carrying a loxP-flanked insulin receptor (IR) gene. After CreERT2-loxP-mediated recombination was induced by tamoxifen injection, mice received insulin pellet or sham (control) implantation, and underwent femoral artery wire injury. Femoral arteries were collected for morphological analysis 28 days after wire injury.
Results:
Tamoxifen-treated Tie2-Cre+ mice showed lower IR expression in ECs, but not in SMCs, than Tie2-Cre− mice. Insulin treatment reduced neointimal area after arterial injury in Tie2-Cre− mice, but had no effect in Tie2-Cre+ mice. Tamoxifen-treated SMMHC-Cre+ mice showed lower IR expression in SMCs, but not in ECs, than SMMHC-Cre− mice. Insulin treatment reduced neointimal area in SMMHC-Cre− mice, whereas unexpectedly, it failed to inhibit neointima formation in SMMHC-Cre+ mice.
Conclusion:
Insulin action in both ECs and SMCs is required for the “anti-restenotic” effect of insulin in vivo.
Introduction
Individuals with diabetes and even those with insulin resistance without diabetes are at increased risk for atherosclerosis. Percutaneous intervention (PCI, i.e. transluminal angioplasty plus stenting) is widely performed for the treatment of atherosclerotic vessels; however, patients with diabetes or insulin resistance are at increased risk for restenosis despite the introduction of drug-eluting stents (DESs). 1
Although various anti-diabetic agents have been developed, insulin remains a cornerstone of treatment for patients with diabetes, especially those with insulin deficiency. Insulin is an essential hormone in the regulation of metabolism via its action in classical insulin target organs such as liver, skeletal muscle, and adipose tissue. In addition to its metabolic actions, insulin has been shown to directly exert multiple actions in various tissues including the cardiovascular system. 2
We previously reported that in rodent models of atherosclerosis, insulin treatment has a protective effect. 3 We also found that, in rodent models of restenosis, systemic or local administration of insulin suppressed neointimal hyperplasia, which is the main factor for in-stent restenosis.4–8 Furthermore, insulin treatment enhanced re-endothelialization. 4 These findings suggest that insulin deficiency or insulin resistance may have adverse vascular effects, potentially implicating a beneficial role for insulin in selected patients with diabetes undergoing PCI.
Insulin exerts multiple actions in vascular endothelial cells (ECs) and smooth muscle cells (SMCs), both of which are involved in the process of neointimal hyperplasia. 9 In vitro studies have revealed that effects of insulin on ECs are mainly vasculoprotective due to insulin’s activation of endothelial nitric oxide synthase (eNOS) via phosphatidylinositol-3 kinase (PI3K)/Akt. 10 In contrast, the in vitro effects of insulin on vascular SMCs are potentially adverse, as insulin increases proliferation and migration via mitogen-activated protein kinase (MAPK), although it also stimulates differentiation via PI3K.11,12 However, neointima is formed upon complex interactions between ECs, SMCs, and even inflammatory cells and platelets. 9 Thus, in vivo studies are required to clarify the cell-specific mechanisms underlying the effect of insulin on neointimal growth.
In the present study, we aimed to determine the roles of ECs and SMCs in the overall “anti-restenotic” effect of insulin in vivo by using the CreERT2-loxP-mediated recombination system, which can be used to selectively and conditionally knock down insulin receptor (IR) expression in ECs or SMCs without affecting tissue development.13,14
Materials and methods
Chemical agents
We obtained chemical agents as follows: tamoxifen from Sigma-Aldrich (Oakville, Ontario, Canada), insulin Lin Bit from Lin Shin Canada (Toronto, Ontario, Canada), and regular insulin Humulin R from Eli Lilly Canada (Toronto, Ontario, Canada).
Generation of mice with conditional knockdown of IR specific to ECs or SMCs
To knock down IR selectively in ECs and SMCs without affecting tissue development or inducing chronic compensatory mechanisms (e.g. overexpression of IGF-1 receptors), we employed the CreERT2-loxP-mediated recombination system.13,14 Mice carrying an IR gene, in which exon 4 was flanked by loxP sites (floxed IR gene), were provided by one of us (C.R.K.). 15 Mice with transgene CreERT2 under the control of EC-specific Tie2 promoter/enhancer (Tie2-Cre) or SMC-specific myosin heavy chain promoter/enhancer (SMMHC-Cre) were provided by Dr. Stefan Offermanns.13,14 By crossbreeding them, we generated heterozygous Tie2-Cre or SMMHC-Cre-positive mice carrying a homozygous floxed IR gene (Cre[+]-IRf/f mice), and Cre-negative littermate mice carrying IRf/f (Cre[−]-IRf/f mice) as Cre-negative controls. In a subset of experiments, heterozygous Tie2-Cre or SMMHC-Cre-positive mice with wild-type IR gene (Cre[+]-IRw/w mice) were used as Cre-positive controls.
Animal models
All procedures were approved by the Animal Care Committee of the University of Toronto. All invasive procedures were conducted under general anesthesia using isoflurane. The mice were housed in the Department of Comparative Medicine at the University of Toronto. Male mice were used as the SMMHC gene is carried by the Y chromosome.
Cre(+) and Cre(−) mice at 5 weeks of age received intraperitoneal injections of tamoxifen (1 mg/day, dissolved in corn oil) for five consecutive days to induce recombination mediated by CreERT2-loxP in Cre(+) mice.13,14 Nine days after the final tamoxifen injection, mice were assigned to insulin pellet or sham (control) implantation treatment as in our previous study. 6 The insulin pellets used in the present study (one per mouse) have been shown to release insulin at a rate of 0.1 U/day for more than 30 days after subcutaneous implantation, which increases plasma insulin levels by approximately six-fold. 6 To avoid hypoglycemia, insulin-treated mice were given a subcutaneous injection of 0.12 mg glucagon in saline and intraperitoneal injection of 0.25 ml 50% glucose solution in water, and drinking water was replaced with 40% glucose water throughout the experimental period. 6 We have previously shown this not to interfere with neointimal growth. 5 Two days after the implantation, mice underwent femoral artery wire injury as described previously. 6 Briefly, the left femoral artery and its muscle branch were carefully isolated from surrounding connective tissues. A straight-spring guide wire (C-SF-15-15; Cook Medical, Bloomington, IN, USA) was retrogradely inserted into the femoral artery through a cut made on the muscle branch, and withdrawn after 1 min. Twenty-eight days after arterial injury, the mice were sacrificed to collect vessel samples.
Blood glucose measurements
Fasting and random plasma glucose levels were assessed once a week after the wire injury. For the measurement of fasting plasma glucose levels, mice had restricted access to chow overnight, but were allowed 40% glucose water to avoid hypoglycemia. Blood samples were obtained from tail prick, and plasma glucose levels were measured using a glucometer (OneTouch UltraMini Blood Glucose Meter; LifeScan Canada Ltd., Burnaby, British Columbia, Canada).
Morphological analysis
Injured and uninjured femoral arteries were collected following perfusion fixation with PBS and 10% neutral formalin. Injured femoral arteries were divided into proximal and distal parts by cutting the artery midsection, and cross-sections taken from each part were stained with elastic Van Gieson (EVG) or picrosirius red (PSR).4,6 Neointimal and medial area were defined as the area between the lumen and internal elastic lamina and the area between the internal and external elastic laminas, respectively (Supplemental Figure 1). The ratio of intima to media (I/M ratio) was calculated by dividing neointimal area by medial area. The cross-sections were analyzed using a computer-assisted morphometric system (NIS-Elements BR 3.0; Nikon Corporation, Minato, Tokyo, Japan) by an investigator blinded to genotype and treatment. 6 The average of two cross-sections was used as the single value of each mouse.
Immunohistochemistry and immunofluorescence
For immunohistochemistry, cross-sections taken from paraffin-embedded femoral arteries were treated with BOND Epitope Retrieval Solution 2 (Leica Biosystems, Nussloch, Germany) for 10 min at 100°C, incubated with anti-IRβ antibody (Santa Cruz, Dallas, TX, USA, catalog number sc-711, RRID: AB_631835, 1/100 dilution) for 1 h at room temperature, then stained with the secondary antibody (Leica Biosystems, Refine Anti-Rabbit IgG Polymer kit, catalog number DS 9800) for 10 min at room temperature. For immunofluorescence, cross-sections were treated with BOND Epitope Retrieval Solution 2 for 10 min at 100°C or TRIS-EDTA (pH 9.0) for 7 min in a pressure cooker and additional 10 min at room temperature. Subsequently, the cross-sections were incubated with the primary antibodies under the indicated conditions, and stained with the secondary antibodies for 1 h at room temperature. The following antibodies were used as primary antibodies: anti-IRβ antibody (Santa Cruz, catalog number sc-711, RRID: AB_631835, 1:100 dilution, 2-h incubation at room temperature); anti-α-SMA antibody (Abcam, Cambridge Biomedical Campus, Cambridge, UK, catalog number ab202295, RRID: AB_ 2890884, 1:300 dilution, 90-min incubation at room temperature); anti-α-CD31 antibody (Abcam, catalog number ab28364, RRID: AB_726362, 1:100 dilution, overnight incubation at 4°C); anti-Ki-67 antibody (Abcam, catalog number ab16667, RRID: AB_302459, 1:150 dilution, 2-h incubation at room temperature); anti-CD45 antibody (Biolegend, San Diego, CA, USA, catalog number 103102, RRID: AB_312967, 1:500 dilution, 2-h incubation at room temperature). The secondary antibodies used were: anti-Rat IgG + Alexa Fluor 555, catalog number A21434, 1:200 dilution, from Invitrogen; anti-Rabbit IgG + Alexa Fluor 488, catalog number ab150081, 1:200 dilution, from Abcam; anti-Rabbit IgG + Alexa Fluor 555, catalog number A21428, 1:200 dilution, from Invitrogen; anti-Rabbit IgG + Alexa Fluor 647, catalog number A21247, 1:200 dilution, from Invitrogen. The specificity of staining was checked on serial sections using IgG isotype controls (Invitrogen, Waltham, MA, USA, catalog number 08-6199), instead of the primary antibodies (Supplemental Figures 2 and 3). Nuclei were stained with DAPI (Sigma-Aldrich, Catalog number D9542, 100 ng/ml). Stained cross-sections were digitized using Olympus VS-120 slide scanner (Olympus, Shinjuku, Tokyo, Japan), and analyzed using OlyVIA software (Olympus) and Image J software (National Institutes of Health, Bethesda, MD, USA).
Insulin tolerance test
In a subgroup of mice that did not receive insulin treatment, insulin tolerance test was conducted 6 weeks after the final injection of tamoxifen, which mimics the time of the assessment of neointimal growth. However, this subgroup was not subjected to femoral artery wire injury to avoid the influence of surgical stress. After 6 h of fasting, mice received an intraperitoneal injection of regular insulin (0.8 U/kg). 16 Plasma glucose levels were assessed 0, 30, and 60 min after the insulin injection.
Statistical analysis
Data are shown as means ± SD. Unpaired t-test and one-way ANOVA followed by Tukey’s test were used for comparisons of two groups and more than two groups, respectively. Statistical calculation was conducted using the JMP software (version 13; SAS Institute Inc., NC, USA). The significance level was defined as p < 0.05.
Results
Effects of EC-specific IR knockdown on insulin’s anti-restenotic effect
First, we assessed IR expression in ECs and SMCs of the uninjured femoral arteries in tamoxifen-treated Tie2-Cre(+)-IRf/f and Tie2-Cre(−)-IRf/f mice. Immunohistochemistry for IR revealed that Tie2-Cre(+)-IRf/f mice had a decrease in IR expression in ECs, but not in SMCs, compared with Tie2-Cre(−)-IRf/f mice (Figure 1(a)–(c)). This was confirmed using immunofluorescence for IR (Figure 2(a)–(d)). Positive and negative controls for both IHC and IF are shown in Supplemental Figures 2 and 3.
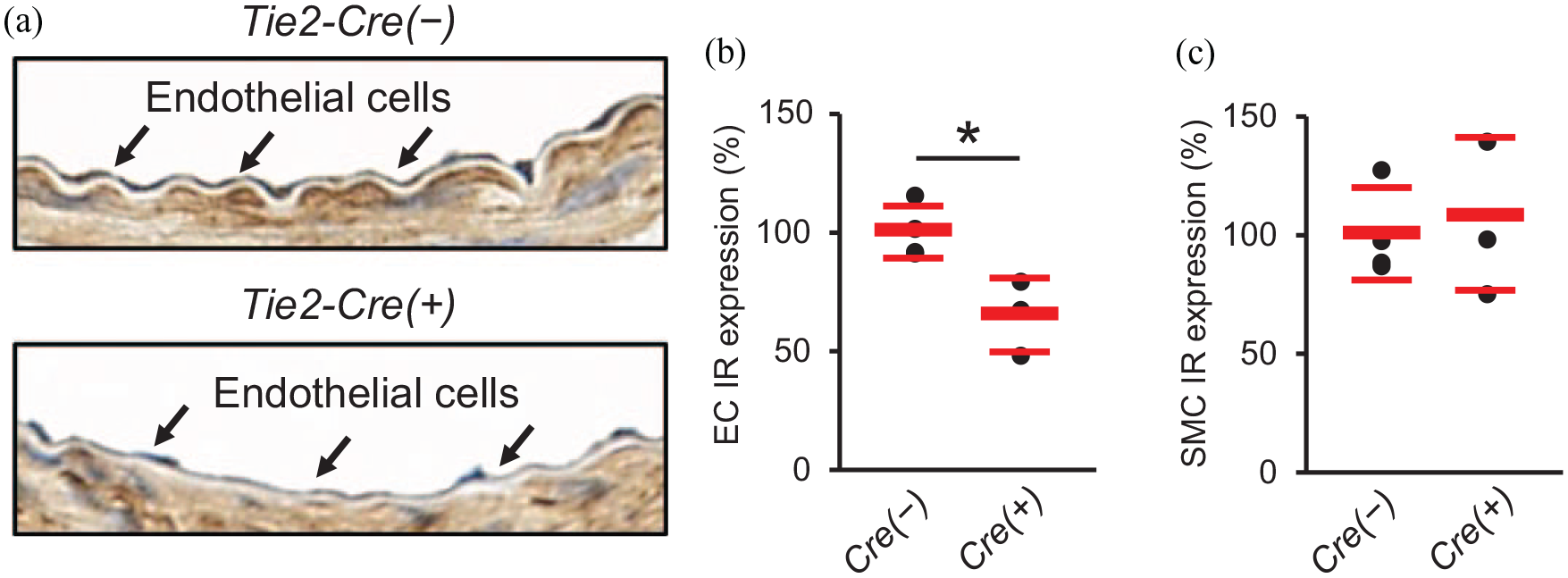
Effects of EC-specific IR knockdown on vascular expression of insulin receptor in immunohistochemical analysis. Uninjured femoral arteries were collected to assess IR expression in ECs and SMCs by immunohistochemistry. (a) Representative immunohistochemistry images for IR (×800). (b) Percentage reduction of IR in endothelial cells. (c) Percentage reduction of IR in smooth muscle cells.

Effects of EC-specific IR knockdown on vascular expression of insulin receptor in immunofluorescence analysis. Uninjured femoral arteries were collected to assess IR expression in ECs and SMCs by immunofluorescence. (a) Representative immunofluorescence images for IR and CD31 (×2000). (b) Representative immunofluorescence images for IR and α-SMA (×400). (c) Percentage reduction of IR in endothelial cells. (d) Percentage reduction of IR in smooth muscle cells. n = 3 per group.
Subsequently, we evaluated the effect of insulin on neointimal growth after arterial injury in tamoxifen-treated Tie2-Cre(+)-IRf/f and Tie2-Cre(−)-IRf/f mice. Metabolic parameters are presented in Supplemental Table 1. There were no differences in food intake, water intake, or body weight. Fasting plasma glucose levels were significantly lower in the insulin treatment groups than in the control groups as expected. Conversely, plasma glucose levels obtained in the random fed state, which represents the physiological state of mice, were comparable between the groups, indicating that hypoglycemia was not responsible for the effect of insulin as previously shown. 5
Representative EVG-stained images of uninjured and injured arteries are shown in Supplemental Figure 1. The degree of neointimal formation is consistent with previous publications in which the wire injury was conducted by the same investigator (YM).17–20 The morphological analysis of injured femoral arteries revealed that there was no difference in neointimal area and intima/media (I/M) ratio between Tie2-Cre(−)-IRf/f and Tie2-Cre(+)-IRf/f mice (Figure 3(a)–(c)) in the absence of insulin treatment. In Tie2-Cre(−)-IRf/f mice, insulin treatment decreased neointimal area (Figure 3(b); means ± SD; Control, 0.019 ± 0.004 mm2, Insulin, 0.013 ± 0.003 mm2, p = 0.02) and I/M ratio (Figure 3(c); means ± SD; Control, 1.57 ± 0.58, Insulin, 0.82 ± 0.22, p < 0.001). In contrast, insulin treatment changed neither the neointimal area nor the I/M ratio in Tie2-Cre(+)-IRf/f mice (Figure 3(a)–(c); means ± SD; neointimal area: Control, 0.019 ± 0.005 mm2, Insulin, 0.017 ± 0.005 mm2, p = 0.72; I/M ratio: Control, 1.46 ± 0.46, Insulin, 1.21 ± 0.41, p = 0.44). In a subset of animals, we evaluated whether Tie2-Cre expression per se affected the effect of insulin on neointimal formation. Insulin treatment reduced the neointimal area and the I/M ratio in Tie2-Cre(+)-IRw/w mice, similar to our observations in Tie2-Cre(−)-IRf/f mice (Supplemental Figure 4(a)–(c)).
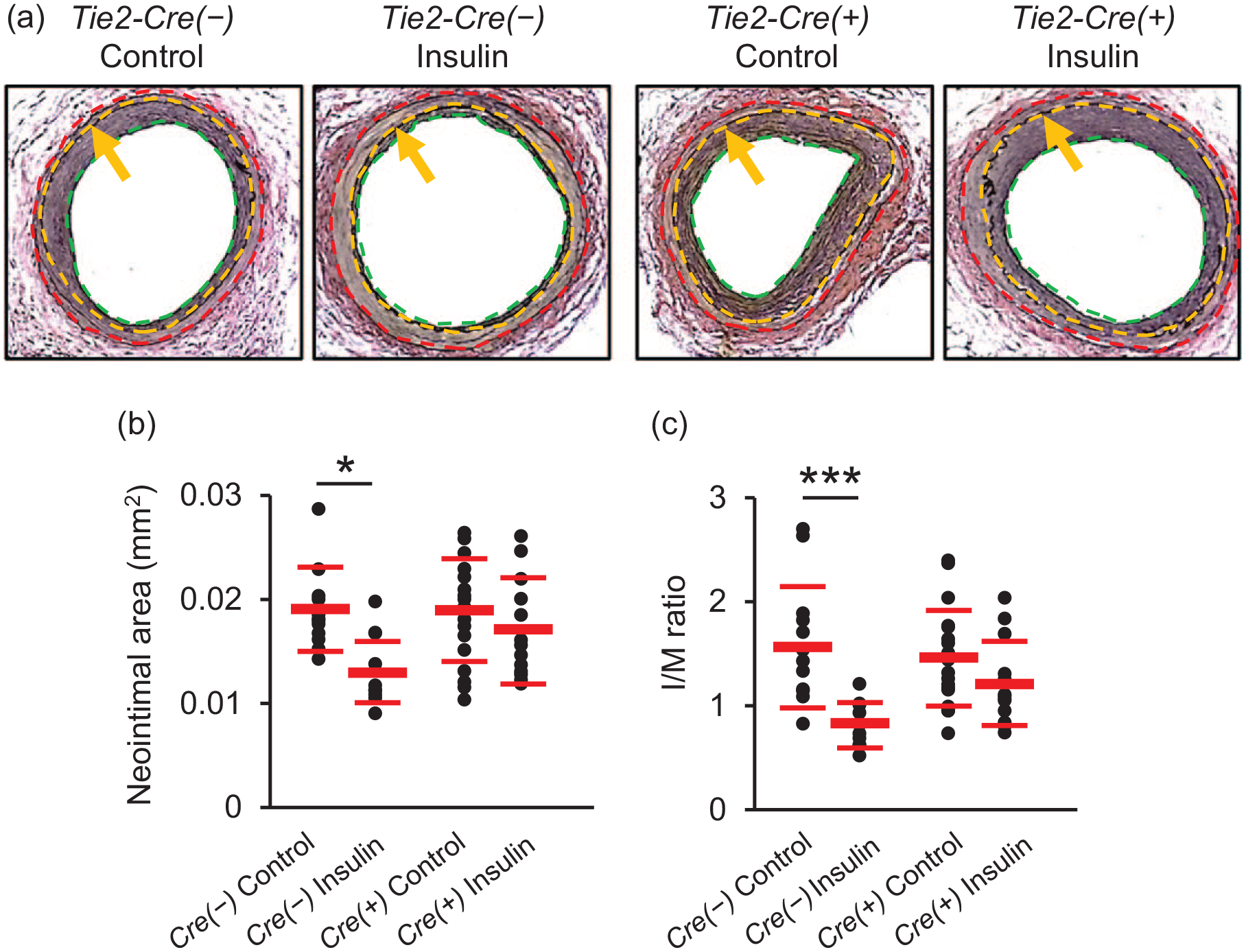
Effects of EC-specific IR knockdown on insulin’s “anti-restenotic” effect. Injured femoral arteries were collected 28 days after wire injury. (a) Representative images of femoral arteries stained with EVG (×200). Green, yellow, and red dotted lines show lumen, internal elastic lamina, and external elastic lamina, respectively. Arrows also show internal elastic lamina. Neointima is the area between green and yellow dotted lines and media is the area between yellow and red dotted lines. (b) Neointimal area. (c) I/M ratio.
We further assessed characteristics of the neointima in Tie2-Cre(−)-IRf/f and Tie2-Cre(+)-IRf/f mice. Insulin treatment significantly increased neointimal expression of α-SMA, a marker of SMC differentiation, in both control mice (Tie2-Cre(−)-IRf/f and Tie2-Cre(+)-IRw/w, Figure 4(a) and (b), Supplemental Figure 4(d) and (e)) and Tie2-Cre(+)-IRf/f mice (Figure 4(a) and (b)), whereas insulin treatment did not affect neointimal expression of ki-67, a marker of cell proliferation, in either Tie2-Cre(−)-IRf/f or Tie2-Cre(+)-IRf/f mice (Figure 4(a) and (d)). In addition, we evaluated characteristics of the neointima. Cell density (DAPI positive nuclei/neointimal area) was not affected by insulin in either group (Supplemental Figure 5(a)). Insulin treatment increased the percentage of collagen-positive area, effect abolished by EC-specific IR knockdown (Figure 5(a) and (b)). Few or no cells in the neointima were positive for CD45 in either Tie2-Cre(−)-IRf/f or Tie2-Cre(+)-IRf/f mice (Supplemental Figure 6(c)).
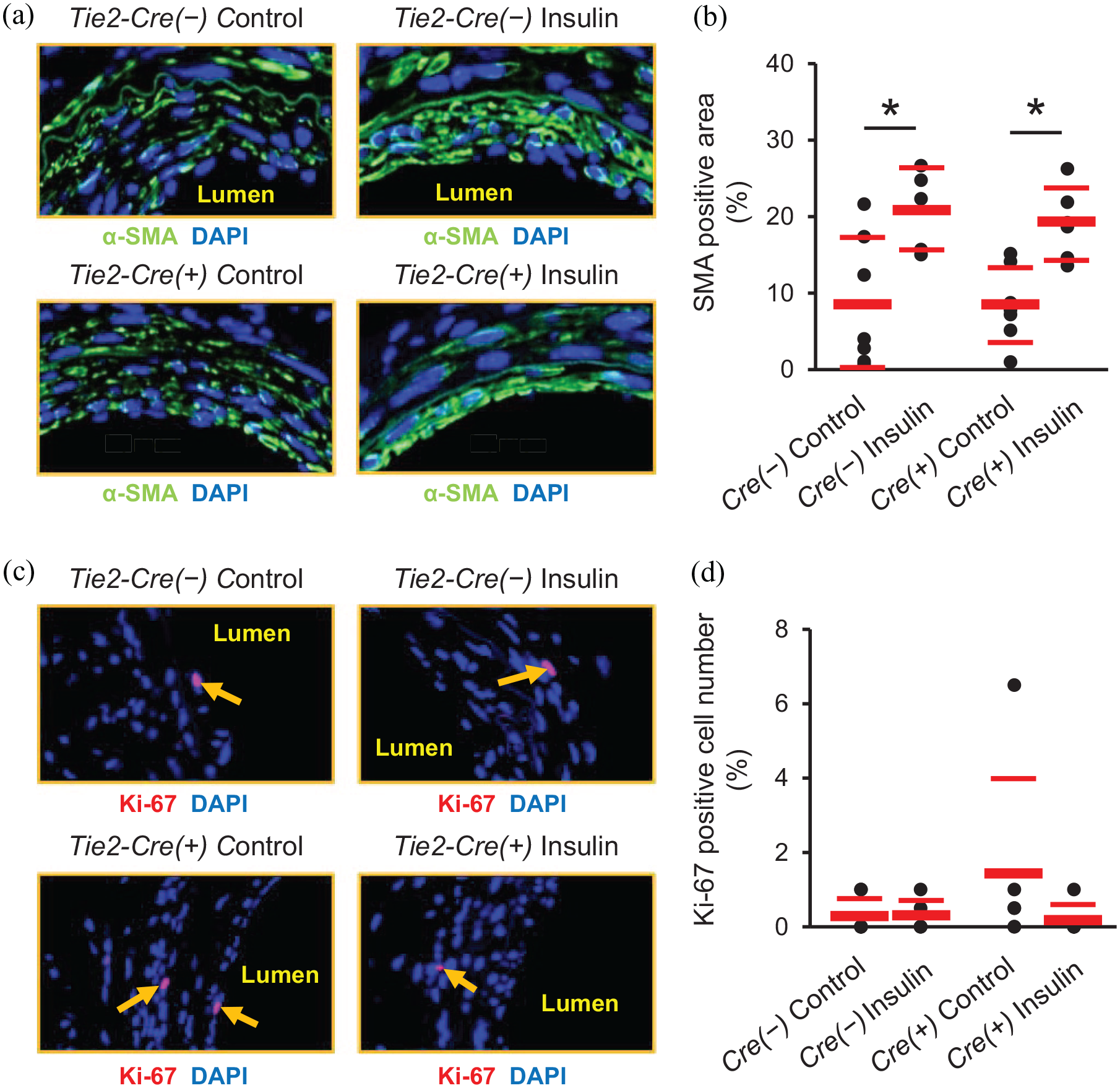
Effects of EC-specific IR knockdown on SMA differentiation and cellular proliferation in neointima. Representative immunofluorescence images of injured femoral arteries stained for α-SMA (a) and ki-67 (b). Magnification: (a), ×1600; (b), ×1200. α-SMA, green; ki-67, red; DAPI, blue. Arrows show ki-67 positive cells. (c) Percentage α-SMA positive area of neointimal area. (c) Percentage ki-67 positive cells of total cells in neointima. n = 3–8 per group.
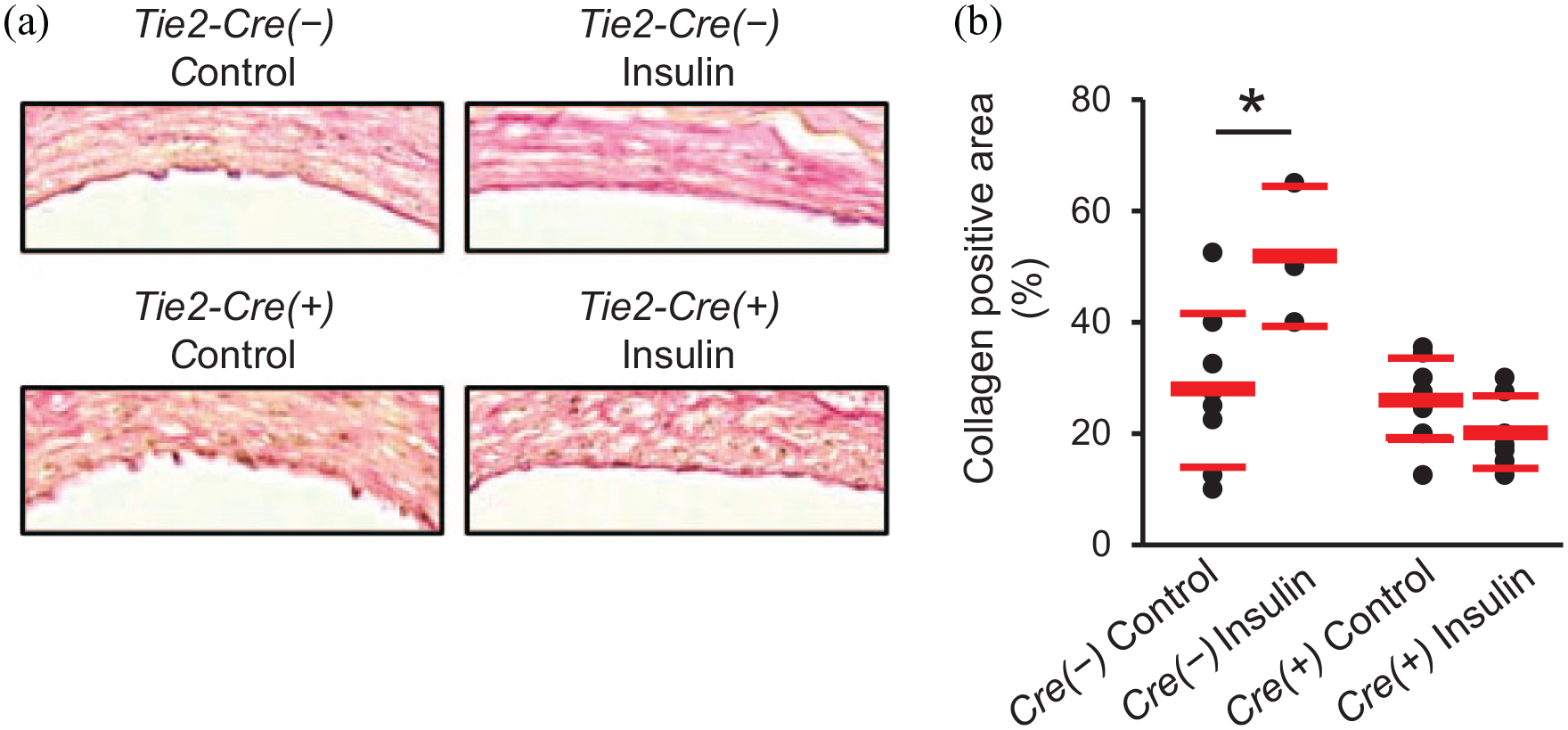
Effects of EC-specific IR knockdown on percentage collagen positive area in neointima. (a) Representative images of injured femoral arteries stained with PSR (×1600). (b) Percentage collagen positive area in neointima. n = 3–8 per group.
Effects of SMC-specific IR knockdown on insulin’s anti-restenotic effect
We assessed IR expression in ECs and SMCs of the uninjured femoral arteries in tamoxifen-treated SMMHC-Cre(+)-IRf/f mice and SMMHC-Cre(−)-IRf/f mice. Immunohistochemical analysis showed that IR expression in SMCs was lower in SMMHC-Cre(+)-IRf/f mice than SMMHC-Cre(−)-IRf/f mice, whereas IR expression in ECs was similar between these mice (Figure 6(a)–(c)). Immunofluorescence for IR showed similar changes to those of immunohistochemistry (Figure 7(a)–(d)).
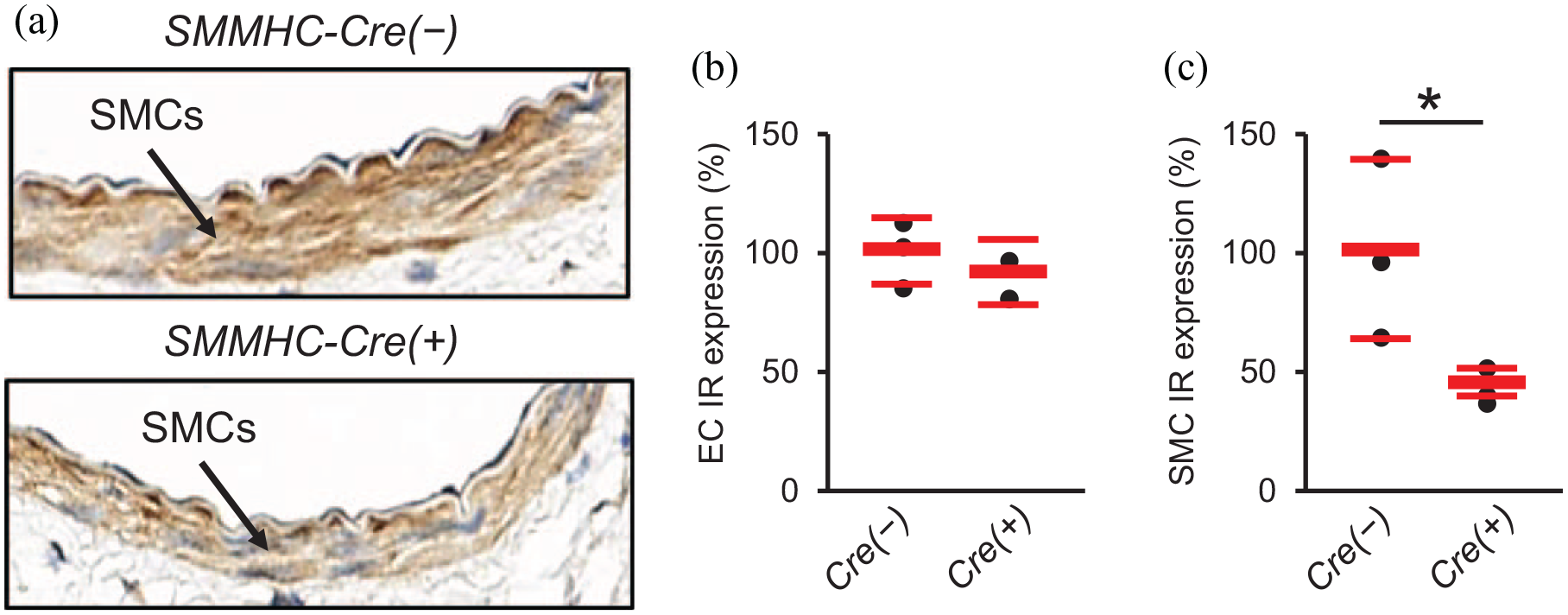
Effects of SMC-specific IR knockdown on vascular expression of insulin receptor in immunohistochemical analysis. Uninjured femoral arteries were collected to assess IR expression in ECs and SMCs by immunohistochemistry. (a) Representative immunohistochemistry images for IR (×800). (b) Percentage reduction of IR in endothelial cells. (c) Percentage reduction of IR in smooth muscle cells.
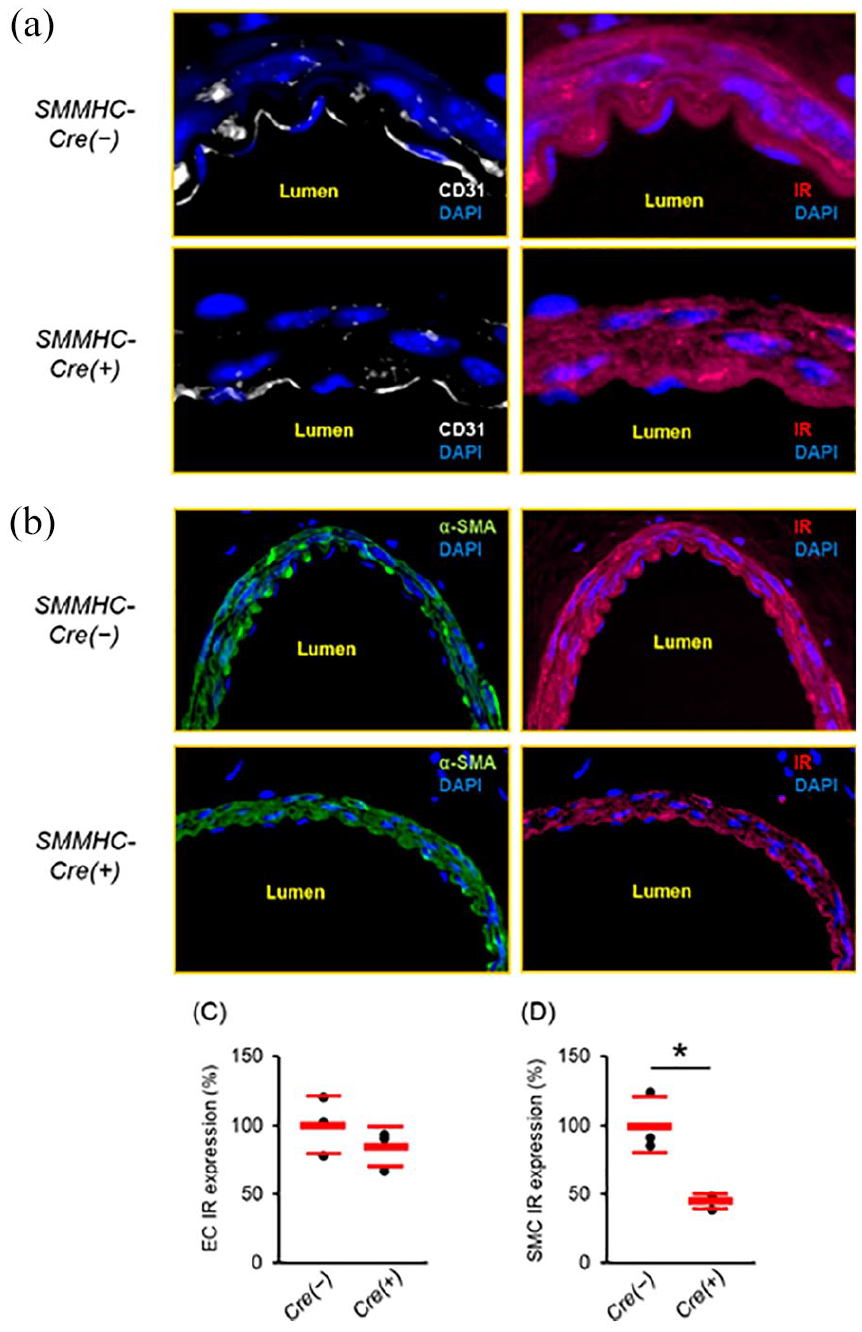
Effects of SMC-specific IR knockdown on vascular expression of insulin receptor in immunofluorescence analysis. Uninjured femoral arteries were collected to assess IR expression in ECs and SMCs by immunohistochemistry and immunofluorescence. (a) Representative immunofluorescence images for IR and CD31 (×2000). (b) Representative immunofluorescence images for IR and α-SMA (×400). (c) Percentage reduction of IR in endothelial cells. (d) Percentage reduction of IR in smooth muscle cells. n = 3 per group.
Metabolic parameters of SMMHC-Cre(−)-IRf/f and SMMHC-Cre(+)-IRf/f mice are shown in Supplemental Table 2. Food intake, water intake, and body weight were similar between the groups. Fasting plasma glucose levels were decreased by insulin treatment in SMMHC-Cre(−)-IRf/f mice and tended to be decreased in SMMHC-Cre(+)-IRf/f mice, whereas random fed plasma glucose levels were similar between the control and insulin groups.
Next, we evaluated the effect of insulin on neointimal growth after arterial injury in mice with SMC-specific IR knockdown. Neointimal area appeared to be greater in SMMHC-Cre(+)-IRf/f mice than SMMHC-Cre(−)-IRf/f mice in the absence of insulin treatment (Figure 8(a)–(c)), but there was no significant difference. I/M ratio was unchanged. In insulin-treated SMMHC-Cre(−) mice, the neointimal area and I/M ratio were lower than those in mice receiving control treatment (Figure 8(b) and (c); means ± SD; neointimal area: Control, 0.018 ± 0.004 mm2, Insulin, 0.011 ± 0.004 mm2, p = 0.03; I/M ratio: Control, 1.50 ± 0.36, Insulin, 0.74 ± 0.33, p < 0.001). Unexpectedly, insulin treatment failed to decrease neointimal area and I/M ratio in SMMHC-Cre(+)-IRf/f mice (Figure 8(b) and (c); means ± SD; neointimal area: Control, 0.024 ± 0.008 mm2, Insulin, 0.020 ± 0.008 mm2, p = 0.73; I/M ratio: Control, 1.36 ± 0.41, Insulin, 1.50 ± 0.49, p = 1.00). In contrast, insulin treatment reduced the neointimal area and the I/M ratio in SMMHC-Cre(+)-IRw/w mice as observed in SMMHC-Cre(−)-IRf/f mice (Supplemental Figure 7(a)–(c)).
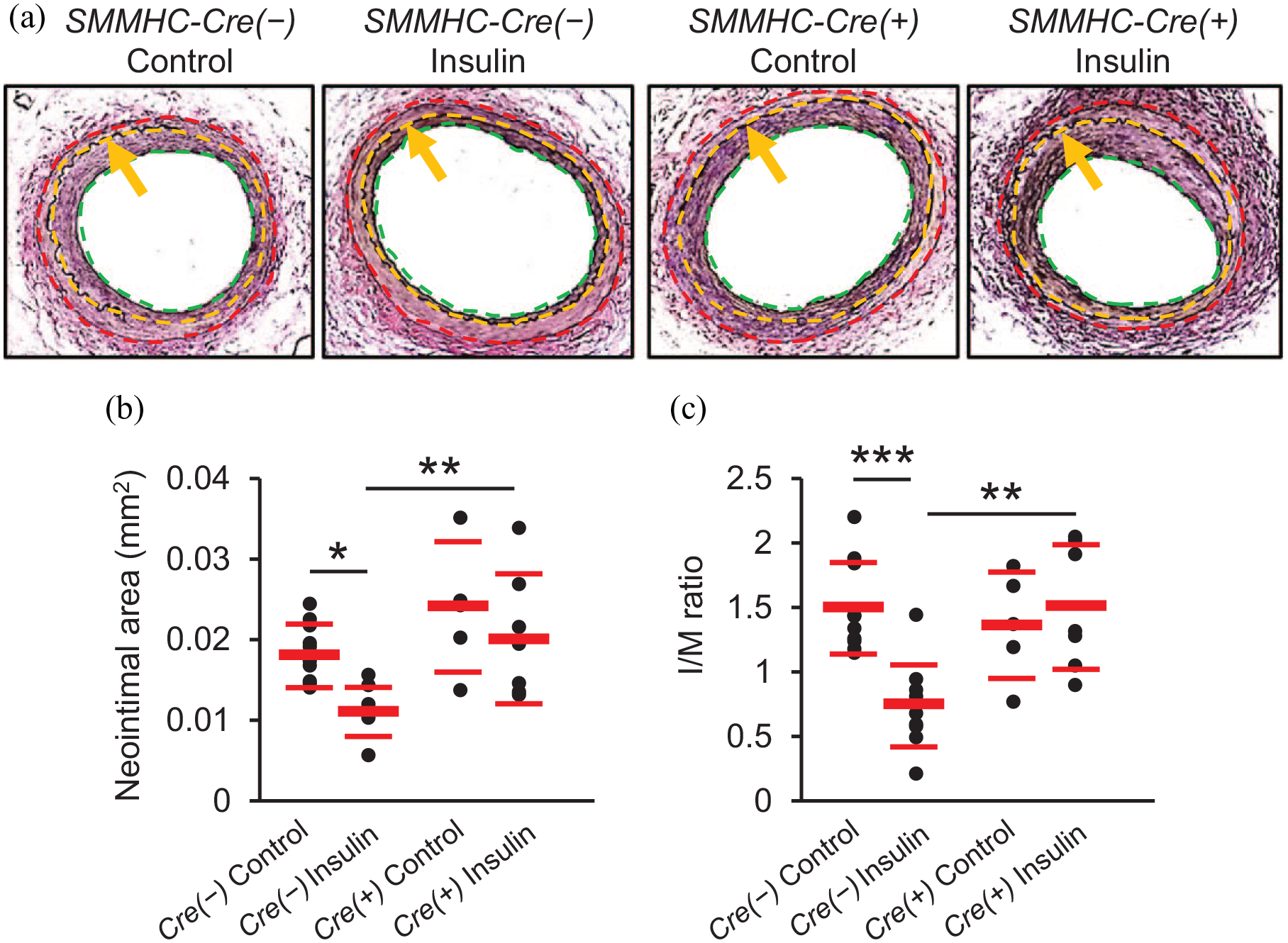
Effects of SMC-specific IR knockdown on insulin’s “anti-restenotic” effect. Injured femoral arteries were collected 28 days after wire injury. (a) Representative images of femoral arteries stained with EVG (×200). Green, yellow, and red dotted lines show lumen, internal elastic lamina, and external elastic lamina, respectively. Arrows also show internal elastic lamina. Neointima is the area between green and yellow dotted lines and media is the area between yellow and red dotted lines. (b) Neointimal area. (c): I/M ratio.
Insulin treatment significantly increased α-SMA expression in the neointima of control mice (SMMHC-Cre(−)-IRf/f and SMMHC-Cre(+)-IRw/w; Figure 9(a) and (b) and Supplemental Figure 7(d) and (e)) and this effect of insulin was not observed in SMMHC-Cre(+)-IRf/f mice (Figure 9(a) and (b)). Compared with SMMHC-Cre(−)-IRf/f mice, neointimal expression of ki-67 was increased in SMMHC-Cre(+)-IRf/f mice and in these mice with SMC knockdown but intact EC IR, ki-67 expression was suppressed by insulin treatment (Figure 9(c) and (d)). Cell density was not affected by insulin in either group (Supplemental Figure 5(b)). The effect of insulin treatment to increase the collagen positive area was not seen in mice with SMC-specific IR knockdown (Figure 10(a) and (b)). CD45-positive cells were not or were rarely observed in the neointima of either SMMHC-Cre(−)-IRf/f or SMMHC-Cre(+)-IRf/f mice (Supplemental Figure 6(d)).
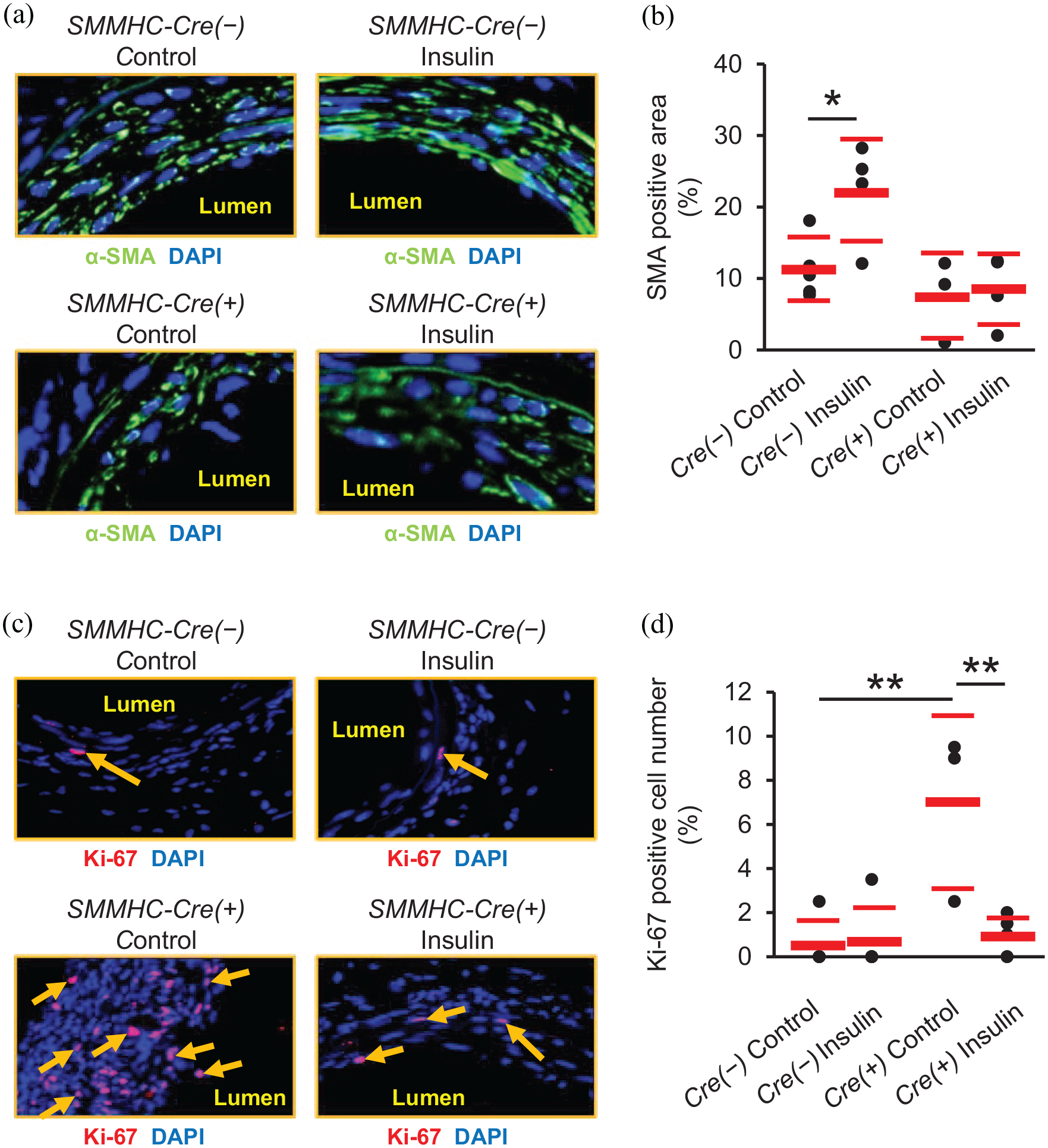
Effects of SMC-specific IR knockdown on SMA differentiation and cellular proliferation in neointima. Representative immunofluorescence images of injured femoral arteries stained for α-SMA (a) and ki-67 (c). Magnification: (a), ×1600; (b), ×1200. α-SMA, green; ki-67, red; DAPI, blue. Arrows show ki-67 positive cells. (c) Percentage α-SMA positive area of neointimal area. (d): Percentage ki-67 positive cells of total cells in neointima. n = 3–6 per group.
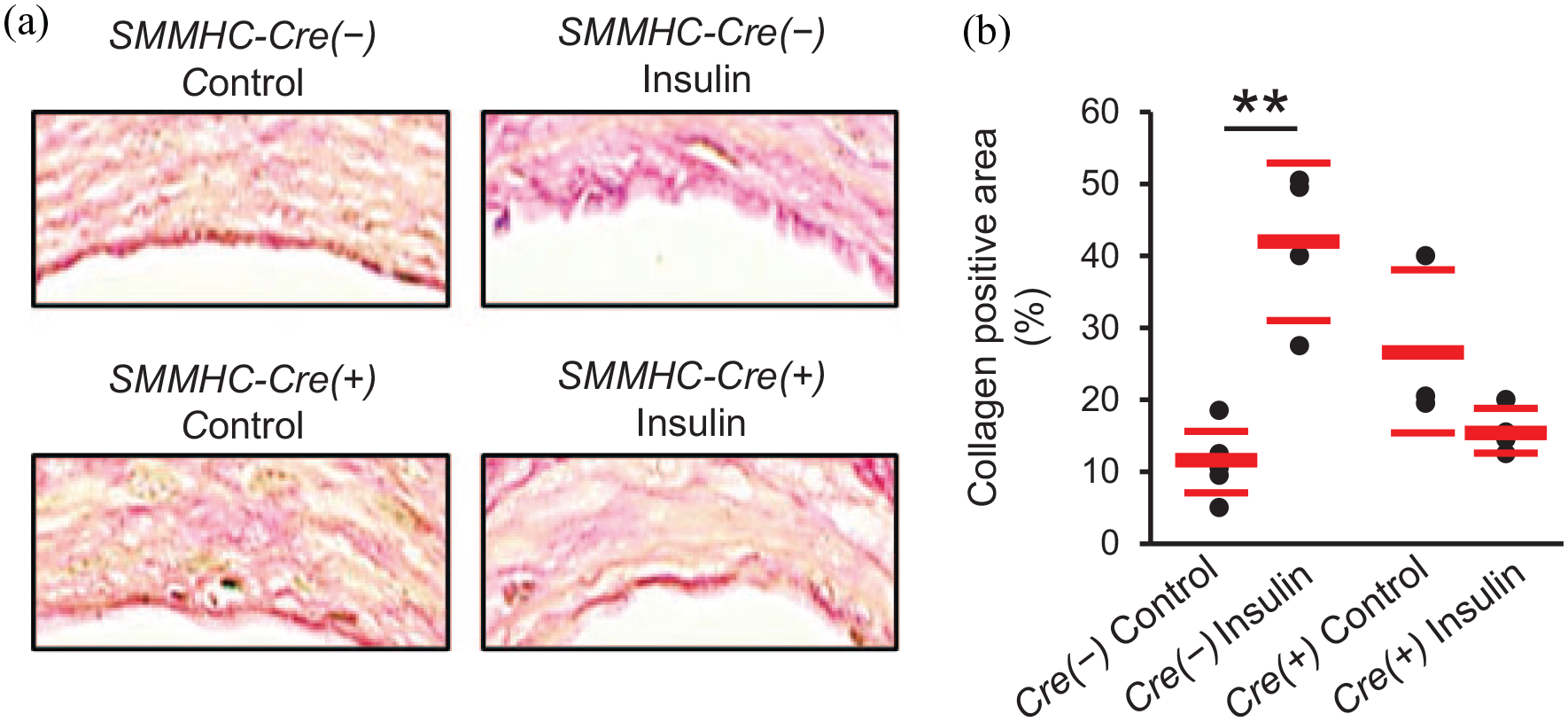
Effects of SMC-specific IR knockdown on percentage collagen positive area in neointima. (a) Representative images of injured femoral arteries stained with PSR (×1600). (b) Percentage collagen positive area in neointima. n = 3–6 per group.
Effects of EC- or SMC-specific IR knockdown on insulin sensitivity
To examine potential systemic effects of EC- or SMC-specific IR knockdown, insulin tolerance test was conducted in a subset of mice that were not subjected to the wire injury. There was no difference in plasma glucose levels after the intraperitoneal injection of insulin between Tie2-Cre(−)-IRf/f and Tie2-Cre(+)-IRf/f mice (Supplemental Figure 8(a)). Plasma glucose levels after the insulin injection were not significantly different also between SMMHC-Cre(−)-IRf/f and SMMHC-Cre(+)-IRf/f mice (Supplemental Figure 8(b)).
Discussion
In the present study, we demonstrated that, in a mouse model of restenosis, the protective effect of insulin against neointimal hyperplasia was abolished by IR knockdown in either ECs or SMCs. Although the deletion of IR in ECs and SMCs was incomplete, the models achieved sufficient IR deficiency to generate differences in neointimal growth in response to insulin treatment as compared to controls. Conditional (i.e. inducible) gene knockout models often achieve only partial deletion compared to non-inducible models; however, inducible models are not susceptible to potential developmental effects of IR deficiency and are less susceptible than non-inducible models to potential compensatory upregulation of IGF-1 receptors. 21
Vascular SMCs mainly play a facilitative role in the process of neointimal hyperplasia via phenotypic switching from differentiated to de-differentiated phenotype, which is prone to migration and proliferation. 9 Conversely, ECs play a primarily protective role via production of nitric oxide (NO), which inhibits migration and proliferation of SMCs. 22 Insulin has been shown to stimulate NO production via the phosphatidylinositol 3 (PI3K)/Akt pathway in cultured ECs. 10 We previously reported that insulin treatment suppressed neointimal hyperplasia, which was abolished by a NOS inhibitor in rats and by genetic ablation of eNOS in mice, indicating that NO produced from ECs is a key mediator of the vasculoprotective effect of insulin. 6 However, insulin also has MAPK-mediated potentially adverse effects on ECs such as the upregulation of endothelin-1, 23 therefore endothelial IR knockdown may not necessarily recapitulate the effect of eNOS knockout. In addition, global eNOS lack in bone marrow precursors, macrophages, and platelets rather than ECs may be responsible for the lack of effect of insulin in the eNOS knockout model. In contrast to ECs, in vitro studies have reported that insulin induces both favorable and unfavorable effects in SMCs. Insulin stimulates proliferation and migration via the MAPK pathway in cultured vascular SMCs,11,12 whereas insulin inhibits growth-factor-induced phenotypic switching of SMC from the quiescent to the proliferative phenotype via the PI3K pathway. 12 The present findings provide evidence that the in vivo insulin action on both ECs and SMCs is mainly beneficial and plays an essential role in insulin’s “anti-restenotic” effect.
In the present study, we found that insulin treatment increased neointimal expression levels of α-SMA, a marker of quiescent, differentiated SMCs, in control mice, which is consistent with our previous report using the balloon injury model in rats. 4 This effect of insulin was similarly observed in mice with EC-specific IR knockdown, whereas it was abolished in mice with SMC-specific IR knockdown. Since de-differentiation of SMC plays an essential role in the pathogenesis of neointimal formation after wire injury, 9 this finding suggests that insulin action on SMC IR decreases neointimal growth via an effect on SMC differentiation. The greater proliferation of SMC in IR deficient SMC mice may suggest that SMC IR also decreases proliferation in vivo. Decreased SMC proliferation in these mice upon insulin treatment may suggest that SMC proliferation could also be decreased by IR in ECs, perhaps via NO. However, proliferation was not evaluated at its peak and was very low and unchanged by insulin in controls. Previously, we observed that insulin either does not affect 4 or increases SMC proliferation in the rat carotid artery balloon injury model, 7 depending on dose. We have also previously shown that insulin stimulates re-endothelialization, 4 which is likely mediated by the EC IR, but the investigation of this would require vessel collection at a much earlier time-point because re-endothelialization is complete at 28 days in this model. 18
In the present study we found that insulin increases the collagen content in the neointima and this was apparently due to IR in both ECs and VSMCs, possibly representing contribution of both receptors to a more mature myofibroblast phenotype. This is in accordance with our previous study on atherosclerosis in mice. 3 In contrast, we found that insulin tended to decrease the collagen content in the carotid artery balloon injury model. 4 However, in that study insulin also decreased cell density, which was unaffected in the present study. Further studies are required to clarify these discrepancies that could be due to the different species or the different vessel and injury model. In accordance with our previous data in rats, 4 we detected few or no cells in the neointima which were positive for CD45, a marker of hematopoietic precursors. Thus, cells in the neointima appear to be mainly derived from medial SMC.
There are several limitations in the present study. First, we employed Tie2-Cre to induce IR knockdown in ECs. Tie2 is a cell-surface receptor for angiopoietins, which are expressed in ECs. 24 However, Tie2 is also expressed in a subpopulation of bone marrow cells and macrophages in addition to ECs. 24 Thus, we cannot exclude potential action of insulin in bone marrow precursors contributing to re-endothelialization 4 or in macrophages,25,26 although macrophage insulin action is unlikely given the lack of positive CD45 cells in the neointima. Second, in contrast to our findings,4–8 a recent study showed that the deletion of IR gene in SMCs lead to the suppression of neointimal hyperplasia in high-fat fed mice. 27 In the present study, we did not observe any insulin-induced acceleration of neointimal hyperplasia regardless of IR expression in ECs or SMCs. As mentioned above, we found that high-dose insulin can induce SMC proliferation also in an in vivo model.5,7 We also found that the proliferative effect of insulin on SMC in vivo is still present in a high fat diet 7 or oral sucrose 5 induced model of insulin resistance, where the effect of insulin to decrease neointimal growth is markedly reduced (but interestingly, not reversed).5,7 The present study was performed in an insulin sensitive model, the chow-fed mouse, but should be extended to models of diabetes and insulin resistance, where insulin treatment is clinically relevant.
In conclusion, we demonstrated that insulin action on both ECs and SMCs is required for the anti-restenotic effect of insulin in vivo. The present findings provide a better understanding of the physiology of the vasculoprotective effects of insulin in an in vivo model. Our results may further imply that insulin treatment has beneficial effects not only on ECs but also on SMCs in patients with diabetes undergoing PCI, at least in those who are not insulin resistant.
Supplemental Material
sj-pdf-1-dvr-10.1177_14791641211027324 – Supplemental material for Roles of vascular endothelial and smooth muscle cells in the vasculoprotective effect of insulin in a mouse model of restenosis
Supplemental material, sj-pdf-1-dvr-10.1177_14791641211027324 for Roles of vascular endothelial and smooth muscle cells in the vasculoprotective effect of insulin in a mouse model of restenosis by Yusaku Mori, Marel Gonzalez Medina, Zhiwei Liu, June Guo, Luke S Dingwell, Simon Chiang, Carl Ronald Kahn, Mansoor Husain and Adria Giacca in Diabetes & Vascular Disease Research
Footnotes
Acknowledgements
We wish to thank Loretta Lam for her excellent technical support and Dr. Stefan Offermanns for his kind gift of the Tie-CreERT2(+) mice and the SMMHC-CreERT2(+) mice.
Author contributions
Conception and design: AG. Acquisition of data: YM, MGM, ZL, SC, LSD, and JG. Analysis of data: YM, MGM, and AG. Drafting the article: YM. All the authors contributed to data interpretation and manuscript revision and approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: YM received a Showa University Research Grant for Young Researchers. AG was supported by a grant from The Heart and Stroke Foundation of Canada (#G-18-0022151).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.