
Abstract
Raynaud’s phenomenon, which is characterized by episodic digital pallor, cyanosis and rubor upon exposure to cold environment or to stress, is relatively common, although the prevalence depends on the climate. Still, it is under-diagnosed, under-treated, and often confused with other conditions. Primary Raynaud’s phenomenon (i.e., Raynaud disease) must be distinguished from secondary Raynaud’s phenomenon (i.e., Raynaud syndrome) as long-term morbidity and outcomes differ vastly between the two conditions. Additionally, the practitioner must differentiate between Raynaud’s phenomenon and related vascular disorders, such as acrocyanosis, pernio, and livedo reticularis. In this article, we review differences between the conditions and suggest an approach to diagnosis and treatment strategy for these disorders.
This opportunity is made possible through a joint partnership with University of Virginia School of Medicine. For instructions, please visit the Society for Vascular Medicine’s website at
The University of Virginia School of Medicine (UVASOM) is accredited by the ACCME to provide continuing medical education for physicians. UVASOM designates this journal-based CME activity for a maximum of one AMA PRA Category 1 Credit™. Participants should claim only the credit commensurate with the extent of their participation in the activity. UVASOM, as an accredited provider, awards 1 hour of participation (consistent with the one AMA PRA Category 1 Credit™) to participants who successfully complete this educational activity. Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to one MOC II point in the American Board of Internal Medicine’s (ABIM) Maintenance of Certification (MOC) program. It is the CME provider’s responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC II credit. UVASOM maintains a record of participation for 6 years.
The faculty, staff, and planning committee of the University of Virginia Office of Continuing Medical Education have no financial affiliations to disclose. The CME planning committee disclosed the following: Aditya Sharma is supported by National Institutes of Health, AstraZeneca, and Vascular Medcure; Heather Gornik and Valerie Clark have no financial affiliations to disclose. The authors have disclosed no financial relationships or interests with a proprietary entity that produces healthcare goods or services related to this article.
Introduction
Raynaud’s phenomenon (RP) is a relatively common condition with an overall prevalence of 1–8% in the general population, with higher prevalence in the colder climates. 1 It is important to distinguish RP from other disorders that cause digital discoloration, including acrocyanosis, pernio, and livedo reticularis (LR), as prognosis and treatment differ between conditions. The evaluation of an underlying cause of vasospastic disorders is challenging due to the usually gradual onset of symptoms. Not all conditions associated with digital vasospasm are benign and some are related to systemic conditions that may require urgent treatment. RP, acrocyanosis, and pernio are generally characterized by discoloration in hands or feet, with or without paresthesia. On the other hand, LR may involve lower extremities, the abdomen, or the chest. While vasospasm implies a reversible constriction of vasculature, ischemic changes seen in some of these conditions may be episodic, persistent or persistent with episodic attacks of a more severe nature depending on their etiology. 2 In this review, we discuss the pathophysiology, diagnosis, and treatment strategies of these disorders that are commonly encountered in vascular clinical practice.
Raynaud’s phenomenon
Raynaud’s phenomenon, named after the French physician Maurice Raynaud (1834–1881), is caused by deregulated constriction of precapillary arterioles that leads to changes in skin color, swelling, and paresthesia. It generally affects the fingers and toes, but can also affect other sites such as the nose, ears, and nipples. Primary (or idiopathic) RP, termed Raynaud disease, has a relatively benign course. Conversely, secondary RP, termed Raynaud syndrome, occurs as a consequence of a systemic disease process and may include digital pitting, ulceration or dry gangrene. 3 Raynaud syndrome is indeed a syndrome of signs and symptoms related to many possible etiologies. The secondary causes include connective tissue disorders, arterial occlusive disorders, neurological disorders, compression disorders, trauma, drugs, hematologic malignancy and others (Table 1). In a study of 307 RP patients, the transition rate from primary to secondary RP was 1% per year. 4 However, the participants were primarily patients with moderate to severe symptoms who were referred to a specialty clinic and may not represent the general population. In a retrospective study of 497 patients with primary RP in whom nailfold capillaroscopy, anti-centromere, and anti-topoisomerase I antibodies were available, 32% of patients evolved to systemic sclerosis and 49% evolved to other connective tissue disorders over the next 2.3 ± 2.9 years of follow-up. 5 Scleroderma pattern on capillaroscopy was the most sensitive marker of scleroderma development, while absence of anti-topoisomerase I antibodies was the most specific finding. In a prospective study of 586 patients with RP, 12.6% developed systemic sclerosis during the median follow-up period of 4 years. 6 An abnormal capillaroscopy finding at baseline with presence of systemic scleroderma-specific antibodies (anti-centromere, anti-topoisomerase I, anti-Th/To, anti-RNA polymerase I/III antibodies) indicated high probability of progression to systemic sclerosis.
Secondary causes of Raynaud’s Phenomenon. 118
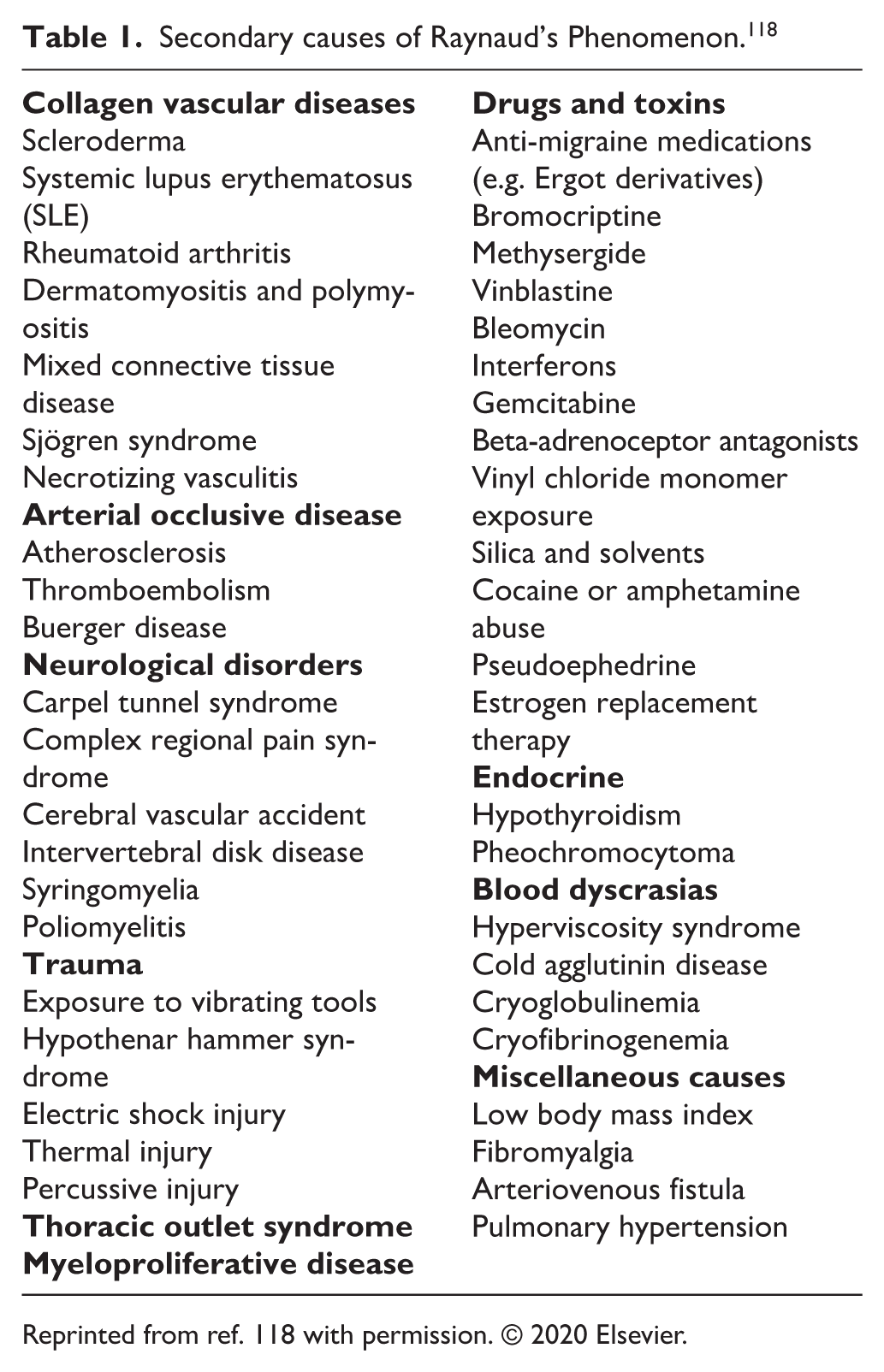
Reprinted from ref. 118 with permission. © 2020 Elsevier.
Clinical presentation
The typical manifestations of RP include a characteristic ‘triphasic’ color pattern, numbness, and swelling in affected digits. 7 The three color phases are pallor caused by vasospasm-induced ischemia, cyanosis caused by tissue hypoxia, followed by rubor caused by tissue reperfusion (Figure 1). It is important to note that some patients only show biphasic response with pallor and cyanosis. The color changes are generally well demarcated. RP can be precipitated by exposure to cold, emotional stress, medications, trauma, and repetitive use of digits. Smoking is associated with higher prevalence of RP. 8
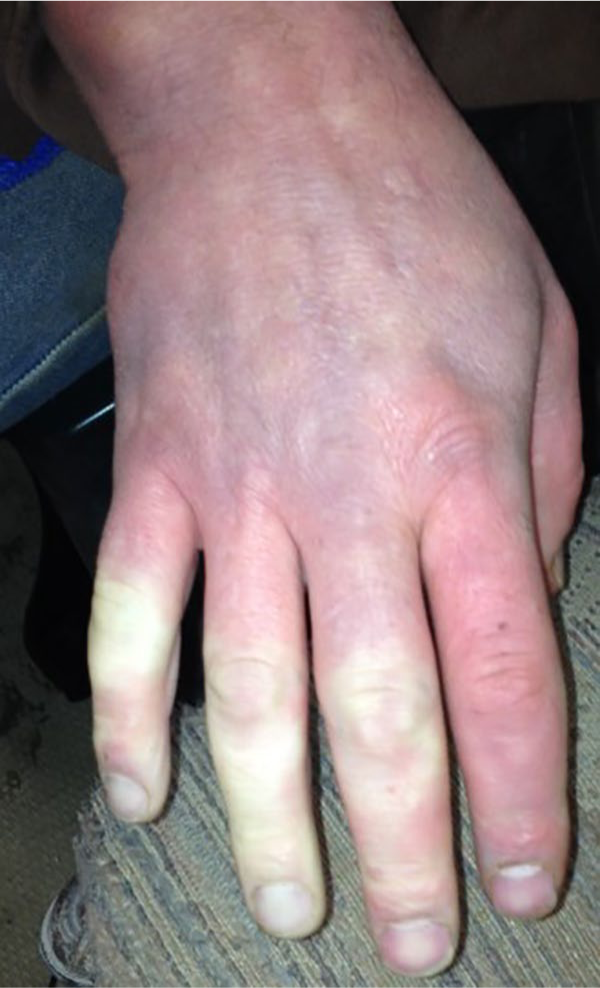
Color change associated with Raynaud’s phenomenon. The third, fourth, and fifth digits are exhibiting pallor associated with Raynaud’s phenomenon.
Epidemiology
Primary RP affects up to 8% of the general population in the United States and is more common in women (7.8% in women vs 5.8% in men). 1 Across different countries, the lowest prevalence was seen in Japan (2.1% in women, 1.1% in men) and the highest prevalence was seen in France (11.7% in women, 6.3% in men).1,9,10 Cold climates are associated with higher prevalence of RP.10,11 Symptoms generally begin before the fourth decade of life; symptom onset later in life should alert the provider to look for secondary causes of RP. Family history may be helpful as more than a third of individuals with RP report a relative with RP.12,13 However, a study exploring an association between known polymorphisms of vasoactive mediator genes and RP found no significant association. 14
Pathophysiology
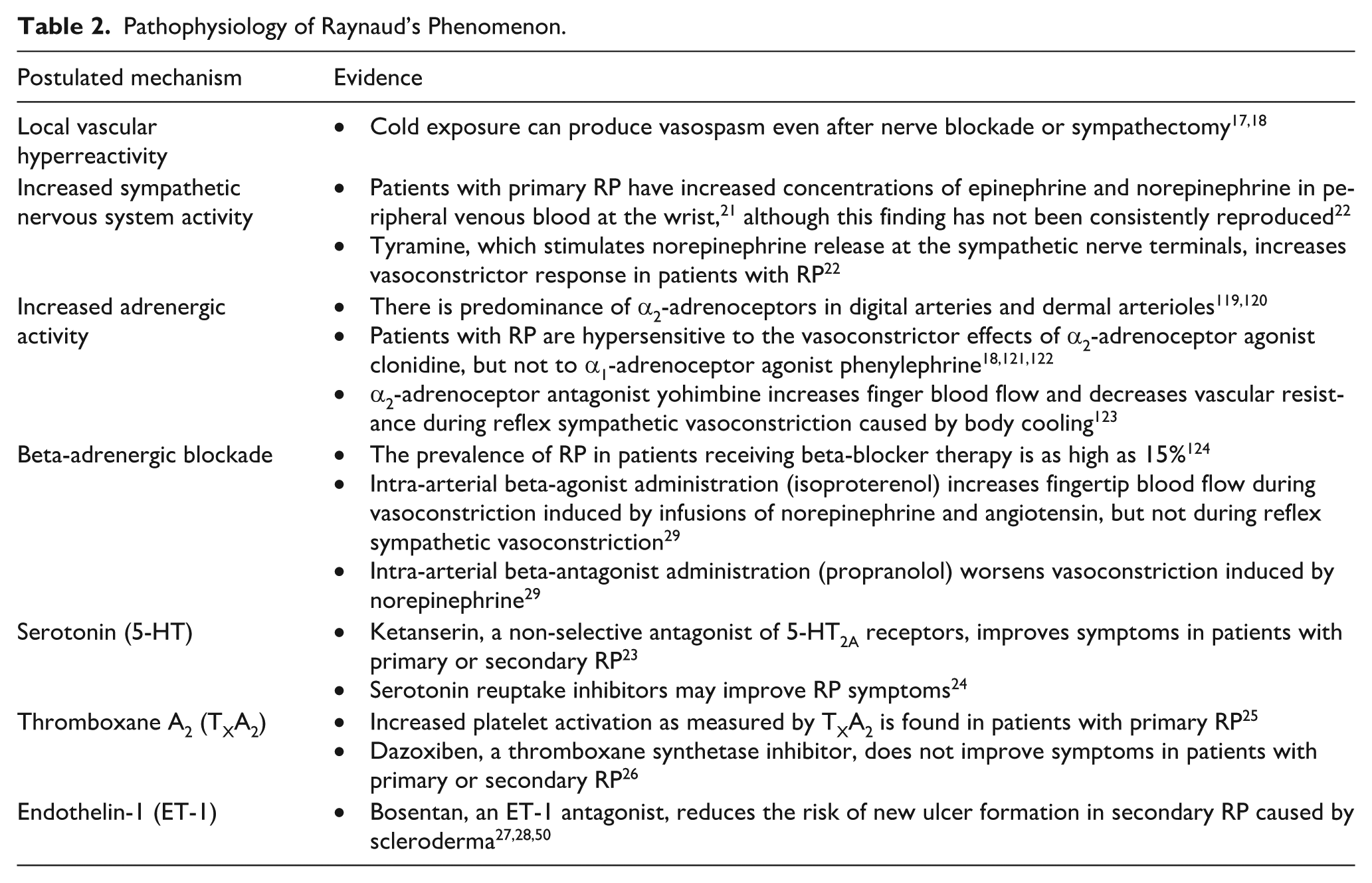
While the precise mechanism of RP is not clearly understood, the episodic attacks of vasospasm are likely the result of an interplay of various factors that regulate peripheral blood flow. Normal regulation of peripheral blood flow depends on intrinsic vascular tone, sympathetic nervous system activity, blood viscosity, and circulating hormonal substances. Unlike other regional circulations that are supplied by both vasoconstrictor and vasodilator sympathetic fibers, the cutaneous vessels of the hands and feet are innervated only by sympathetic vasoconstrictor fibers. Thus, vasodilation is achieved by withdrawal of such stimulus that causes vasoconstriction. Vasospasm is an excessive vasoconstrictor response causing obliteration of the vascular lumen. The balance between arterial wall tension (favoring closure of vessel) and intravascular distending pressure (favoring opening of vessel) is important in maintaining patency of a blood vessel. Interestingly, compared to controls, individuals with RP have lower brachial and digital artery pressure.15,16 These findings are more apparent in individuals with secondary RP and may be a result of arterial occlusive disease, vasculitis, or trauma, rather than a mechanism for primary RP. Additionally, disorders that increase viscosity of blood, such as polycythemia vera, Waldenström macroglobulinemia, cryoglobulinemia, and cold agglutinin disease, may reduce blood flow velocity in digital vessels with subsequent decrease in intravascular pressure.
There are multiple theories as to potential mechanisms of RP, described in Table 2; however, the exact mechanism is unknown. Theories include local vascular hyperreactivity;17,18 potentiation of vascular adrenoceptor response by cold exposure;19,20 increased sympathetic nervous system activity;21,22 and role of serotonin,23,24 thromboxane A2,25,26 and endothelin 1.27,28 Effects of beta-agonists and beta-blockers on peripheral blood flow have been studied as a response to vasoconstriction, induced exogenously (i.e., norepinephrine) or environmentally (i.e., cold). 29 Beta-agonists increase peripheral blood flow while beta-blockers potentiate vasoconstriction, but only during exogenously induced vasoconstriction. These findings recognize the role of beta-adrenergic vasodilation in human digits but propose that its antagonism may not be the primary mechanism by which episodic vasospasm occurs in the setting of environmental cooling.
Pathophysiology of Raynaud’s Phenomenon.
Diagnosis
The diagnosis of RP is primarily based on a careful history and physical examination. Allen and Brown’s classical criteria for diagnosis of primary RP include: (1) episodes precipitated by cold or emotion; (2) bilateral involvement of the extremities; (3) absence of gangrene, or, if present, limited to the skin of the finger tips; (4) no evidence of an underlying disease that could be responsible for vasospastic attacks; and (5) history of symptoms for at least 2 years. 30 Primary RP diagnosis is supported by age of onset (most often before the fourth decade), normal capillaroscopy, and absence of exam findings suggestive of secondary causes.
On the other hand, secondary RP should be suspected if onset of symptoms occurs later in life (generally older than 40 years of age), course is progressive, capillaroscopy is abnormal, or physical exam and/or laboratory findings are more consistent with a secondary cause (Figure 2). Additionally, evaluation of digital pressures, pulse volume waveforms, or laser Doppler skin perfusion with cold exposure and/or rewarming may be helpful, although provocative testing may not be widely available (Figure 3A–B). In primary RP, digital pressures and laser Doppler perfusion decrease with cold exposure and normalize with rewarming; conversely, in secondary RP, or fixed ischemia, findings may be abnormal at rest, and do not improve with rewarming (Figure 3C–D). Additionally, RP should be differentiated from benign blue finger syndrome (BBFS), characterized by benign, painless discoloration of digits; and Achenbach syndrome, characterized by benign but painful discoloration of digits.31–33 Unlike RP, BBFS and Achenbach syndrome affect an older population (mean age 57 years), do not affect the tips of digits, and do not respond to changes in temperature.34,35
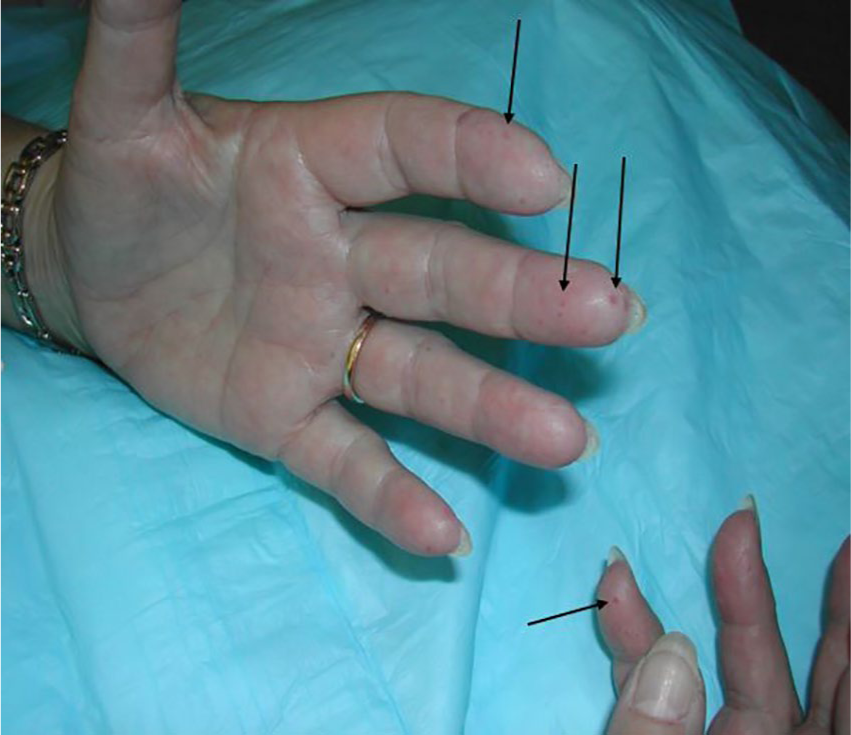
Digital findings concerning for secondary Raynaud’s Phenomenon. Arrows are pointing to telangiectasias in a patient with positive anticentromere antibody and diagnosis of limited scleroderma.
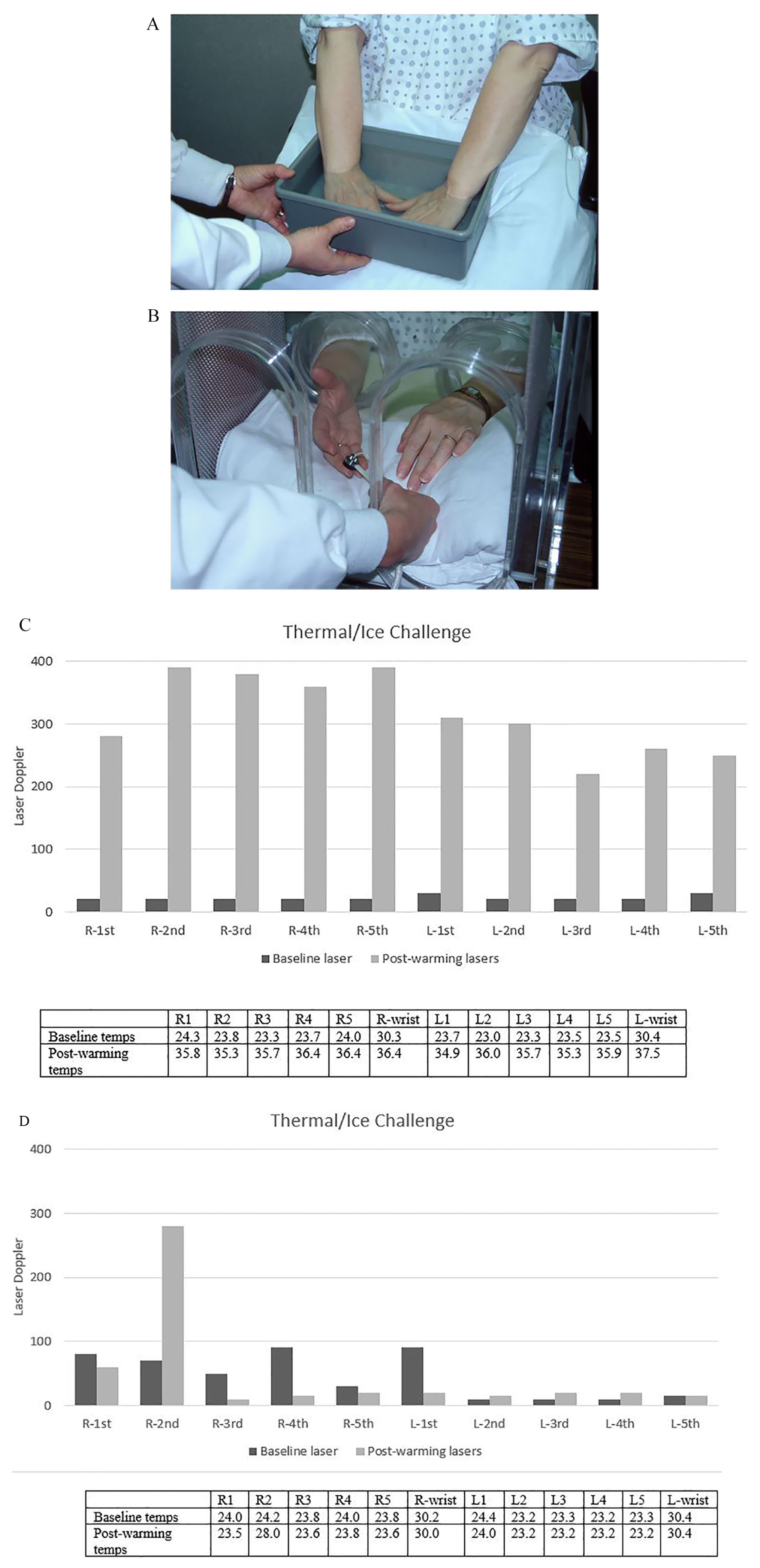
Evaluation of primary versus secondary Raynaud’s phenomenon with measurement of digital temperatures and laser Doppler signals before and after rewarming (courtesy of Dr Paul Wennberg, Mayo Clinic, Rochester, MN, USA).
Serological studies evaluating secondary RP should screen for vasculitis, collagen vascular disorders, and blood dyscrasias. For example, laboratory may be used to evaluate for evidence of systemic inflammation (erythrocyte sedimentation rate, C-reactive protein); scleroderma (anti-nuclear antibody, anti-centromere antibody, anti-Scl70 antibody, anti-nuclear ribonucleoprotein antibody, anti-nucleolar antigens); systemic lupus erythematosus (SLE; anti-nuclear antibody, anti-double-stranded DNA), rheumatoid arthritis (rheumatoid factor, anti-citrullinated protein peptide), dermatomyositis/polymyositis (creatinine kinase, aldolase, serum glutamic oxaloacetic transaminase, lactic acid dehydrogenase), primary Sjögren syndrome (anti-SSa/Ro antibody), and mixed connective tissue disease (anti-U1-RNP antibody). Individuals presenting with RP should be asked about history of medication changes, trauma or use of illicit substances. Medications such as beta-blockers, ergot alkaloids, certain chemotherapy drugs (vinblastine, bleomycin, gemcitabine) and methysergide can cause secondary RP. Trauma to digits, such as vibratory trauma, electric injury, thermal injury or hypothenar hammer syndrome, can precipitate secondary RP. Other conditions associated with secondary RP are listed in Table 1. Angiography is rarely indicated as diagnosis can often be made with careful history, physical examination, and selective serological studies. A diagnostic approach to RP is presented in Figure 4A–B.
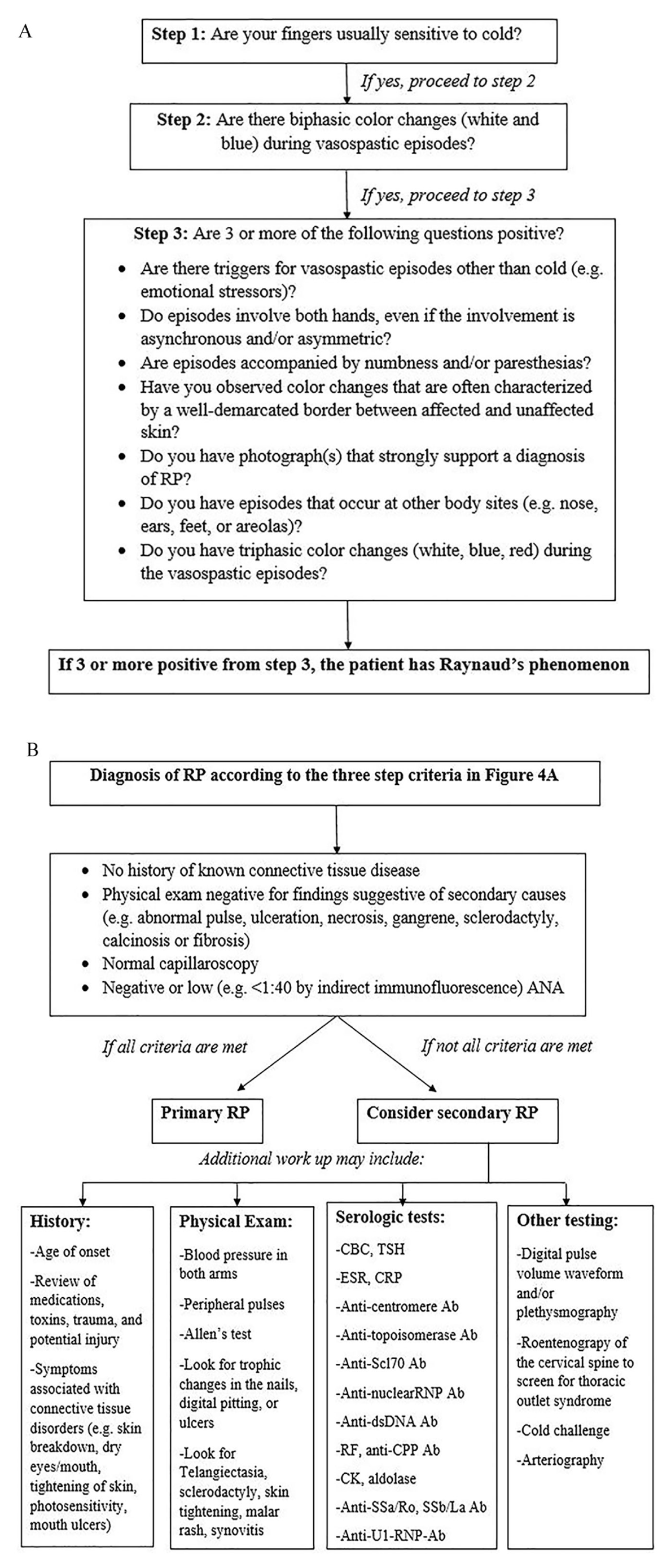
Diagnostic approach to RP.
Treatment
Treatment options for RP vary and depend on severity of symptoms and underlying cause. Patients with primary RP should be educated and reassured regarding the benign nature of the disease process. Importantly, all individuals with RP should be counseled about cold avoidance and the use of appropriate clothing, gloves, mittens, heat packs, and footwear to maintain core temperature within the appropriate range. 36 Warm water may be advised for rewarming. 37 However, use of hot water for rewarming should be avoided as prolonged exposure for rewarming may results in burns. Smoking cessation should be strongly encouraged as nicotine enhances vasoconstriction. 38
Pharmacological intervention is often initiated when conservative approaches are ineffective in improving quality of life. Nonetheless, strong data, such as large randomized controlled trials (RCTs), supporting specific drug therapy is lacking and choice of agent is often dependent on patient-specific factors. The most commonly used drug class for the treatment of RP is a long-acting dihydropyridine calcium channel blocker (CCB). Most trial data pertained to nifedipine, but amlodipine is also commonly used in practice. A meta-analysis of 18 small RCTs performed by Thompson and Pope found that CCB use in primary RP decreases the weekly number of attacks, up to five fewer attacks per week, as well as improved symptom severity. 39 However, the authors indicate possible bias as many of the included trials were crossover trials, in which order effects were not studied. They also note the effect size may have been small due to different dosing in studies. A Cochrane review of 38 low-to-moderate quality RCTs suggested that patients with primary or secondary RP who were prescribed CCB had fewer RP episodes and improved symptom severity, as measured by visual analogue scale (moderate quality evidence). 40 However, there were more withdrawals in the CCB treatment group due to side effects such as headache, dizziness, nausea, palpitations, and ankle edema. 40
Alpha1-adrenoceptor antagonists (prazosin, doxazosin, and terazosin) have been shown to reduce the severity of vasospastic attacks in patients with RP. 41 In a double-blind, placebo-controlled, crossover study of 24 patients, a low dose prazosin therapy reduced the number and duration of attacks and improved finger blood flow during a finger cooling test in both primary and secondary RP groups. 42 Two randomized controlled crossover studies have shown that prazosin is more effective than placebo in the treatment of RP secondary to scleroderma. 43 However, its use may be limited by side effects such as fatigue, dizziness, and headache.
In cases where CCBs or α1-adrenoceptor antagonists cannot be used due to side effects, or are ineffective, a phosphodiesterase type 5 (PDE-5) inhibitor or a topical nitrate may be used. It is important to note that data for PDE-5 inhibitors is limited to studies of individuals with secondary RP, and use in individuals with primary RP is based on extrapolation from these studies. 44 A 2013 meta-analysis of 296 secondary RP patients showed a significant improvement in RP symptoms, including severity, frequency, and duration of episodes. 44 A recent randomized, placebo-controlled study in patients with systemic sclerosis showed a significant decrease in the number of digital ulcers in patients receiving sildenafil compared to placebo. 45
A meta-analysis of topical nitrate therapy in 346 patients with primary and secondary RP showed a moderate-to-large treatment effect as compared to placebo, 46 although use of nitrates was associated with more headaches. Additionally, there has been no standardized topical nitrate across different studies. Topical nitrates seem to increase perfusion at both distal digital ulcer and extensor digital ulcer cores as compared to placebo in patients with systemic sclerosis when evaluating with laser Doppler imaging (LDI). 47 A recent study revealed that application of a nitroglycerin patch to the hand dorsum led to an increase in blood flow and hand temperature in patients with systemic sclerosis after cold challenge using LDI. 48
Bosentan, an endothelin receptor antagonist, has been used to treat refractory secondary RP and digital ulcers in patients with scleroderma.49,50 Other endothelin receptor antagonists, such as macitentan or ambrisentan, do not improve digital blood flow.51,52
While oral prostaglandins have not shown benefit, intravenous iloprost has been shown to decrease the frequency, duration, and severity of attacks and promote wound healing in secondary RP.53,54
Botulinum injection into the perineurovascular tissue of the wrist, the distal palm, or along the digits has been reported to relieve symptoms in uncontrolled, open-label case series.55,56 Digital periarterial sympathectomy may be considered when there is critical digital ischemia or resistant ulceration despite aggressive vasodilatory therapy. A long-term retrospective study of 35 patients with primary or secondary RP who underwent thoracoscopic sympathectomy had a positive response in 77%; however, symptoms recurred in 60% at a median follow-up of 5 months. 57 Therefore, there is no clear role for thoracoscopic upper limb sympathectomy, unless no other reasonable options exist.
In regards to pharmacologic treatment of primary RP, no matter the agent, we recommend starting with a low dose, and titrating the dose while monitoring for both response to therapy and side effects. Given the described limitations, it is reasonable to start with α1-adrenoceptor antagonists or long-acting dihydropyridine CCBs in younger patients (usually < 60 years old) with symptoms not well controlled with lifestyle modifications alone. The initial choice for the specific agent depends on shared decision-making between the provider and the patient, based on discussion of the side-effect profile of each agent. In older patients, or those with higher risk of developing side effects to α1-adrenoceptor antagonists, CCBs are preferred. We do not recommend using α1-adrenoceptor antagonists and CCB simultaneously due to the higher risk of side effects and lack of data with this approach. Regular follow-up, every 3–6 months, assessing the response and side effects of therapy, is vital.
In patients with secondary RP, CCBs, α1-adrenoceptor antagonists, or PDE-5 inhibitors are a reasonable first choice. In patients with persistent digital ischemia or symptoms, topical nitrate and/or intravenous prostacyclin therapy can be added. Recent meta-analysis has suggested that other drug classes do not have a significant impact on symptoms of secondary RP. 58 Digital sympathectomy may be considered when there is threatening digital ischemia or persistent ulcers despite maximal vasodilatory therapy.
Acrocyanosis
Acrocyanosis was originally described by Crocq in 1896 as a persistent blue and red discoloration of the digits, sometimes extending as far as the wrists or the ankles. The blue discoloration in acrocyanosis is relatively persistent, less episodic, and not associated with discomfort when compared to RP. Similar to RP, it is often exaggerated by exposure to cold or emotional stress; and may be idiopathic (primary) or associated with a variety of systemic disorders that require timely diagnosis and treatment (secondary). The primary form is often benign and has a favorable prognosis, while prognosis of secondary acrocyanosis depends on the underlying disorder.
Clinical presentation
The classic presentation of primary acrocyanosis is described as a symmetric, painless, and persistent bluish discoloration of hands and feet in a thin young woman. Other body parts such as the nose tip, ears, lips, or nipples can be affected, but are less common. While individuals with acrocyanosis may report a sensation of coolness in affected extremities, they typically do not experience pain or tissue injury. If pain, ulceration or gangrene is seen, an alternative diagnosis should be considered. 59 The discoloration seen in acrocyanosis may have different hues, such as bluish-pink or brownish-yellow. The affected areas may have mild swelling and frequently have hyperhidrosis. The discoloration may be exacerbated by cold or emotional stress and diminished by rewarming. Another characteristic of acrocyanosis is the slow and irregular return of the blood from the periphery to the center in the area of blanching when pressure is applied to the skin (Crocq’s sign). Acrocyanosis is distinguished from RP by the absence of episodic digital pallor, rubor, or pain.
Epidemiology
While there is limited information on the precise incidence and prevalence of the disease, acrocyanosis is known to affect younger adults (second to third decades of life), and women more frequently than men. 60 The incidence of acrocyanosis decreases with age, with almost no cases seen after menopause, suggesting hormonal influences on the underlying mechanism.59,61 It is associated with a low body mass index, anorexia nervosa, malignancy, outdoor occupations, and cooler climates. 62 There has been a report of genetic predisposition, with a report of first degree family history in up to 10% of patients with acrocyanosis. 62
Pathophysiology
Proposed pathophysiological mechanisms of primary acrocyanosis are similar to those of RP and include dysregulated arteriolar tone, autonomic nervous system imbalance, and response to circulating vasoconstrictive mediators. The fundamental abnormality in primary acrocyanosis is the persistent arteriolar vasoconstriction which leads to compensatory venular dilatation in the postcapillary vascular beds.63,64 Hypoxia caused by precapillary vasoconstriction increases the release of vasodilatory mediators, which in turn leads to postcapillary dilation and possible arteriovenous subpapillary plexus shunting. 65 Other potential mediators of altered vasoconstriction in acrocyanosis include epinephrine, norepinephrine, serotonin, and endothelin-1.59,66 As rewarming of a part of the affected limb produces a sharp demarcation, this suggests that the primary vessel defect is localized to distal and smaller arteries.61,67,68
Diagnosis
The diagnosis is made clinically and typically does not require laboratory or imaging studies. 69 A careful history and physical examination should be obtained in order to eliminate an underlying disease as a potential cause of discoloration. In primary acrocyanosis, arterial pulses and pulse oximetry are normal. Elevation of a cyanotic limb over the heart produces pallor, suggesting there is no venous obstruction.
Secondary acrocyanosis can be seen with anorexia nervosa, connective tissue disorders (scleroderma, SLE, rheumatoid arthritis, mixed connective tissue disease, antiphospholipid antibody syndrome (APS)), arterial occlusive disease (including atheromatous/septic embolism), coagulopathy (disseminated intravascular coagulation, heparin-induced thrombocytopenia), malignancy (lymphoma), infection (hepatitis C), and presence of cryoglobulins and cold agglutinins. 60 Medications and toxins associated with acrocyanosis include tricyclic antidepressants, interferon-α, interferon-β, amphotericin B, arsenic, and butyl nitrite.70,71 Chronic arsenic toxicity accompanied by acrocyanosis is known as ‘black foot disease’. This condition is thought to be caused by inhibition of prostacyclin synthetase, formation of intravascular clots, and inflammation of the vascular wall leading to subsequent gangrene and amputations of the limbs.60,72,73 A cyanotic appearance may also be a result of systemic hypoxemia with increased deoxyhemoglobin; or due to accumulation of methemoglobin and sulfahemoglobin due to exposure to oxidizing agents (e.g. aniline, benzocaine, dapsone, phenazopyridine, nitrates, and naphthalene).74,75 Therefore, a careful review of medications and toxins is important to investigate the cause of cyanosis. In secondary acrocyanosis, abnormalities, such as asymmetric limb involvement, hypoxemia, weak/absent pulse, pain or trophic skin changes, may be seen depending on the underlying disease. When a secondary cause is suspected, laboratory and imaging work-up is similar to that of secondary RP, as previously described. Capillaroscopy may be performed to assess capillary morphology, distribution, and blood flow. 60 Nonspecific findings such as abnormal capillary morphology, pericapillary edema, and hemorrhage may be present. Mean capillary density is reduced in patients with presumed primary acrocyanosis compared to normal subjects, although is higher than that of patients with systemic sclerosis. 76
Treatment
Primary acrocyanosis is a benign condition that usually only requires conservative measurements such as reassurance, avoidance of cold exposure, and maintenance of appropriate body temperature. Several agents such as CCBs, beta-blockers, and other adrenergic blocking agents have been tried but without clear success. 60 Nicotinic acid derivatives have been used in a clinical trial with some effect but with variable responses.77,78 Bromocriptine has been reported to relieve acrocyanosis but may cause RP. 79 Sympathetic nerve block and sympathectomy have been used for severe cases but these are rarely appropriate for primary acrocyanosis. 60 Treatment of secondary acrocyanosis is directed against the primary cause.
Pernio
Pernio (chilblains) is an inflammatory cutaneous condition involving localized swelling and erythema caused by exposure to cold, damp, and non-freezing conditions. Hands, feet, ears, and face are most commonly affected. There are acute and chronic forms of pernio. While it is possible to have pernio without underlying vascular pathology, secondary pernio associated with other conditions also exists. Various names used in the literature to describe pernio or a pernio-like condition include dermatitis hiemalis, erythrocyanosis, nodular vasculitis, or cold panniculitis.
Clinical presentation
The initial presentation of pernio is acute pernio, where the lesions appear during the cold months and disappear when the weather warms up. Lesions vary in size, shape, color, and number, and are associated with itching, burning or pain. 80 The lesions may be red-to-purple macules, papules, or plaques on one or more body parts exposed to cold. 81 Acute lesions are generally self-limiting once the exposure stops. Although infrequent, complications of acute pernio include skin breakdown, ulceration, and infection. Chronic pernio may occur when there is repeated and prolonged exposure to cold during the acute phase and/or the individual goes through several seasons of acute pernio. The chronic lesions may be associated with scarring, atrophy, permanent discoloration, and ulceration. 80 Adults tend to have more severe forms of pernio and, if untreated, could develop microvascular occlusive disease. 82 On the other hand, children may have recurrent acute pernio over several seasons. 83 Conditions associated with pernio include anorexia nervosa, RP, acrocyanosis, macroglobulinemia, dysproteinemias, leukemia, cryoproteinemia, SLE, antiphospholipid syndrome, peripheral artery disease, erythromelalgia, complex regional pain syndrome, and use of sulindac.84–88 It is important to note that RP and acrocyanosis can coexist with pernio, cause secondary perniosis or clinically manifest after the initial diagnosis of pernio. 84
Epidemiology
An epidemiologic study dating back to the Second World War reported a pernio prevalence of 50% in military participants. 89 In a more recent case series of 104 patients with pernio, a female predominance (79% of cohort) with mean age of 38 years and frequent smoking was noted. 90 While 17% of patients also had RP, only 4% had concomitant autoimmune connective tissue disease and 3% had hematologic malignant disease. Pernio is most commonly seen in the northern and western parts of the United States and during a wet, near-freezing season.82,91 In general, the disease is becoming less common with higher standards of home and workplace heating.
Pathophysiology
While the exact pathogenesis of pernio is unknown, it has been suggested that persistent cold exposure leads to vasoconstriction, hypoxemia, and a subsequent inflammatory reaction in the affected tissue. Many studies have described histological findings of perivascular lymphocytic infiltration, and vessel wall and papillary dermal edema.92–94 Humidity in cold temperatures is an important factor as it increases air conductivity and further promotes heat loss from the skin. In response to such conditions, the cutaneous vasculature exhibits arteriolar constriction, venular relaxation and increased blood viscosity, leading to ischemia of the tissue. 80 Individual susceptibility to cold exposure also plays a role in development of pernio, as affected individuals have been shown to have cooler skin for longer durations than controls.82,95
Diagnosis
Diagnosis is based on the recognition of lesions and chronological correlation between the onset of symptoms and cold exposure. Extensive laboratory or imaging studies are not required to make the correct diagnosis unless a secondary cause is suspected. As with other conditions, careful physical examination is key. The differential diagnosis of pernio includes a variety of diseases, but one disorder that requires careful consideration is chilblain lupus erythematosus (CHLE) due to its association with SLE and implications of familial genetic inheritance. 96 Additionally, with the recent coronavirus (COVID-19) pandemic, there has been a report of ‘COVID toes’, which clinically appears similar to pernio. A recent study evaluated patients with CHLE compared to COVID-19. 97 Interestingly, cutaneous histologic findings of both groups are similar, with elevated expression of type-I interferon-induced protein (IFN-I). Individuals with more significant COVID-19 cutaneous symptoms had higher levels of IFN-I, suggesting that ‘COVID toes’ may occur due to an exaggerated immune response.
Although not required, biopsies may be performed to support the diagnosis.85,93 The characteristic histopathological features of pernio include edema of the papillodermis, perivascular infiltration by mononuclear and lymphocytic cells, thickening and edema of the vascular walls, fat necrosis, and chronic inflammatory reaction with giant cell formation. 80 In a study of nine patients with pernio, histologic findings demonstrated lymphocytic vasculitis in eight of nine specimens. 81 These histologic changes were consistent regardless of the clinical appearance of the lesions.
Treatment
The primary focus of treatment is prevention. Avoidance of cold and damp conditions is critical in the prevention and recurrence of pernio. A dihydropyridine CCB shortens duration of lesions, reduces severity of symptoms, and prevents development of new lesions in some studies.98,99 Other agents such as topical nitroglycerin, oral pentoxifylline, and hydroxychloroquine have been used with variable results.100–102 Some clinicians use topical corticosteroids (0.025% fluocinolone acetonide) but benefits are unclear. 103
Livedo reticularis
Livedo reticularis (LR) is a physical exam finding characterized by reddish purple reticular mottling that forms a net-like pattern (Figure 5). In many cases, primary LR is a benign finding aggravated by cold exposure and alleviated by warming and/or elevation. Unlike the benign presentation of LR that is often reversible, livedo racemosa is a permanent condition that is always pathologic.104,105 Systemic diseases such as APS, Sneddon’s syndrome, connective tissue disorders, cryopathies, malignancy, neurologic disorders, infections, and embolic disease (atherosclerotic/septic emboli) are associated with secondary LR or livedo racemosa. 106 Medications associated with the development of LR include catecholamines, amantadine, quinidine, and bismuth. 107 LR is found in up to 40% of patients taking amantadine. 108 Ulceration formation can be seen with continued use of amantadine, but ulcers typically resolve quickly with discontinuation of the medication. 108 Long-term therapy with gemcitabine may lead to thrombotic microangiopathy manifested by LR and hemolytic uremic syndrome. 109 In heparin-induced thrombocytopenia and thrombosis, disseminated intravascular coagulation and thrombotic occlusion of cutaneous blood vessels may manifest as LR. 110
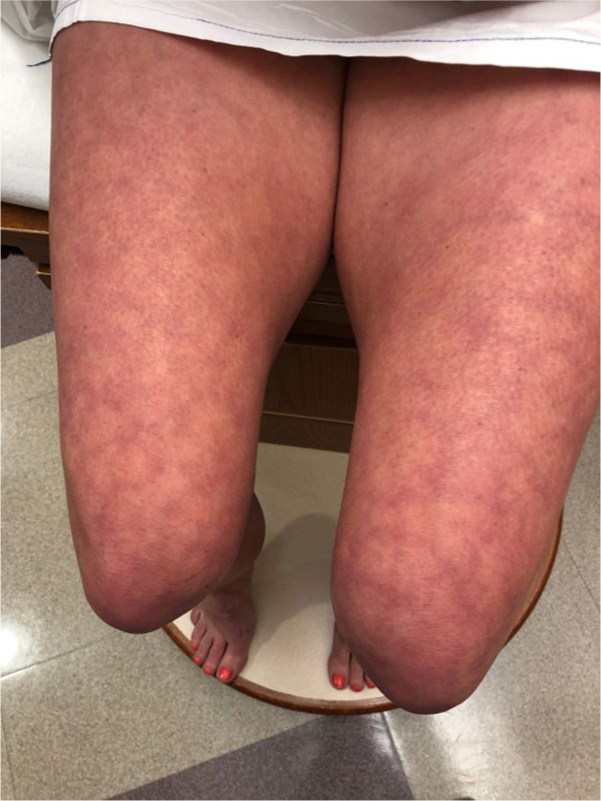
Skin discoloration consistent with livedo reticularis. There is bilateral symmetric lower extremity net-like mottling, consistent with livedo reticularis. (Please note the acrocyanosis in the feet.)
Clinical presentation
LR can present in primary or secondary form. In LR, the net-like mottling is symmetrically distributed, painless, often reversible, limited to the extremities, and rarely associated with other skin abnormalities. In contrast, livedo racemosa, a condition that is always pathologic and associated with an underlying disease, has asymmetrical distribution of irregular mottling with diffuse involvement of the trunk, buttocks, extremities, and/or face; is irreversible; can be painful; and may be associated with other skin abnormalities (e.g. purpura, nodules, or ulcers).
Epidemiology
Both physiologic and primary LR are commonly seen in young women, especially those with fair skin, between the ages of 20 and 60 years. 111 LR is a common dermatologic finding in patients with APS. It is present in approximately 25% of patients with APS and in up to 70% of patients with SLE-associated APS.112,113
Pathophysiology
LR results from changes within the cutaneous microvascular system. The dermis is supplied by a network of perpendicularly oriented arterioles that form a capillary bed at the surface. These capillary beds then empty into a conical-appearing, peripherally located subpapillary venous plexus. Anything that increases the visibility of the venous plexus can result in net-like venous mottling. Thinning of the papillary dermal layer alone can make this venous network visible. 114 Decreased arteriolar perfusion from vasoconstriction or other systemic condition (hyperviscosity, inflammation, embolic disease) can cause deoxygenation of the venous plexus, which in return leads to net-like mottling. Venodilatation of the venous plexus secondary to hypoxia or autonomic dysfunction is another mechanism by which mottling occurs.
Diagnosis
Similar to other vasospastic disorders, the first diagnostic step is a comprehensive history and physical exam. The location of LR, exacerbating and alleviating factors, duration of attacks, associated skin findings, and risk factors for autoimmune connective tissue disorders or other secondary causes are important factors to consider. Of note, reversible LR can be seen in RP flare. On physical examination, it is important to note the location of LR as typical mottling seen outside of extremities is consistent with livedo racemosa and is always pathologic. The presence of ulcerations or nodules is suggestive of vasculitis. 115 Serologic testing should be directed based on the patient’s history, physical exam, and risk factors. However, an antiphospholipid antibody panel should be obtained in all patients presenting with livedo racemosa as it has been identified as a marker for predicting multisystem thrombosis in APS.116,117 A skin biopsy is not required for diagnosis of LR but punch biopsies of a central blanched area or a peripheral bluish area may be helpful in differentiating a vasculitis, vasculopathy or normal tissue. 115
Treatment
For primary LR, cold avoidance and limb elevation are usually sufficient. In rare cases, vasodilator therapy may be used if a patient is significantly affected by the cosmetic appearance of LR. Treatment of secondary LR and livedo racemosa is directed towards the underlying disease. Patients with livedo racemosa and APS complicated by thrombosis should be treated with therapeutic anticoagulation. However, currently there is no evidence-based practice on how to treat patients with livedo racemosa and APS without a history of thrombosis. 106
Summary
Comparison of clinical characteristics of primary RP, primary acrocyanosis, period, and primary LR is summarized in Table 3, and key points to remember are summarized in Table 4. A comprehensive history and physical exam remain the most important diagnostic tool for differentiating various vasospastic disorders and determining the secondary cause if one is present. Routine laboratory tests and imaging tests are not necessary in many patients with RP and related vasospastic disorders. Diagnostic tests and treatment should be guided by the individual patient’s medical history, risk factors for a secondary cause, and the extent of digital ischemia. Making the correct diagnosis is imperative in understanding the trajectory of the disease and identifying the correct treatment strategy.
Clinical characteristics of primary RP, primary acrocyanosis, pernio, and primary LR.
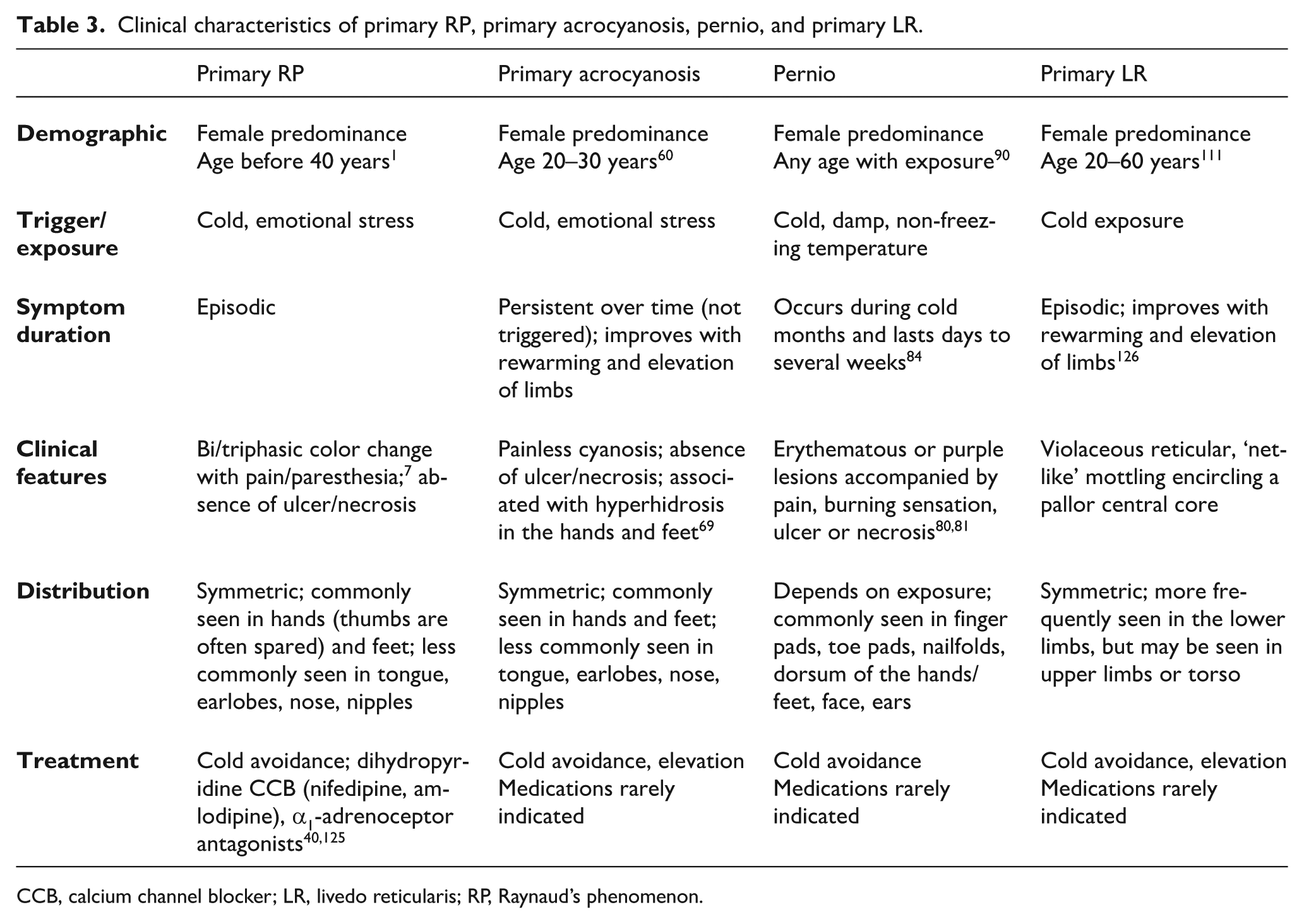
CCB, calcium channel blocker; LR, livedo reticularis; RP, Raynaud’s phenomenon.
Key points to remember.

LR, livedo reticularis; RP, Raynaud’s phenomenon.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.