
Abstract
With the ability to cause massive epidemics that have consequences on millions of individuals globally, the Chikungunya virus (CHIKV) emerges as a severe menace. Developing an effective vaccine is urgent as no effective therapeutics are available for such viral infections. Therefore, we designed a novel mRNA vaccine against CHIKV with a combination of highly antigenic and potential MHC-I, MHC-II, and B-cell epitopes from the structural polyprotein. The vaccine demonstrated well-characterized physicochemical properties, indicating its solubility and potential functional stability within the body (GRAVY score of –0.639). Structural analyses of the vaccine revealed a well-stabilized secondary and tertiary structure (Ramachandran score of 82.8% and a Z-score of –4.17). Docking studies of the vaccine with TLR-2 (−1027.7 KJ/mol) and TLR-4 (−1212.4 KJ/mol) exhibited significant affinity with detailed hydrogen bond interactions. Molecular dynamics simulations highlighted distinct conformational dynamics among the vaccine, “vaccine-TLR-2” and “vaccine-TLR-4” complexes. The vaccine’s ability to elicit both innate and adaptive immune responses, including the presence of memory B-cells and T-cells, persistent B-cell immunity for a year, and the activation of TH cells leading to the release of IFN-γ and IL-2, has significant implications for its potential effectiveness. The CHIKV vaccine developed in this study shows promise as a potential candidate for future vaccine production against CHIKV, suggesting its suitability for further clinical advancement, including in vitro and in vivo experiments.
Introduction
Chikungunya is an emerging mosquito-borne disease caused by the Chikungunya virus (CHIKV), which belongs to the alphavirus genus and the Togaviridae family. 1 Mosquito vectors, Aedes aegypti and Aedes albopictus, often spread this viral disease by their bites from an infected individual to a healthy one. 2 The virus poses high susceptibility to premature infants, the elderly, and immunosuppressed populations, which can cause severe health issues, including vasculitis, fulminant hepatitis, encephalitis, myocarditis, prerenal failure, cranial nerve palsies, hemorrhage or even death.3,4 The CHIKV infection can cause symptoms such as fever, intense joint pain, a rash, and muscle discomfort. 4 Although the illness usually resolves on its own, there are instances where the joint pain can last for an extended period, ranging from months to even years.
From 1999 to 2019, 25 chikungunya outbreaks were recorded in 16 countries globally.5,6 Besides, 5 documented epidemics in Europe have been attributed to the chikungunya infection transmitted by Aedes mosquitos, such as Ae. aegypti, and Ae. albopictus.5,7,8 Between 1999 and 2020, 13 chikungunya outbreaks occurred in eleven African countries. 5 During this period, multiple epidemics were reported in 6 Asian countries: Bangladesh, India, Indonesia, Malaysia, the Philippines, and Thailand. 5 Besides, Bangladesh had a significant epidemic in 2017, followed by 2 barely noticeable outbreaks in 2008 and 2011. Although, sporadic CHIKV infection cases were reported in 2013, 2015, and 2016. 9 In April to September 2017, Bangladesh experienced a significant outbreak of CHIKV infection. Many positive cases were reported from 23 nationwide districts during this time, while Dhaka city alone documented over 13,000 clinically confirmed cases.10-12 In 2019, Ethiopia experienced a significant CHIKV outbreak, where a total of 41,162 suspected cases were reported. 13 There was another outbreak in Congo in 2019, with 6,149 cases suspected. 14 Several outbreaks of CHIKV were reported in Europe, America, and Latin America. In the year 2017, Italy had again another epidemic, this time with a total of 499 suspected cases and 270 confirmed cases. 15 In the United States, there were 296 fatalities and more than 2.9 million reported cases in 2016. 16 The Paraguayan Ministry of Health recorded a total of 81,037 cases of CHIKV between October 1, 2022, and March 11, 2023, while, out of these cases, 75,911 (94% of the total) occurred in 2023. 17 Brazil has seen over 1.2 million cases of CHIKV in the past decade, making it the country in the Americas with the highest number of infections. It has accounted for 85% of Latin America’s nearly 1 million CHIKV infections since 2018. 18 According to the Ministry of Health, there has been a significant increase in the number of CHIKV cases reported in Argentina. Between January 1, 2023, and March 12, 2023, 593 cases were recorded, 11 times higher than last year. 17 Genomic surveillance of such outbreaks revealed that the CHIKV isolates of these outbreaks were unlikely the strain identified in the preceding outbreak. 19 Subsequent phylogenetic analysis showed that the strain formed a new cluster within the Indian Ocean clade, indicating that they are novel variants. 20 These numbers underscore the urgent need for effective preventive measures.
The frequency and spread of these outbreaks further emphasize the pressing need for a comprehensive vaccine strategy. Various therapeutic strategies and supportive treatments are available to manage the symptoms of CHIKV infection, although there is no specific antiviral treatment. 21 Research is ongoing to develop antiviral therapies against CHIKV infection, including ribavirin, favipiravir, sofosbuvir, chloroquine, suramin, defluorinated favipiravir, mycophenolic acid, prostratin, tocilizumab, and interferon therapy.22,23 Recent advancements in CHIKV vaccine development have shown promising progress, with several candidates using different platforms, including live-attenuated, chimeric viral vector, virus-like particles, and mRNA-based vaccines. The U.S. Food and Drug Administration (FDA) recently authorized a live-attenuated vaccine called VLA1553 (IXCHIQ) against CHIKV in November 2023. 24 Apart from this, several vaccine candidates for CHIKV are progressing through preclinical and clinical stages. Hallengärd et al 25 developed a live-attenuated vaccine against CHIKV, called D5nsP3, which showed promising results in their phase I clinical trial. In a joint venture, Takeda Pharmaceuticals and Zydus Cadila are developing a live-attenuated viral vaccine against CHIKV, namely, TAK 507.26-30 Contrarily, Chattopadhyay et al. (2013) developed a chimeric viral vector (CVV) vaccine through the utilization of chimeric vesicular stomatitis virus (VSV), targeting the entire CHIKV envelope polyprotein (E3-E2-6K-E1). The vaccine candidate, however, showed potent antibody responses in mouse models. 31 Rossi et al. (2019) 32 developed another CVV using a measles virus vector (MVV), which showed promising results in phase I clinical trial. In addition, there have been multiple CVV vaccines developed against CHIKV utilizing the replication-deficient chimpanzee adenovirus (rAd), including Ad-CHIKV-SG, 33 Ad-CHIKV-E3/E2/E1, 33 Ad-CHIKV-E3/E2/6K, 33 ChAdOx1 sCHIKV, 34 and ChAdOx1 sCHIKV ΔC, 34 which are still under in vivo trial and sought to further preclinical trial.
Besides CVV, the National Institute of Health (NIH) Vaccine Research Center has completed a phase I clinical trial of a virus-like particle (VLP) based vaccine against CHIKV using FDA-approved HEK293 cells. However, phase II trials of the vaccine are ongoing, where the immunization showed potent neutralizing antibody responses. 35 Saraswat et al. (2016) 36 developed a CHIK-VLP, which showed promising immunological responses in both in vitro and in vivo trials. Chen et al. (2020) 37 also developed a VLP-based vaccine against CHIKV, which showed potent immunogenicity, safety, and well tolerated in a phase 2 trial. Themis Bioscience later developed a measles vector-based CHIKV vaccine, which passed a phase 2 trial. 38 However, the challenge of this vaccine was to integrate the viral antigen into measles vector and ensure robust immune response in diverse populations. 38 Apart from these, mRNA vaccines offer several advantages over other types of vaccines, particularly in terms of safety, effectiveness, and efficient production. 39 First, they are developed using a synthetic approach, which removes the relevance of live pathogens, ensuring a safer manufacturing and storage process. Furthermore, mRNA vaccines have the advantage of being easily and quickly developed and produced, enabling faster responses to emerging pathogens or variants. Also, they elicit dominant immune responses, encompassing the production of antibodies and cellular immunity, resulting in an effective barrier for infection. On top of that, mRNA vaccines are not incorporated as a component of the host genome, addressing any concerns about potential long-term effects or genetic changes. 40 Hence, mRNA vaccines have emerged as a prominent method of immunization for dealing with infectious disease epidemics within 3 decades, owing to their inherent simplicity, expedited production cycle, and less constrained manufacturing conditions. 41 Multiple mRNA vaccines have received approval for commercial distribution, while others are still undergoing clinical trials. 41 With this in mind, the first vaccines against the coronavirus disease 2019 (COVID-19) approved for use worldwide were mRNA-1273 from Moderna and BNT162b2 from BioNTech. Both of these vaccines have shown a wide range of immunogenicity.42-46 Following the notions, various vaccine candidates have been developing against CHIKV utilizing the mRNA-based platform. Shaw et al. (2023) 47 developed an mRNA vaccine against CHIKV, known as mRNA-1388, which proved to be a safe, well-tolerated, and immunogenic vaccine candidate in phase 1 dose-ranging trial.
The virion of CHIKV, however, contains a single-stranded RNA encapsulated by a protective envelope and icosahedral capsid. 48 The genome of the virus consists of 4 nonstructural protein-coding genes (nsP1, nsP2, nsP3, and nsP4) located at the 5′ end, and 5 structural protein-coding genes (E1, E2, E3, 6K, and C) at the 3′ end.49,50 Therefore, the E1, E2, and E3 proteins are mainly translated from the 3′ open reading frame (ORF) of the viral genome. The E1 and E2 proteins are integrated into the viral membrane via short trans-membrane sections, while the E3 protein resides on the outermost layer of the viral membrane and engages with the E2 protein.51,52 The E1 protein is involved in the fusion of the viral envelope with the host cell membrane during viral entry, while the E2 protein plays a crucial role in viral attachment to host cells. The function of E3 in the viral entry needs to be clearly understood. 53 However, upon infection, the E1, E2, and E3 proteins of CHIKV are synthesized as a single polypeptide and subsequently undergo proteolytic processing to become separate proteins.1,54,55 Considering the importance of these structural proteins in the CHIKV pathogenesis, an effective mRNA vaccine can be formulated against the virus utilizing the E1, E2, and E3 proteins. This type of mRNA vaccine comprises multiple epitopes, including MHC-I, MHC-II, and B-cell epitopes from the structural polyprotein of the CHIKV. MHC-I molecules serve as immunological stimulants against intracellular microbes or tumorigenic antigens; hence, they initiate the cytotoxic T cell (TC) response to the interaction between cytotoxic T lymphocytes (CTLs) and MHC-I molecules. The vaccines in consideration are thus developed based on MHC-I binding epitopes that induce CD8+ T cell responses, which significantly impact the elimination of viral infections. 56 Furthermore, MHC-II molecules are responsible for presenting immunogenic processed peptides to the T cell receptor (TCR) on CD4+ T cells, an essential step to start cellular and antibody-mediated immune responses. The association between MHC-II molecules, which contain processed peptides, and TCR is of the utmost significance in avoiding microbial infections, identifying growing malignancies, and rejecting transplants.57-60 Consequently, the identification of CD4+ and CD8+ T cells is an indispensable process throughout the vaccine development process.61-63 Moreover, B-cell epitopes have been recognized as an essential aspect to be considered during vaccine development since they have a crucial role in the interactions between antigens and antibodies. 61 ,64-66
This study aimed to design an mRNA-based CHIKV vaccine utilizing reverse vaccinology and immunoinformatics approaches. The vaccine comprises multiple epitopes, including MHC-I, MHC-II, and B-cell epitopes, which are from the structural polyprotein of the CHIKV. Following epitope predictions, the potential epitopes were connected to an adjuvant, such as 50S ribosomal protein L7/L12, and 4 linkers, including EAAAK, AYY, AK, and KFER. We hope this study will help establish the groundwork for developing an mRNA vaccine targeting CHIKV in humans. The proposed CHIKV vaccine was based on a computational approach, therefore it requires further experimental validation to confirm its accuracy and applicability in real-world scenarios.
Materials and methods
Sequence retrieval
The structural polyproteins’ amino acid sequence of CHIKV (accession number: QGZ98799.1) was obtained from the National Center for Biotechnology Information (NCBI) protein database (https://www.ncbi.nlm.nih.gov/) and stored in FASTA format. 67 However, the E1, E2, and E3 sequences of this polyprotein were then applied for subsequent vaccine construction.
Major histocompatibility complex-I (MHC-I) binding epitope prediction
The MHC-I binding epitopes of the selected region of the structural polyprotein were predicted using the Immune Epitope Database (IEDB) (http://tools.iedb.org/mhci/)68,69 and Net-MHC 4.0 server (http://www.cbs.dtu.dk/services/NetMHC/).70,71 The IEDB is an extensive database that provides information on antibodies, B-cell epitopes, T-cell epitopes, MHC molecules, and MHC binding ligands across humans and various other animal species, including primates and mice.38,68,71-82 By analyzing ligand data and MHC-I binding affinities, the NetMHC 4.0 server can foretell how MHC-I will interact with peptides.68,70,81 Therefore, the epitopes were predicted in 9-mer length and a threshold value of ⩽ 1.00 (percentile rank). The selected epitopes were assessed for antigenic properties using the VaxiJen 2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) with a threshold value of 0.4. 83 The epitopes were further screened for allergenicity with the AllerTOP v. 2.0 server (https://www.ddg-pharmfac.net/AllerTOP/index.html) (threshold value of <0.00) 84 and evaluated for toxicity using the ToxinPred server (https://webs.iiitd.edu.in/raghava/toxinpred/algo.php) (threshold score of <0.00). 85
Major histocompatibility complex-II (MHC-II) binding epitope prediction
The MHC-II binding epitopes of the selected region of the structural polyprotein were predicted using the IEDB (http://tools.iedb.org/mhcii/)68,69 and NetMHCIIpan 4.0 server (http://www.cbs.dtu.dk/services/NetMHCIIpan/).69,86 The NetMHCIIpan 4.0 server uses Artificial Neural Networks (ANNs) for the MHC-II binding epitope prediction.69,86 Subsequently, the epitopes were predicted in 15-mer length and a threshold value of less than or equal to 1.00 (percentile rank). The selected epitopes were evaluated for antigenic characteristics by VaxiJen 2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (threshold value of 0.4), 83 as well as assessed for allergenicity and toxicity by AllerTOP v. 2.0 server (https://www.ddg-pharmfac.net/AllerTOP/index.html) (threshold value of <0.00), 84 and ToxinPred server (https://webs.iiitd.edu.in/raghava/toxinpred/algo.php) (threshold value of <0.00), respectively. 85 The IFNepitope server was also applied to predict the interferon-gamma (IFN-γ) induction capabilities of the selected epitopes. The epitopes that passed all the parameters were then chosen for the vaccine construction.
Molecular docking between T-cell epitopes and MHC molecules
Molecular docking between T-cell (MHC-I and MHC-II) epitopes and MHC molecules, including MHC-I and MHC-II, is essential for evaluating immune responses and advancing immunological research. 87 Prior to docking, the tertiary structures of human MHC-I (PDB id: 2XPG) and MHC-II (PDB id: 1DLH) were retrieved from the PDB (Protein data bank) database (www.rcsb.org). Subsequently, the HDOCK server was used to perform the docking where both the tertiary structures of MHC-I and MHC-II were uploaded along with their respective T-cell epitopes. All the parameters were kept at default.88,89
Linear B-cell epitope prediction
The linear B-cell epitopes of the targeted polyprotein, were predicted by the IEDB (http://tools.iedb.org/bcell/) and the ABCpred server (https://webs.iiitd.edu.in/raghava/abcpred/). 90 IEDB employs computational approach to estimate linear B-cell epitopes based on the sequence properties of the antigen. 91 Conversely, the ABCpred server uses an HMM and a propensity scale approach to compute the linear B-cell epitopes. 90 The selected epitopes were, therefore, screened for antigenic characteristics by VaxiJen 2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (threshold value of 0.4), 83 allergenicity by AllerTOP v. 2.0 server (https://www.ddg-pharmfac.net/AllerTOP/index.html) (threshold value of <0.00), 84 and toxicity by ToxinPred server (https://webs.iiitd.edu.in/raghava/toxinpred/algo.php) (threshold value of <0.00). 85
Vaccine mapping
The highly prioritized epitopes from the structural polyprotein were applied to construct the vaccine map. The vaccine map was completed by connecting the selected epitopes and the adjuvant (50S ribosomal protein L7/L12) with the linkers, include EAAAK, AYY, AK, and KFER.92-94 The efficacy of a vaccine can be enhanced by adding L7/L12 of Mycobacterium tuberculosis adjuvants, as demonstrated in laboratory trials. 93 ,95-97 Hence, the L7/L12 was selected as an adjuvant for the proposed vaccine. Linkers are used to ligate epitopes and maintain the flexibility and stability of the epitopes.93,96,97 The immunogenicity of an epitope vaccine can be improved by using AYY and EAAAK linkers, as these allow for intramolecular hydrogen bonding and preserve the functional properties of the individuals. 93 ,96-99 In addition, the AK linker retains MHC-II epitopes active independently within the immune system.93,96,97,100 Moreover, the KFER linker provides flexibility between linked linear B-cell epitopes, allowing effective interaction with B-cell receptors.93,96,97,101
Physicochemical properties analysis
The physicochemical properties of the vaccine, such as molecular weight, total amino acid count, instability and aliphatic index, isoelectric point (pI), grand average of hydropathicity (GRAVY), the total number of positively and negatively charged residues, and the total atom count, were predicted using the Expasy ProtParam server (http://web.expasy.org/protparam/). 102 Following that, the solubility of the vaccine was predicted by the SOLpro and SOSUI server.103-106 The allergenicity of the vaccine was predicted using the AllergenFP v.1.0 (https://ddg-pharmfac.net/AllergenFP/), 107 AllerTOP v. 2.0 (https://www.ddg-pharmfac.net/AllerTOP/index.html) 84 and AlgPred server (http://crdd.osdd.net/raghava/algpred/). 108 The antigenicity of the vaccine was also calculated by ANTIGENpro (https://scratch.proteomics.ics.uci.edu/) 109 and VaxiJen 2.0 server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). 83
Secondary structure prediction
The secondary structure of the vaccine was predicted by the GOR4 (https://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_gor4.html), SOPMA (https://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) and the PSIPRED (https://academic.oup.com/bioinformatics/article/16/4/404/187312).110-113 The GOR4 employs information theory and Bayesian statistical analysis to predict the secondary structure of a protein. 114 In contrast, the PSIPRED server employs 2 feed-forward neural networks to analyze the output from PSI-BLAST (Protein Specific Iterated Basic Local Alignment Search Tool) for secondary structure prediction. 115 However, the SOPMA server can predict approximately 69.5% of amino acids from a protein sequence regarding a 3-state illustration (alpha-helix, beta-sheet, and coil) of the secondary structure. 112
Tertiary structure prediction, refinement, and validation
The tertiary structure (3D) of the vaccine was predicted by the I-TASSER server (https://zhanggroup.org/I-TASSER/). The server performs multiple threading alignments and iterative template fragment assembly simulations to predict 3D structure of a protein.82,116 The 3D structure of the vaccine was applied for further structural refinement by the GalaxyWEB server (https://galaxy.seoklab.org/). 117 Further structural validation of the vaccine was carried out by the SAVES v6.0 server (https://saves.mbi.ucla.edu/), which defines the stereochemical quality of the predicted vaccine model.118-122 Finally, the ProSA web server (https://prosa.services.came.sbg.ac.at/prosa.php) was used to detect potential errors in the predicted 3D model, with the calculated Z-score indicating the model’s accuracy.123-125
Discontinuous B-cell epitope prediction
The discontinuous B-cell epitopes of the vaccine were predicted by the Disco Tope 2.0 (https://services.healthtech.dtu.dk/services/DiscoTope-2.0/) 126 and Ellipro server (http://tools.iedb.org/ellipro/). 127 The Disco Tope 2.0 server predicted the epitopes by surface accessibility computation, 126 where the ElliPro server predicts through a combination of 3 algorithms. 127
Disulfide engineering of the vaccine
Disulfide engineering is applied in proteomics to get a more stable folded protein structure. This allows an increased denatured state free energy and a decreased conformational entropy of a protein structure. 128 Using the Design 2.13 algorithm, we identified potential cysteine disulfide bonding pairs within the vaccine structure (http://cptweb.cpt.wayne.edu/DbD2/). 129 The residue pair was selected for performing mutation based on the lowest energy and χ3 angle in a range of –87° or +97° ± 30.97,129,130
Molecular docking study
Toll-like receptors (TLRs), also known as pattern recognition receptors (PRRs), are primarily responsible for identifying infection-causing agents and activating both innate and adaptive immune cells to eliminate the infection promptly. However, these receptors have been crucial in vaccine-mediated immune responses, especially the human TLRs; human TLR-2 (Toll-like receptor 2) and human TLR-4 (Toll-like receptor 4). 131 These receptors identify diverse PAMPs, considering them adaptable targets for several vaccine antigens. 132 Furthermore, the binding of these receptors induces strong immunological responses, which are crucial for the effectiveness of vaccination.132,133 Therefore, the tertiary structure of human TLR-2 (PDB: 2Z7X) and human TLR-4 (PDB: 3FXI) were retrieved from the PDB (Protein data bank) database (www.rcsb.org) to perform molecular docking between the vaccine and aforementioned TLRs. Finally, PyMOL (https://pymol.org/2/) and PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html) were used to analyze and visualize docked complex structures. It is worth mentioning that the complete molecular docking study was carried out by the CLUSPRO 2.0 (https://cluspro.org/login.php?redir=/queue.php) server.134-137
Molecular dynamic simulation
The GROningen MAchine for Chemical Simulations (GROMACS) (version 2022.3) (https://www.gromacs.org/) (Abraham et al) software was used for the molecular dynamic studies of the “vaccine-TLR-2” and “vaccine-TLR-4” complexes along with the vaccine apo structure. The CHARMM36 m force field 138 was used for the simulation run, where transferable intermolecular potential with 3 points (TIP3P) water model was used to build a water box with its edges at a 1 nanometer (nm) distance from the protein surface. The TIP3P water model is one of the most widely used models in molecular dynamics simulations for water, which provides a simulation run with better accuracy, transferability, and efficiency. 139 In addition, the systems were neutralized using the Na + ions. 140 After energy minimization, the system underwent isothermal-isochoric (NVT) and isobaric (NPT) equilibration. A 100-nanosecond (ns) molecular dynamics simulation was then conducted using periodic boundary conditions and a time step of 2 femtoseconds (fs), with snapshot intervals set at 100 picoseconds (ps) for trajectory analysis. Upon completing the simulation, the root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and solvent accessible surface area (SASA) were calculated using the modules integrated into the GROMACS software.
The RMSD values quantify the mean distance between the atoms of a reference structure and the corresponding atoms in the trajectory snapshots of the simulated system and are often used to evaluate the convergence of MD simulations, to measure the similarity between distinct conformations, and to detect substantial structural alterations such as protein folding or binding events.141,142 Moreover, the RMSF measures the degree of flexibility or variation of individual atoms or residues within a biomolecular system throughout the simulation, which also provides valuable insights into the functional dynamics of proteins, including fluctuations in the active site and movements within protein domains.143,144 In addition, the Rg measures the degree of compactness or spatial arrangement of atoms in a biomolecular system. Therefore, this analysis has been applied to investigate protein folding, aggregation, and conformational changes. Alterations in the Rg values can suggest compression or expansion of the molecule, potentially linked to shifts in structure or interactions with other molecules. 145 Meanwhile, the SASA calculates the biomolecule’s total solvent-accessible surface area. It reveals details on the molecule’s hydrophobicity, binding surfaces, or conformational changes as they pertain to its solvent exposure. The binding of ligands to proteins, interactions between proteins, and folding of proteins are all the most common subjects for SASA analysis. Observations of changes in SASA may provide information about which residues or domains have been exposed to solvent, which might provide evidence on binding kinetics or structural transitions. 146 Finally, the ggplot2 package in RStudio was used for generating the graphical representation of each analysis.
Generalized Born and surface area solvation paired with molecular mechanics for computing free energy (MM-GBSA)
The free energy of the interactions between “vaccine-TLR-2” and “vaccine-TLR-4” was calculated using molecular mechanics and Generalized Born methods, specifically through the MM-GBSA approach. This involved analyzing various factors, including binding and electrostatic interactions, van der Waals forces, polar and nonpolar components, and the effects of bound interactions. To perform these calculations, the HawkDock server (http://cadd.zju.edu.cn/hawkdock/) was used, which applies the Generalized Born equation.147,148
Normal Mode Analysis (NMA)
The functional dynamics of macromolecules can be effectively analyzed through normal mode analysis (NMA).149-151 Accordingly, the functional movements of the macromolecular complexes “vaccine-TLR-2,” and “vaccine-TLR-4” were investigated using the iMODS server (http://imods.chaconlab.org/). 151 A variety of motion configurations, such as affine-model arrows, vector fields, and modal animations, are made available by this platform. Furthermore, it is capable of calculating a variety of attributes, including mobility (B-factor), deformability, eigenvalues, covariance maps, and linkage matrices. 151 The B-factor measures the extent to which atoms deviate from their equilibrium positions in a structure, while the deformability plot visually illustrates protein flexibility, emphasizing regions such as coils or domain linkers. In addition, the eigenvalue serves as an indicator of the complex’s stability, with higher values signifying greater structural robustness.150,151
Codon optimization and in silico cloning
The Java Codon Adaptation Tool (JCat) server (http://www.jcat.de/Start.jsp) was used for codon optimization of the vaccine utilizing the Escherichia coli strain K12. The server determines codon adaptation index (CAI) and GC contents of a protein to evaluate its expression level. However, the CAI value ⩾ 0.8 is considered a good score, while the value ⩾ 1.0 is the best, and the GC contents, ranging from 30% to 70%.61,152 During the process of cloning the optimized vaccine gene sequence into the E coli plasmid vector pUC19, the restriction sites EcoRI and AhdI were ligated to the N-terminal and C-terminal regions of the vaccine sequence, respectively. The pUC19 has a high copy number (~100–500 copies per cell), making it ideal for robust expression and amplification of the inserted gene sequences in both computational and experimental setups. 153 In addition, this vector contains extensive multiple cloning sites (MCS) within lacZα, which facilitates the insertion of foreign DNA sequences and supports a wide range of restriction enzyme compatibility. 153 Finally, the optimized vaccine sequence was introduced into the plasmid vector pUC19 using the SnapGene program (https://www.snapgene.com/free-trial/).
Immune simulation
Immune simulation of the vaccine was carried out by the C-ImmSim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/). 154 This platform models both humoral (antibody-driven) and cellular (T-cell mediated) immune responses in mammals following vaccination.61,155 The vaccination protocol involved 3 doses, administered at 4-week intervals. Default settings were used for the simulation, including 100 adjuvant doses and 1000 antigen doses. The time points for analysis were set at days 1, 84, and 168. In addition, the simulation volume was set at 50, with 1000 steps in total, and a random seed value of 12 345 was applied. Lipopolysaccharides (LPS) were not included in the simulation.
MRNA prediction of the vaccine
The secondary structure of the vaccine mRNA was predicted using the RNAfold server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). 156 This tool calculates the minimum free energy (MFE) of the mRNA structure based on thermodynamic principles.157,158 The optimized DNA sequence obtained from the JCat server was subsequently transcribed into a potential RNA sequence through the DNA-RNA-Protein conversion process using the platform available at http://biomodel.uah.es/en/lab/cybertory/analysis/trans.htm.
Results
Sequence retrieval
The amino acid sequence of the CHIKV polyprotein was retrieved from the NCBI database and used for further analysis in this study. A summary of the study is presented in Figure 1.

The summary of the mRNA vaccine design study.
MHC-I binding epitope prediction
The chosen segment of the structural polyprotein (including the E1, E2, and E3 regions) was used to predict potential MHC-I binding epitopes using the IEDB database and the Net-MHC 4.0 server. Peptides of 9-mer length were selected based on their affinity scores and a percentile rank of less than or equal to 1.00 (Table 1). These peptides were further evaluated to ensure they were antigenic, nonallergenic, and nontoxic, making them suitable for inclusion in the vaccine design.
List of chosen MHC-I epitopes from the polyprotein sequence and their antigenic, allergic, and toxic properties.

MHC-II binding epitope prediction
Using the IEDB and NetMHCIIpan 4.0 servers, the MHC-II binding epitopes of the structural polyprotein were predicted. Six 15-mer peptides synthesized from the polyprotein were chosen referring to their affinity and percentile rank. The chosen peptide sequences were also passed the antigenic, nonallergenic, and nontoxic criteria. Furthermore, the epitopes were shown to possess the ability to induce IFN-γ, but no stimulation of IL-10 was seen. Following the screening process, all of these peptides were used to develop prospective vaccines (Table 2).
List of chosen MHC-II epitopes from the polyprotein sequence and their antigenic, allergic, and toxic properties, allergenicity, IFN-γ and IL-10 inducing capability, and toxicity.

Molecular docking between T-cell epitopes and MHC molecules
The docking between the human MHC-I and MHC-II and T-cell epitopes was carried out using the HDOCK server. For each docking, HDOCK generated 10 models where the model with the highest confidence and lowest possible docking score was selected from each epitope. In the docking between MHC-I molecule and selected MHC-I epitopes, the docking scores were −161.05, −188.42, −247.11, −184.86, −230.39, −172.98, −277.26, −234.40, and −177.02 KJ/mol for STKDNFNVY (Figure 2A), LLYPDHPTL (Figure 2B), KVFTGVYPF (Figure 2C), KARNPTVTY (Figure 2D), VMRPGYYQL (Figure 2E), VTLEPTLSL (Figure 2F), YYYELYPTM (Figure 2G), HPHEIILYY (Figure 2H), and ATVPFLLSL (Figure 2I) epitopes, respectively.

The docking between the human MHC molecules (MHC-I and MHC-II) and T-cell epitopes (MHC-I and MHC-II), where “MHC-I protein-MHC-I epitopes” (A-I), and “MHC-II protein-MHC-II epitopes” (J-O) are depicted in surface view model.
Subsequently, in the docking between MHC-II molecule and selected MHC-II epitopes, the docking scores were: −189.64, −193.62, −161.07, −165.99, −220.32, and −177.94 KJ/mol for VTGYACLVGDKVMKP (Figure 2J), VGVPYKTLVNRPGYS (Figure 2K), TGYACLVGD KVMKPA (Figure 2L), RCITPYELTPGATVP (Figure 2M), KVTGYACLVGDKVMK (Figure 2N), and GVPYKTLVNRPGYSP (Figure 2O), respectively.
Linear B-cell epitope prediction
Using the IEDB and ABCpred servers, 7 peptide sequences from the polyprotein were chosen to serve as linear B-cell epitopes. Vaccines were developed using peptide sequences that were shown to be safe, nonallergic, and antigenic (Table 3).
List of chosen B-cell epitopes from the polyprotein sequence and their toxicity, allergenicity, and antigenicity.
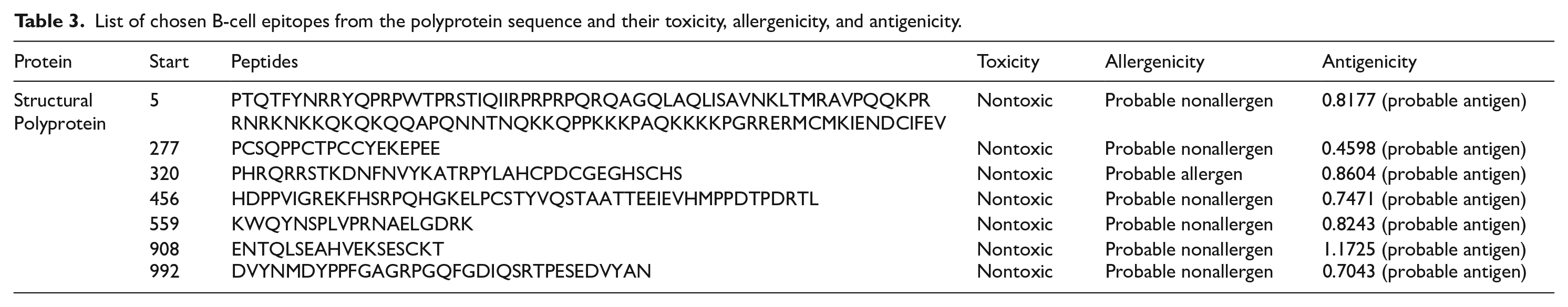
Vaccine mapping
The vaccine map was constructed using the most relevant epitopes derived from the structural polyprotein. The chosen epitopes and adjuvants in the designed vaccine were connected using linkers comprising EAAAK, AYY, AK, and KFER. Through the EAAAK linker, the adjuvant—50S ribosomal protein L7/L12—was joined to the MHC-I epitopes. The AK linkers were used to further connect the MHC-II epitopes. While the KFER linkers connected the linear B-cell epitopes to the rest of the vaccine design and each other, they also connected their locations (Figure 3).
A potential vaccine against CHIKV using mRNA technology. A schematic representation of a vaccine sequence is provided, showing various components such as adjuvant, epitopes, and linkers. The adjuvant is depicted in yellow, MHC-I epitopes in orange, MHC-II epitopes in green, and B-cell epitopes in blue. The linkers are illustrated in different shades of blue, purple, and maroon.
Physicochemical properties analysis
The physicochemical characteristics of the vaccine were analyzed using the ExPASy ProtPram server. The analysis indicated that the vaccine is composed of 657 amino acids, with a molecular weight of 73 640.52 Da. Its isoelectric point (pI) is 9.38, which classifies it as acidic (pH < 7). The GRAVY score of −0.639 suggests that the vaccine is hydrophilic and water-soluble. However, with an instability index of 44.59, the protein is predicted to be unstable (Table 4). The aliphatic index of 65.11 indicates that the vaccine is hydrophobic, containing aliphatic side chains. In addition, it was predicted to have 72 negatively charged and 104 positively charged residues. According to the SOSUI server, the vaccine is classified as a soluble protein with a hydrophobicity value of −0.345. The SOLpro server predicted a high likelihood of successful solubility when expressed in E coli, with a score of 0.998. Allergenicity predictions from AllerTOP v. 2.0, AllergenFP v.1.0, and AlgPred servers determined that the vaccine was nonallergenic (Table 4). Finally, antigenicity predictions from Vaxijen 2.0 and ANTIGENpro servers identified the vaccine as a probable antigen (Table 4).
The physicochemical and immunological aspects of the vaccine.

Secondary structure prediction
The GOR4, SOPMA, and PSIPRED servers were used to forecast the vaccine’s secondary structure. With 52.36% being random coil, 31.66% being alpha helix, and 15.98% being extended strands (beta sheet), according to the GOR4 server. Alternatively, the secondary structure of the vaccine was predicted by the SOPMA server to have an alpha helix at 32.42%, extended strands at 15.68%, and a random coil at 43.53% (Supplementary Table 1). In addition, the PSIPRED server supplied the secondary structure of the protein, including its coil, helix, and strand properties, using 3-state prediction (Figure 4 and Supplementary Figure 1).

The secondary structure of the vaccine is predicted using PSIPRED. The confidence in the forecast is expressed by the first bar (Conf), with each consecutive bar’s size signifying a separate degree of confidence. The yellow hue in the second bar (Cart) symbolizes the beta-sheet, the pink color denotes the helix, and the gray color reflects the vaccine’s coil structure. The fourth bar (AA) and the third bar (Pred) reflect 3 unique amino acid sequences and structural characteristics, respectively.
Tertiary structure prediction, refinement, and validation
Out of the 5 models predicted by the I-TASSER server, the first model was chosen due to its highest C-score of −0.87, a TM-score of 0.74 ± 0.12, and an RMSD of 7.5 ± 3.5 Å. The refined 3D structure, obtained from the GalaxyWEB server, had an RMSD of 0.240, a MolProbity score of 1.105, and 93.5% of residues in the favored regions of the Ramachandran plot, indicating a well-optimized 3D model. When the I-TASSER predicted model was superimposed with the GalaxyWEB refined model, an RMSD of 0.575 Å was observed (Supplementary Figure 2), confirming the refined model’s reliability for further analysis. The Ramachandran plot of the energy-minimized model showed 82.8% of residues in the most favored regions, 12.7% in the additionally allowed regions, and 0.9% in the generously allowed regions (Figure 5A). In comparison, the unrefined model had 53.8%, 35.0%, and 5.8% in these regions, respectively. The ProSA server confirmed the quality of the refined model with a Z-score of −4.17, significantly better than the unrefined model’s score of −2.25 (Figure 5B). In addition, the refined vaccine model had an ERRAT score of 75.949 (Figure 5C).

Validation of the I-TASSER predicted model of the constructed vaccine following Ramachandran plot refinement (A), Z-score (B), and ERRAT (C) analysis.
Discontinuous B-cell epitope prediction
The Discotope 2.0 server identified 398 discontinuous B-cell epitope residues from the vaccine’s total amino residues (657) (Figure 6 and Supplementary Table 2). These epitopes have different scoring ranges from 0.557 to 0.672 with an individual number of residues (Supplementary Table 2).

Three-dimensional illustration of discontinuous B-cell epitopes of the vaccination design (A-D). Yellow-colored surfaces are used to depict the discontinuous B-cell epitopes, and gray sticks are used to represent the whole vaccine part.
Disulfide engineering of the vaccine
A total of 42 amino acid pairs were determined to be eligible for disulfide bonds using the Disulfide by Design 2.13 server. However, when energy and χ3 values were incorporated, the number of couples decreased. Therefore, 2 mutations were introduced at the SER 203-CYS 547 and PHE 522-GLY 642 pairs of residues (Supplementary Figure 3). The χ3 values between −87 and +97 and energy values below 2.2 were suggested for disulfide engineering (Supplementary Table 3).
Molecular docking study
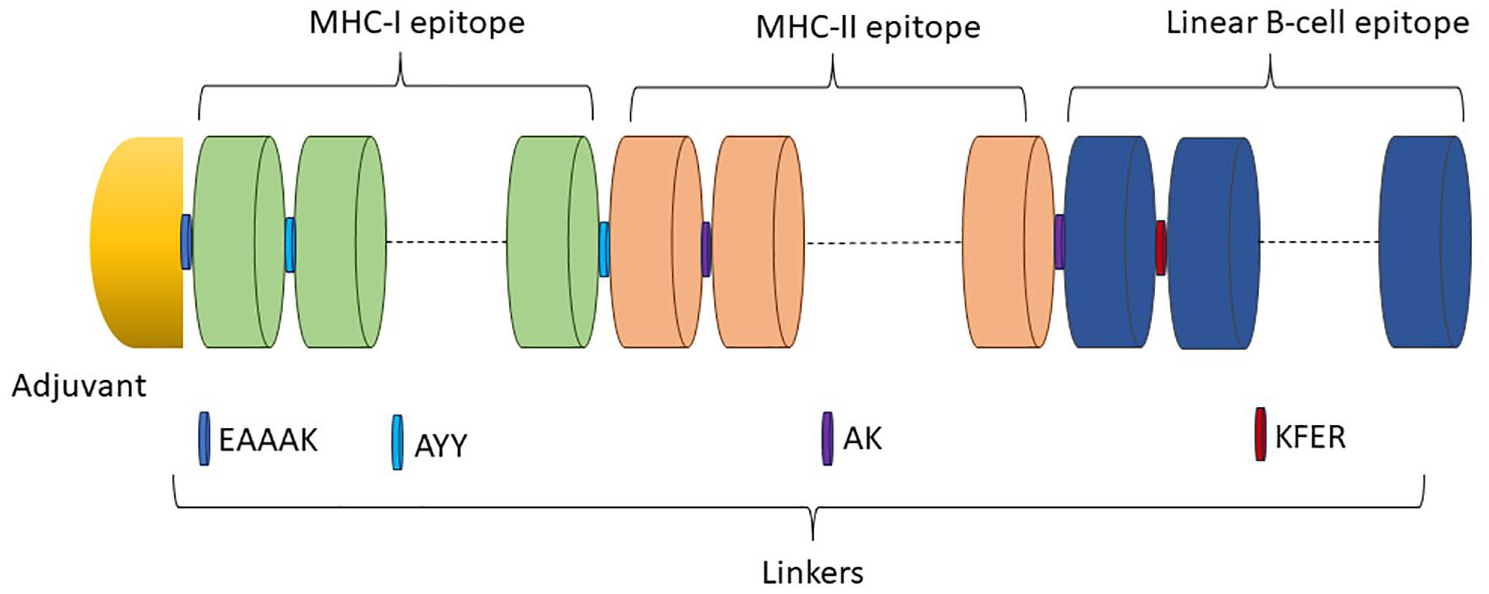
The docking of the vaccine construct with human receptors TLR-2 and TLR-4 was carried out using the Cluspro 2.0 server. For each receptor, Cluspro generated 30 models. The selected model for the “vaccine-TLR-2” complex had a center and lowest energy scores of −1005.1 and −1027.7, respectively, while the chosen “vaccine-TLR-4” complex had scores of −1100.1 and −1212.4 (Figure 7 and Table 5). The docked structures were further analyzed and visualized using PyMOL and PDBsum. According to PDBsum, the “vaccine-TLR-2” complex had 26 hydrogen bonds, 13 salt bridges, and 280 nonbonded interactions (Figure 8A and Table 5), while the “vaccine-TLR-4” complex had 22 hydrogen bonds, 2 salt bridges, and 235 nonbonded interactions (Figure 8B and Table 5).
The interaction of human receptors (cyan) with the mRNA vaccine (green) construct.
The docking scores and interactions between the vaccine-receptor complexes.


The interaction between “vaccine-TLR-2” (A) and “vaccine-TLR-4” (B) complexes. The vaccine and the receptors are portrayed as red and violet, while their interactions are marked with numerous colors reflecting the peculiarities of the bonds.
Molecular dynamic simulation
The Root Mean Square Deviation (RMSD) of the “vaccine,” “vaccine-TLR-2,” and “vaccine-TLR-4” were calculated to evaluate the stability of the systems. Protein conformational changes are correlated with variations in the RMSD value. However, after 25 ns, the RMSD value for the vaccine steadily increased (Figure 9A), while the RMSD values for “vaccine-TLR-2” (Figure 9B) and “vaccine-TLR-4” (Figure 9C) gradually increased throughout the simulation. In addition, the Root Mean Square Fluctuation (RMSF) was calculated for the structures to assess the regional flexibility. The higher the RMSF, the higher the flexibility of a given amino acid position. Throughout the simulation, the motility pattern of the “vaccine-TLR-2” (Figure 9E) is significantly different than “vaccine-TLR-4” (Figure 9F) and vaccine apo (Figure 9D), especially at the C and N terminals. The motility pattern in RMSF provides valuable insights into the functional dynamics of a protein or protein-protein complex, thereby providing the fluctuations states in the active site and their movements within protein domains. Thus, the lower the RMSF value, the better the structural integrity of a protein and protein-protein complex in a simulated physiological environment. 159

The RMSD and RMSF analysis of the vaccine, “vaccine-TLR-2” and “vaccine-TLR-4” structures. The plot represents the RMSD of vaccine (A), “vaccine-TLR-2” (B), and “vaccine-TLR-4” (C) with maroon, green, and blue color lines, respectively. Similarly, the RMSF of vaccine (D), “vaccine-TLR-2” (E), and “vaccine-TLR-4” (F) were depicted in maroon, green, and blue color line plots, respectively.
The radius of gyration (Rg) measures a protein’s compactness, where a stable Rg value indicates consistent protein folding and higher Rg values suggest reduced rigidity in the biological system. Fluctuations in Rg typically point to protein unfolding. The compactness of the “vaccine-TLR-4” complex remained stable after 25 ns, with consistent Rg values (Figure 10C). The Rg values of the vaccine alone showed a gradual increase over time (Figure 10A), while for the “vaccine-TLR-2” complex, the Rg decreased after 50 nanoseconds (Figure 10B). In addition, Solvent Accessible Surface Area (SASA) is used in molecular dynamics simulations to assess how much of a protein’s hydrophobic core is exposed to solvents. Higher SASA values indicate more protein surface exposed to water, while lower values suggest the protein is buried within its hydrophobic core. After 25 ns, the SASA values for the vaccine (Figure 10D), “vaccine-TLR-2” (Figure 10E), and “vaccine-TLR-4” (Figure 10F) showed minimal variation throughout the simulation.

The Rg and SASA analysis of the vaccine, “vaccine-TLR-2” and “vaccine-TLR-4” structures. The plot represents the Rg of vaccine (A), “vaccine-TLR-2” (B), and “vaccine-TLR-4” (C) with maroon, green, and blue color lines, respectively. Similarly, the SASA of vaccine (D), “vaccine-TLR-2” (E), and “vaccine-TLR-4” (F) were depicted in maroon, green, and blue color line plots, respectively.
Generalized Born and surface area solvation paired with molecular mechanics for computing free energy (MM-GBSA)
The HawkDock server determined that the total binding free energy for the “vaccine-TLR-2” complex was −132.27 kcal/mol. The individual contributions from VDW, ELE, GB, and SA were −193.07, −2481.15, 2571.16, and −29.21 kcal/mol, respectively. In the case of the “vaccine-TLR-4” complex, the total binding free energy was found to be −140.72 kcal/mol, with VDW, ELE, GB, and SA contributions of −188.38 kcal/mol, −4873.39 kcal/mol, 4944.61 kcal/mol, and −23.56 kcal/mol, respectively (Supplementary Table 4).
Normal Mode Analysis (NMA)
The NMA studies on the “vaccine-TLR-2” and “vaccine-TLR-4” docked complexes were conducted using the iMODS server, providing a comprehensive assessment of their structural integrity and alterations. The deformability graph emphasizes the flexible areas of the docked complexes by using graphical peaks (Supplementary Figure 4). In terms of structural flexibility and stiffness, the eigenvalues of the “vaccine-TLR-2” and “vaccine-TLR-4” complexes were found to be 2.839671e-05 and 4.557879e-05, respectively (Supplementary Figure 4). In this analysis, the covariance map illustrates the relationships between residue movements within the structures, with red, white, and blue colors representing correlated, uncorrelated, and anticorrelated movements, respectively (Supplementary Figure 4).
The B-factor graph further demonstrated the simulation of the docked complexes using NMA and the PDB sector. The B-factor values represent the extent of atomic displacements, revealing that the “vaccine-TLR-2” and “vaccine-TLR-4” complexes show a higher level of deformability, indicating greater flexibility (Supplementary Figure 5). The variance plot depicts the sum of the variance in cyan and the individual variance in purple. The first 3 modes out of 20 in the docked complexes combine to explain 80% of the overall variation (Supplementary Figure 5). The elastic map of the docked complexes, which comprises darker gray patches, reveals the interactions among atoms, therefore implying the existence of areas with higher rigidity (Supplementary Figure 5).
Codon optimization and in silico cloning
To optimize the codons for the vaccine, we used the Java Codon Adaptation Tool (JCat) to ensure that the protein is expressed at its highest level in the E coli strain K12. The server generated an optimized codon sequence that is 1966 nucleotides long. In addition, the value of the codon optimization index (CAI) and the average GC content of the adapted sequence were determined to be 1.0% and 51.45%, respectively. These findings indicate that the protein expression in the E coli host will be maximized. Finally, with the help of SnapGene software, we successfully incorporated modified codon sequences into the plasmid vector pUC19, resulting in the creation of a recombinant plasmid sequence (Figure 11).

In silico pUC19 plasmid cloning. SnapGene software free-trial (https://www.snapgene.com/free-trial/) allowed the expression of pUC19 plasmid vector conjugated with the vaccine sequence. The red section represents the vaccine’s gene coding, while the black circle represents the vector backbone.
Immune simulation
Following vaccination (after a 3-dose regime), there was a notable increase in the B-cell population, especially elevated amounts of memory B cells. The total amounts of B cells remained active, indicating a sustained year-long immunity (Figures 12A and 12B). T cell responses, including helper T cell (TH), cytotoxic T cell (TC), and regulatory T cell (Treg), were evident in the postvaccine regime. The study also showed elevated expression of both active and memory TH cells on day 60, which gradually decreased though but was sustained for about a year (Figures 12C and 12D). Furthermore, active TCs exhibited high-level expression after a postvaccination regime and remained active for a year around (Figures 12E and 12F).
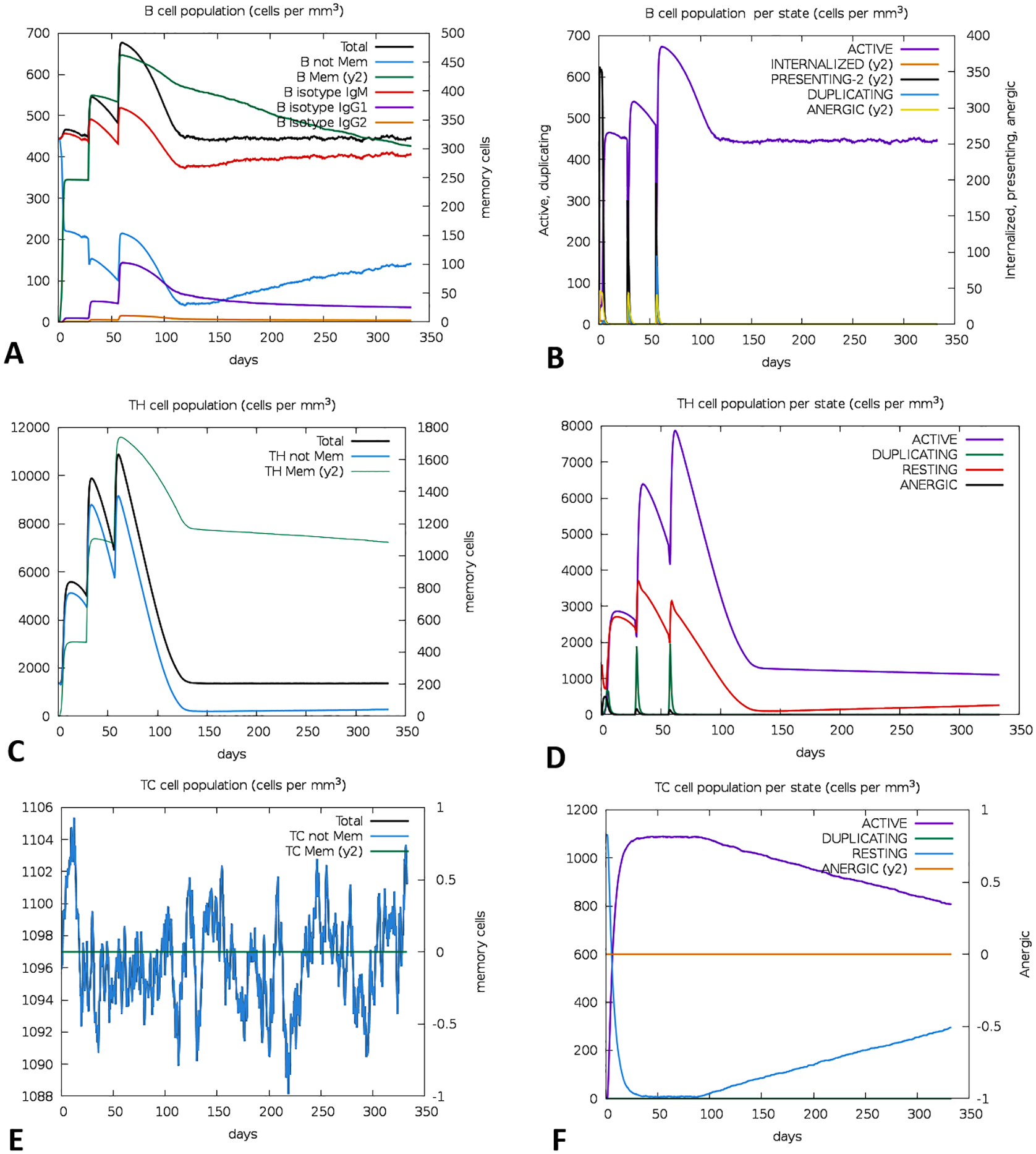
An immunological simulation of the vaccine design was supposed to be predicted using C-ImmSim. The progression of B-cell populations (A, B), the progression of TH-cell populations (C, D), and the progression of TC-cell populations (E, F) after 3 consecutive injections.
MRNA prediction of the vaccine
The RNAfold server revealed the secondary structure of the vaccine mRNA (Figure 13), exhibiting an MFE score of −561.60 kcal/mol for the optimum structure and −328.64 kcal/mol for the centroid structure. The predicted thermodynamic free energy was −591.41 kcal/mol, and the MFE structure showed no correlation with the ensemble. This finding aligns with previous research, which also showed that the vaccine’s mRNA structure remains stable during entry, transcription, and expression in the host.

Predicted mRNA structure of the vaccine using RNAfold web server. The base pair probabilities of the mRNA vaccine with the least free energy (A) and centroid (B) structure; and the positional entropy of the mRNA vaccine with the lowest free energy (C) and centroid (D) structure.
Discussion
One of the greatest scientific triumphs of the modern era is the development of safe and effective vaccinations to prevent infectious and emerging diseases. The most challenging aspect of vaccine design is identifying antigenic areas or epitopes that trigger host immune responses. Over the past 20 years, various approaches have emerged to develop effective vaccines or immunotherapies to combat emerging infectious diseases. 160 With the advancement of sequencing and a well-organized database system, the reverse vaccinology approach has become one of the most intriguing strategies in recent years. 161 This approach has been used in computational vaccine design against infectious diseases, including COVID-19,162-164 as well as for combating various cancers, including colon, lung, liver, etc.165-167 Such an approach results in vaccines with high magnitude in terms of specificity and safety, lower manufacturing costs, and elevated cellular and humoral immune responses. 168 Immunogenicity analysis, vaccine mapping, and epitope mapping are considered as the three core fundamental concepts for developing such in silico–based vaccines. 169 The global emergence of CHIKV heightens the need for more effective interventions targeting this virus. Therefore, the provision of a safe and efficacious in silico–based vaccine should take precedence regarding public health concerns.61,170
There is a diverse range of approaches in the development of CHIKV vaccines and vaccine candidates, each showing potential. These include live-attenuated CHIKV, recombinant CHIKV with nsP3 deletion, virus-like particles expressed from mammalian cells plus adjuvant, recombinant simian adenovirus vaccine expressing CHIKV structural proteins, recombinant measles vaccine expressing CHIKV structural proteins, mRNA vaccine encoding structural proteins, and mRNA encoding a CHIKV neutralizing antibody. Two mRNA vaccines, one producing CHIKV neutralizing antibody and the other encoding structural proteins, are undergoing phase I clinical trials. 47 ,171-174 Several CHIKV vaccines targeting specific epitopes or peptide antigens from the virus’ proteome have been developed using reverse vaccinology techniques. The E1, E2, 6 K, and E3 antigens of CHIKV were used by Mahmoodi et al. (2023) 175 to create a multiepitope-based vaccination against the virus. However, little research has been done on potential multiepitope vaccines against CHIKV. Sánchez-Burgos et al. (2021) 176 designed another multiepitope-based vaccine against CHIKV targeting the CHIKV structural and nonstructural proteins, though their research was restricted to vaccine mapping. Furthermore, by focusing on the structural polyprotein, Tahir ul Qamar et al. (2018) 177 created a peptide-based vaccination against CHIKV, where they docked the vaccine with the HL-B7 allele to analyze binding specificity prior to immune responses. Another multiepitope peptide vaccine targeting the E1 and E2 glycoproteins of CHIKV was created by Zakaria et al. (2024) in 2023. It has been demonstrated to elicit humoral and adaptive immune responses by strong interactions with the TLR-1/2 receptor, as well as increased activation of adaptive immune cells and induction of cytokines that can combat CHIKV. 178 However, Jaan et al. (2022) 179 designed an mRNA vaccine against CHIKV using the envelop glycoprotein of the virus, where they identified the most immunogenic and antigenic T- and B-cell epitopes. While there have been relatively few mRNA-based vaccine candidates developed for CHIKV compared to the multiepitope-based vaccine; nonetheless, the potential of this vaccine platform demonstrated promising results in both in vivo and in silico techniques. Considering this fact, we successfully developed an mRNA vaccine targeting the highly potent antigenic protein of CHIKV, namely structural polyprotein.
This study predicted and used the MHC-I, MHC-II, and B-cell epitopes from the structural polyprotein in subsequent vaccine development. All the selected MHC-I epitopes were further validated as potentially antigenic, nonallergenic, and nontoxic. We also successfully validated the MHC-II epitopes with potential antigenicity and IFN-γ producibility. IFN-γ enhances antigen presentation via the MHC class II pathways, making malignant and virus-infected cells more immunogenic, thus rendering them more accessible for immune surveillance and effector mechanisms to identify and eliminate them.180,181 Also, the selected B-cell epitopes were shown to have antigenic characteristics that were minimized or nonallergenic. Allergenic responses must be carefully considered in vaccine design to prioritize safety and efficacy while maintaining reliance on vaccinations as critical therapeutic agents.182,183 Finally, we used several linkers and adjuvants to construct the final multiepitope vaccine with the selected epitopes. According to physicochemical properties, the vaccine was predicted as a soluble protein. Therefore, it is evident that the protein might be functionally stable under body conditions.61,87,92,184 In addition, the theoretical pI of the vaccine showed it is probably a basic protein (pI of 9.38). The vaccine has an aliphatic index of 65.11, indicating that the vaccine is probably a hydrophobic protein containing aliphatic side chains.61,167,185 In a similar study on human papillomavirus, the mRNA vaccine had a pI of 8.73. The study also included a GRAVY score of −0.648 for the proposed vaccine, whereas the GRAVY score of the CHIKV vaccine was −0.639. 186 However, the instability index of our proposed vaccine was 44.51, whereas the instability index of the papilloma vaccine was 57.24, indicating a comparatively stable structure of our proposed vaccine. 186 The process of protein folding into its secondary and tertiary structures needs to be considered while developing an effective vaccine. Antigens found in unfolded proteins and α-helical coils exert an essential function in protein-specific immune responses. Therefore, these 2 structural antigens are prime targets for antibodies that mount in response to unexpected infections.187,188 Subsequently, we predicted both the secondary and tertiary structure of the vaccine and found a satisfactory outcome. The vaccine’s secondary structure contains substantial amount of alpha helix, beta sheet, and coil structure to be a well-stable protein. In addition, the Ramachandran plot analysis revealed that a substantial amount (82.8%) of the vaccine residues were situated within the preferred regions, conferring its tertiary structure integrity. Also, the Z-score was calculated as −4.17 KJ/mol, indicating the vaccine model’s overall quality. A docking study was performed using human TLR-2 and TLR-4 to assess the potential association between the vaccine and TLRs on immune cells. According to this analysis, the vaccine model exhibited a substantial affinity toward TLR-2 (lowest energy score of −1005.1 KJ/mL) and TLR-4 (lowest energy score of −1027.7 KJ/mL) receptors. Also, 26 hydrogen bonds were found within the “vaccine-TLR-2” complex, whereas 22 hydrogen bonds were found in the “vaccine-TLR-4” complexes. Molecular dynamic simulation was also performed to validate the structural integrity of the vaccine and vaccine-TLRs complexes. The RMSD analysis depicted distinct conformational dynamics among the vaccine, “vaccine-TLR-2,” and “vaccine-TLR-4” complexes throughout the simulation. Considerably, the dynamic simulation showed that the vaccine model had a consistent rise in RMSD after 25 nanoseconds, suggesting structural instability, while “vaccine-TLR-2” and “vaccine-TLR-4” exhibit gradual increases, showing alterations in their conformations. RMSF analysis highlights the unique mobility patterns of the “vaccine-TLR-2,” particularly in the C and N terminals, distinct from the “vaccine-TLR-4” and the vaccine alone. The Rg values of this structure provide additional clarity on the stability. The “vaccine-TLR-4” complex maintains a consistent level of compactness, while the vaccine alone shows a progressive increase in compactness over time. Interestingly, the “vaccine-TLR-4” complex experiences a drop in compactness after 50 nanoseconds. The measurement of SASA reveals that all complexes exhibit similar levels of exposure to solvents after 25 ns, indicating consistent interactions inside the hydrophobic core. This comprehensive evaluation highlights the complex and unique characteristics of the vaccine, “vaccine-TLR-2” and “vaccine-TLR-4” complexes, offering vital insights into their structural stability, flexibility, compactness, and exposure to solvents throughout the simulation.
Codon optimization was used to evaluate the expression of the recombinant vaccine in the E coli cloning vector, particularly the K12 strain. The computational result showed that the vaccine had a high expression level, with a GC content of 51.44% and a CAI score of 1.0. According to the immune simulation, the vaccine had the ability to trigger both innate and adaptive immune responses. Innate responses were evidenced by the involvement of NK cells, dendritic cells (DC), and macrophages (MA). In contrast, adaptive responses were shown through helper T cells (TH), cytotoxic T cells (TC), and B cell activity. Memory B-cells and T-cells were also present, with B-cell immunity lasting up to a year. The activation of TH cells and the release of IFN-γ and IL-2 were notable, with an initial spike in these cytokines following the first dose and sustained elevated levels upon repeated antigen exposure. Immunoglobulins (IgM and IgG) production indicated a strong humoral immune response. According to the RNAfold server, the mRNA secondary structure of the vaccine was predicted to be stable with an MFE score of −561.60 kcal/mol (optimal) and −328.64 kcal/mol (centroid). Thermodynamic free energy was predicted at −591.41 kcal/mol, with 0.00% frequency for the MFE structure, suggesting the postentry stability and possible expression within the host.
The computational studies offer substantial evidence for vaccine efficacy, and it is essential to validate these findings across several preclinical trials. Variations in immune system dynamics and TLR expression between species may impede the execution of in silico results to humans, requiring meticulous selection of models to activate human immunological responses.94,189
Furthermore, the vaccine’s possible side effects and anaphylactic reactions of particular adjuvant-linker combinations must be meticulously evaluated in in vivo experiments. 190 Proactively addressing these concerns during the vaccine development process is essential for surmounting obstacles to clinical use and optimizing the vaccine’s worldwide impact. 190
Finally, this study emphasizes the computationally designed CHIKV vaccine’s potency and its capacity to elucidate strong cellular and humoral immune responses through structural polyprotein epitopes.
Conclusion
Developing an mRNA vaccine targeting the highly potent antigenic protein of CHIKV, the structural polyprotein, demonstrates promising results. The vaccine design incorporates validated MHC-I, MHC-II, and B-cell epitopes, exhibiting antigenic characteristics, non-allergenicity, and well stability. The analyses of physicochemical properties, secondary and tertiary structure analyses, and molecular dynamic simulations further corroborate the vaccine’s structural integrity. Docking studies demonstrate a substantial affinity with TLR-2 and TLR-4 receptors, suggesting potential immune activation. However, the vaccine shows structural instability during molecular dynamics simulations, necessitating further investigation. Codon optimization ensures effective expression in E coli, and experimental results indicate the vaccine’s ability to elicit both innate and adaptive immune responses, including the synthesis of immunoglobulins and persistent B-cell memory. Despite these promising findings, the lack of validation through in vitro, in vivo, and clinical trials poses a significant limitation, emphasizing further research to substantiate the vaccine’s efficacy and safety. Nevertheless, the lowest docking scores and molecular dynamic simulations identified strong and stable interactions between the vaccine candidate and TLRs, providing a computational basis for its potential efficacy. Moreover, the analysis of adjuvant-linker combinations revealed promising candidates for optimizing immune responses. To translate these findings into practical applications, future experimental steps should focus on in vivo validation through binding assays and cytokine profiling, followed by in vivo testing to assess immunogenicity and safety in animal models. However, this novel mRNA vaccine can be a vital supplementary tool in next-generation CHIKV vaccine design, as well as in establishing effective and efficient immunotherapies against the virus.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-docx-1-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-2-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-docx-2-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-3-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-docx-3-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-4-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-docx-4-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Supplemental Material
sj-tif-5-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-tif-5-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Supplemental Material
sj-tif-6-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-tif-6-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Supplemental Material
sj-tif-7-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-tif-7-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Supplemental Material
sj-tif-8-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-tif-8-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Supplemental Material
sj-tif-9-bbi-10.1177_11779322251324859 – Supplemental material for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations
Supplemental material, sj-tif-9-bbi-10.1177_11779322251324859 for Revolutionizing Chikungunya Vaccines: mRNA Breakthroughs With Molecular and Immune Simulations by Md. Habib Ullah Masum, Ahmad Abdullah Mahdeen and Abanti Barua in Bioinformatics and Biology Insights
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.