
Abstract
Dabie bandavirus (DBV), also called severe fever with thrombocytopenia syndrome virus (SFTSV), is a tick-borne virus that leads to a serious illness with high fever, low platelets, bleeding risks, and organ damage. Death rates can reach 5% to 30% in affected areas like China, South Korea, and Japan, where cases continue to rise. No approved vaccines or specific treatments are available, prompting the World Health Organization to mark it as a priority emerging infectious disease. This in silico study designs a multi-epitope mRNA vaccine candidate targeting the virus’s membrane glycoprotein. After reviewing 1042 virus sequences, 9 cytotoxic T-cell (cytotoxic T lymphocyte [CTL]), 6 helper T-cell (helper T lymphocyte [HTL]), and 5 B-cell epitopes were chosen for their high conservation, predicted antigenicity, safety (non-toxic, low allergenicity), and lack of similarity to human proteins, achieving 94.77% global population coverage. The construct included beta-defensin-3 as an adjuvant. Computational predictions indicated favorable features: structural stability, potential binding to immune receptors (Toll-like receptor 3 [TLR3] and Toll-like receptor 4 [TLR4]), consistent behavior in molecular dynamics simulations (100 ns), encouraging patterns in immune response modeling, and good codon optimization for possible expression. These results are hypothesis-generating and based solely on in silico tools and require experimental validation, including in vitro studies, immunogenicity tests, and animal challenge models to assess any real-world potential.
Introduction
Numerous tick vectors are responsible for transmitting a wide range of pathogens to hosts, resulting in various tick-borne viral diseases. 1 Severe Fever with Thrombocytopenia Syndrome Virus (SFTSV) is an emerging tick-borne pathogen that causes viral hemorrhagic fever and is primarily transmitted through tick bites. The SFTSV is caused by the recently identified Dabie bandavirus (DBV). 2 According to the International Committee on Taxonomy of Viruses (ICTV), DBV belongs to the genus Bandavirus, order Bunyavirales, and family Phenuiviridae. The virus is also referred to as the SFTSV, Huaiyangshan virus (HYSV), or novel bunyavirus (NBV). As most members of the Phenuiviridae family are vector-borne, ticks are considered the principal agents responsible for DBV transmission. 3
Among tick species, Haemaphysalis longicornis, also known as the Asian longhorned tick, is considered the primary vector of SFTSV transmission. 3 Other species of ticks were also reported to carry the SFTSV RNA, such as Haemaphysalis flava, Hyalomma asiaticum, Dermacentor nuttalli, Ixodes nipponensis, and Amblyomma testudinarium, which suggests their connection to the circulation of the virus in endemic areas.4-6 In addition to the transmission by the use of vectors, DBV can also be spread by direct contact with infected blood of humans or animals. 6
The SFTSV infection triggers multiple pathogenic processes, including viral replication, immune dysregulation, and inflammatory damage. 7 At the initial stage of the SFTSV infection, mononuclear phagocytic systems, such as monocytes, macrophages, and dendritic cells (DCs), became infected. Viral replication in these cells leads to depressed innate immunity and inhibition of antiviral defense mechanisms, especially interferon-mediated responses. 8 Myeloid DCs have defective maturation and impaired antigen presentation and therefore are limited in the development of effective humoral immune responses. Although the exact role of natural killer (NK) cells in DBV infection is not well defined, differences in the number of NK cells have been linked to disease progression. 9 Moreover, enhanced type I interferon signaling induces the expression of interferon-stimulated genes, contributing to dysregulated immune responses. The dysregulated immune response results in the excessive production of inflammatory cytokines that leads to clinical symptoms. 2
The DBV is an enveloped spherical virus with a diameter of approximately 80 to 100 nm. 10 Its negative-sense RNA genome consists of 3 segments: small (S), medium (M), and large (L). 11 The M segment encodes 2 surface glycoproteins, Gn and Gc, which are major antigenic determinants on the viral envelope. 12 Compared with non-structural proteins encoded by the S segment, the DBV envelope glycoproteins (Gn and Gc) are more exposed to neutralizing antibodies and provide better protective immunity in animal models; thus, preclinical studies have supported the preferential selection of the DBV envelope glycoproteins as vaccine targets. 13 Upon viral entry, infection initially involves skin-resident cells, such as keratinocytes, mast cells, immature Langerhans cells, and epidermal cells. In the early stages of disease, infection of monocytes, macrophages, and DCs results in suppression of first-line immune responses, thereby facilitating viral dissemination. 14
The initial outbreak of SFTSV occurred in rural central China between the end of March and mid-July 2009 and was linked to severe clinical cases and a high mortality rate of an estimated 30%. 15 The virus then spread to several other provinces in China, having over 2500 reported cases and an average case-fatality rate of 7.3%. 16 Later, the illness spread to other nations like Japan, South Korea, and Vietnam. 1 By the end of 2019, China, Korea, and Japan had reported more than 14 500 cases. 17 Patients in the United States were also found to have a genetically related virus, the Heartland virus. 16 The transmission of SFTSV is usually during the period between March and November, with high transmission rates in May to July. More recently, from April 2023 to August 2024, 10 cases of DBV infection were reported in Suzhou City, China, 3 of which resulted in death during hospitalization. 18
Common clinical symptoms associated with an SFTSV infection include hemorrhagic fever, fatigue, vomiting, dizziness, myalgia, nausea, and diarrhea. Severe instances of DBV infection are characterized by thrombocytopenia, leukopenia, liver dysfunction, increased levels of hepatic enzymes, renal impairment, neurological complications, and gastrointestinal disorders.3,19 During the acute phase, patients, particularly those older than 40 years of age, can suffer from hemorrhage, neurological deficits, respiratory failure, and, in some cases, death.19,20 Due to its increasing incidence and persistently high mortality rate, the World Health Organization in 2018 classified the SFTSV as an emerging infectious disease.21,22
Despite the increasing number of reported cases of SFTS, there is currently no approved vaccine or antiviral therapy against SFTSV infection. 23 Previous vaccine development attempts have faced serious challenges such as mortality rates, geographic distribution, nucleotide diversity, and the zoonotic nature of the virus.24,25 Although recent studies have led to an increased understanding of the molecular structure, epidemiology, and pathogenesis of DBV, effective immunization strategies are limited. 26 Certain strains have been suggested as potential vaccine candidates, like HB29;10,26 however, the experimental studies using mouse models, including C57BL/6 mice, have not been able to elicit protective immune responses against nonstructural proteins of the S segment. 27
Vaccine development against tick-borne viruses is faced with further difficulties as compared to directly transmitted viral pathogens. Tick-borne viruses have a vector and animal reservoir, which makes it difficult to identify the antigen. 28 Moreover, immunomodulatory components found in tick saliva are capable of suppressing host immune responses, which facilitates the transmission of the virus. 29 Antigenic diversity between circulating strains also further complicates the development of broadly protective vaccines, and research on the longer-term immune protection against tick-borne viruses is limited.
Given the lack of approved vaccines, the limited availability of high-containment experimental models, and the genetic diversity of circulating DBV strains, immunoinformatics offers a rapid, cost-effective, and hypothesis-driven platform for immunogen prioritization of vaccine candidates at the early stages.2,30,31 In recent years, various approaches have been used for vaccine research, including live-attenuated, inactivated, and subunit vaccines. Nevertheless, only a few vaccines are available against tick-borne viral diseases, although their global distribution is increasing. 32 Epitope-based vaccine approaches have attracted interest because of their potential to generate long-lasting immune responses. Peptide- and DNA-based vaccines have the advantage of speed and flexibility, but they are often less immunogenic and have the risk of genomic integration.33,34 In comparison, mRNA vaccines have demonstrated higher efficacy, improved safety profiles, and reduced adverse effects.35,36 The mRNA vaccination strategy allows for endogenous antigen expression by both major histocompatibility complex (MHC) class I and MHC class II pathways, resulting in activation of CD8+ cytotoxic T lymphocytes and CD4+ helper T cells, as well as the induction of neutralizing antibody responses that are often difficult to obtain using subunit vaccines. Importantly, mRNA vaccines can be developed quickly, without requiring viral culture or protein purification, which makes them especially appropriate for emerging threats posed by tick-borne viruses.37,38 The combination of immunoinformatics and the use of a multi-epitope mRNA vaccine design enable the optimization of B-cell and T-cell epitopes into 1 immunogen, which has been used successfully for the development of vaccines against emerging pathogens, including the novel coronavirus (SARS-CoV-2).39,40
Previous computational studies have been conducted for multi-epitope protein-based vaccine design against DBV, including the selection of epitopes, immune response prediction, and protein interaction analysis. 41 However, these strategies were mostly based on recombinant protein expression systems. Viral glycoproteins are important immunogenic targets in enveloped viruses because of their involvement in host cell attachment and membrane fusion as well as the fact that they contain conserved regions that can trigger a neutralizing antibody and T-cell response. Glycoprotein-based epitopes have been used previously in the fight against other emerging viruses with promising results. 42
In this context, mRNA-based multi-epitope vaccine strategies have a unique advantage because they stimulate both humoral and cellular immune responses by expressing antigens endogenously. To date, this strategy for DBV has not been explored. In the current study, a holistic immunoinformatics framework was used to identify and evaluate appropriate B-cell, helper T lymphocyte (HTL) and cytotoxic T lymphocyte (CTL) epitopes in terms of antigenicity, conservancy, non-toxicity, non-homology, allergenicity, and physicochemical characteristics. The secondary and tertiary structures of the designed vaccine construct were predicted, improved, and validated. Furthermore, molecular docking with Toll-like receptor 3 (TLR3) and Toll-like receptor 4 (TLR4), molecular mechanics with generalized Born surface area (MM/GBSA) analysis, molecular dynamics (MD) and immune simulations, computational cloning, and mRNA structure analyses were conducted to assess the predicted stability, immunogenic potential, and translational feasibility of a proposed mRNA vaccine candidate, generating hypotheses for future experimental validation.
Methodology
Online-based servers and tools were utilized for the development of the multi-epitope mRNA vaccine. The methodology for building an epitope-based mRNA vaccine is summarized in a diagram in Figure 1.

A diagrammatic pathway illustrating the construction of an mRNA vaccine focusing on the diverse epitopes of Dabie bandavirus (DBV).
Obtaining protein sequence and physicochemical properties assessment
The glycoprotein of Banda virus is encoded by the M segment, which is responsible for host entry. We targeted the membrane glycoprotein polyprotein (Gn and Gc), comprising 1073 amino acids (Accession ID: YP_009666134), which was retrieved from the National Center for Biotechnology Information (NCBI) database for vaccine construction. The target protein’s antigenicity was analyzed via version 2.0 of the VaxiJen server. 43 VaxiJen 2.0, an online-based tool that estimates the protein’s or peptide’s antigenicity from various bacterial, viral, and fungal origins, where 0.4 was the cut-off value. 43 Another web server, ExPASy ProtParam, was employed to analyze a protein’s physicochemical characteristics. 44
Mapping of T-cell epitopes
T cells recognize antigens via antigen-presenting cells (APCs) through interactions with MHC molecules. 45 The CTLs detect endogenous peptides presented by MHC class I and eliminate infected cells, while HTLs recognize exogenous peptides on MHC class II and orchestrate adaptive immunity by activating immune cells and secreting cytokines such as interleukin (IL)-4, IL-10, and interferon-gamma (IFN-γ).45,46 Consequently, HTL epitopes play a critical role in vaccine and immunotherapy development. The Immune Epitope Database (IEDB), which compiles experimental data on antibodies and epitopes, was used for T-cell epitope prediction. 47 The NetMHCpan 4.1 EL method within IEDB predicted epitopes for both MHC class I and II molecules. Peptides of 9-mer and 15-mer lengths were selected for CTL and HTL prediction, respectively, as these lengths optimize MHC binding and recognition. 48 A panel of 27 MHC class I and 10 HLA class II alleles (Supplementary File 1), representative of global populations, was used for the analysis.
Mapping of B-cell epitopes
B-cells are involved in humoral immunity and are stimulated by the B-cell receptors (BCRs) to recognize antigens. When B-cell epitopes bind to cell membrane BCRs, lymphocytes produce neutralizing antibodies that assist the immune system in fighting off viral infections. 49 B-cells with the class II molecules deliver antigenic peptides to CD4+ follicular helper T (TFH) cells, thereby producing memory B-cells and plasma cells.49,50 This study utilized the Bepipred Linear Epitope Prediction 2.0 tool available on the IEDB website to identify linear B-cell epitopes. 51 The 7 to 35 amino acid residues comprising the B-cell epitopes were considered for further analysis. Conformational B-cell epitopes were analyzed separately based on the tertiary structure of the final vaccine construct.
Evaluating epitopes according to their physical characteristics
A systematic methodology was employed to identify the most suitable vaccine candidates, focusing on the expected CTL, HTL, and B-cell epitopes. This procedure was carried out following established criteria that encompass immunological relevance, safety considerations, and physicochemical characteristics. The process of identifying T-cell epitopes commenced with an evaluation of their percentile rank, selecting CTL (MHC-I) ⩽ 0.5 and HTL (MHC-II) ⩽ 2.0 as strong MHC binders. 52 All selected epitopes, including both T-cell and B-cell types, underwent a thorough evaluation of their antigenic characteristics using VaxiJen v2.0 (with a threshold of ⩾0.4). Furthermore, the evaluation of their allergenic potential was conducted using AllerTOP v2.1, while toxicity assessments were performed through ToxinPred. Peptides recognized as non-immunogenic, exhibiting less allergenic potential and non-toxic, were subsequently omitted from further examination.43,53,54 The epitopes that met these criteria underwent additional examination concerning their physicochemical characteristics, such as GRAVY, aliphatic index, instability index, and expected half-life. This analysis was conducted using ExPASy ProtParam to evaluate their stability and appropriateness for vaccine development. 44 In conclusion, the evaluation of HTL epitopes was conducted to determine their ability to stimulate IFN-γ, IL-4, and IL-10 through the utilization of IFNepitope, IL4pred, and IL-10pred servers, respectively. This assessment was crucial in confirming their potential to provoke significant adaptive immune responses.55-57 Only those epitopes that fulfilled all established criteria were progressed for subsequent vaccine development.
Epitope conservancy and human proteome homology analysis
Furthermore, epitope conservation percentages were explored utilizing the web-based conservation analysis server of IEDB. 47 In this study, the percentage of conservancy was evaluated against 1042 glycoprotein sequences of DBV retrieved from the NCBI database, which contains a minimum identity threshold of 100%. 58 These sequences represent all publicly available DBV glycoprotein variants reported up to 2025, enabling a comprehensive evaluation of epitope conservation across circulating and emerging strains and addressing the potential for strain-specific immune escape.
The BLAST protein (BLASTp) tool was used to assess human proteome homology by comparing each epitope against the human proteome (taxid: 9606). The E-value threshold was set at 0.05 using default parameters. 59 Epitopes with E-values greater than 0.05 were considered non-homologous to human proteins and were therefore preferred due to their reduced risk of autoimmunity and enhanced immunogenic potential. 60
Population coverage analysis
The IEDB population coverage tool was employed to assess the distribution of anticipated HTL and CTL epitopic alleles among various populations. 61 Due to the high polymorphism of HLA alleles, population coverage was evaluated in regions where DBV is endemic, particularly in East Asian nations such as China, South Korea, and Japan, identified as primary risk areas for infection. 62 Population coverage was also analyzed in non-endemic regions to investigate the wider applicability of the chosen epitopes across varied worldwide populations. 63 This integrated strategy assures that the recommended vaccine candidates are appropriate for both groups at high epidemiological risk and for worldwide use.
Vaccine development and biophysical property assessment
After screening for epitopes, specific linkers (EAAAK, AAY, GPGPG, and KK) and an adjuvant were connected to merge the epitopes. 64 Adjuvants enhance immunogenicity and activate TLRs, thereby improving the vaccine’s immune response. 65 Adjuvant beta-defensin-3 was inserted at the N-terminal end of the constructed sequence. 64 The EAAAK linker added the adjuvant to CTL epitopes to promote structural rigidity and immune response. In addition, to ensure flexibility and preserve immunogenic activity, 3 other linkers were applied to assemble CTL, HTL, and B-cell epitopes. 65 Post-construction evaluations, including antigenicity and allergenicity for the new vaccine sequence, were executed to ascertain the physicochemical stability by using online tools. Afterwards, the ProtParam server examined the instability index, in vivo half-life, molecular weight, instability index, GRAVY, and aliphatic index for high accuracy of the new vaccine. 44
Vaccine secondary structure prediction
The SOPMA and PSIPRED web servers, both well-established and highly accurate computational tools, were employed to predict the secondary structure of the designed vaccine construct.66,67 These methods estimate the relative proportions of α-helices, β-strands, and random coil regions within the amino acid sequence, providing insight into the structural organization of the protein.
Vaccine tertiary structure prediction, refinement, and validation
The algorithm-based Iterative Threading ASSEmbly Refinement (I-TASSER) tool used the amino acid sequences to build the 3-dimensional (3D) structure of the developed vaccine. 68 Then, it used C-scores (confidence scores) to determine which model was more accurate (a higher C-score means higher quality and accuracy). The alignment algorithms identify the template structures from the PDB (Protein Data Bank), and fragment assembly simulations generated some structural models. 69 Subsequently, the designed vaccine models were refined through the refinement tool GalaxyRefine server of GalaxyWEB, 70 which was validated through CASP10. 71 The side chains are primarily reassembled and repacked in this method, and subsequently, relaxation was applied to the overall structure via a molecular dynamic simulation. 71 The PROCHECK suite was used to evaluate the structural quality after each refinement by the visualization of the Ramachandran Plot. 72 The percentages of phi (φ) degree and psi (ψ) degree of amino acids represent the allowed and disallowed areas located at the Ramachandran plot that calculate the radii generated by van der Waals for side chains.70,71 If the number of residues is at a higher level, it would indicate the better structural stability of the most favorable position. In addition, the overall protein structure quality was calculated by the Z-score of the ProSA-web server. It reflects the model attributes by determining the total energy fluctuation level generated by the energy distribution of random conformations. When the Z-score falls within the range typical of native proteins, the model would be considered reliable. 73
Conformational B-cell prediction
The ElliPro server identified the discontinuous or conformational B-cell epitopes. It applies different algorithms to predict and visualize antibody epitopes from the antibody-protein complexes of 3D structures. 74 Default parameters were used, including a maximum distance of 6 Å and a minimum score of 0.5.
Disulfide engineering of the vaccine
Disulfide engineering changes some protein structural residues to cysteine, which makes it easier for new disulfide bonds to form. Lowering the entropy of the unfolded state makes proteins more stable since synthetic disulfide linkages are covalent. 75 DbD2, an online platform, estimated the cysteine residue pairs in a protein’s structure from the tertiary structure of the developed vaccine. 76 In his study, the Cα-Cβ-Sγ angle was default, and it was maintained at 114.6° ± 10, while the χ3 angle was modified to −87° or +97°. Residues were converted to cysteine residues to form disulfide bridges when the level of energies was 2.5 kcal/mol. 76
Molecular docking
Docking studies estimate the stability, binding affinity, and binding orientation of a ligand relative to a receptor when forming a stable complex. 77 In this context, TLRs are essential human immune receptors that identify pathogens and trigger immune responses. The ClusPro 2.0 online tool (https://cluspro.bu.edu) was utilized to get information on the binding molecules between vaccine-TLR complexes, where the PDB IDs of TLR3 and TLR4 were 2A0Z and 4G8A in the PDB (https://www.rcsb.org/). Discovery Studio software was used to preprocess the TLRs’ 3D structures, and the PDBsum tool evaluated the 2-dimensional interactions that occur by the combined vaccine-TLR complex molecules. 78 The 2 most appropriate docked complexes, one associated with TLR3 and the other with TLR4, were selected for further analysis due to their effective docking and low binding energy.
Post-docking molecular mechanics with generalized Born surface area–based free energy calculations
Following molecular docking, MM-GBSA binding free energy calculations were performed using the HawkDock web server to evaluate the stability of the docked protein-peptide complexes. 79 The binding free energy (ΔG) was estimated by integrating key molecular mechanics energy components, including van der Waals interactions (ΔVDW), electrostatic energy (ΔELE), generalized Born solvation energy (ΔGBE), surface area energy (ΔSA), and the total binding free energy (ΔTOTAL). 80
Molecular dynamics simulation of the vaccine-Toll-like receptor complexes
The MD simulation experiments were executed for exploring the configurations of both vaccine-TLR complexes’ stability and the tiny interactions between the molecules. 81 In this study, the YASARA dynamics suite was navigated to calculate conformational change and to predict the interactions. 82 All MD simulations employed AMBER 14 software to conduct simulations at a fixed temperature (298 K) under the NVT ensemble. 83 A 0.9% NaCl solution, supplemented with water molecules (0.998 g/cm3), was added to neutralize the system. A cuboidal simulation box with dimensions 20 Å larger than the complex was applied, and periodic boundary conditions were considered to run the simulation. Long-range electrostatic interaction was calculated by the method named Particle-mesh Ewald (PME), where 8.0 Å was set as the cut-off radius. 84 The total simulation time for each case was fixed to 100 ns with trajectories recorded at 100 ps intervals.
Immune simulation
Machine learning and PSSM (position-specific scoring matrices) were used in the C-ImmSim server that calculates the immune response profile of the vaccine. 85 This online tool provides the mechanism of specific immune epitopes’ functions. 86 During the process, 3 injections were scheduled at 15-day intervals, and 1-45-90 were the time steps, when each step represents 8 hours. Moreover, 1050 simulation steps were set up over a 1-year period to analyze the immunologic responses of the administered vaccine. The C-ImmSim models immunological responses with an agent-based framework that incorporates immunoinformatics and epitope prediction methodologies. 85 Similar to previous agent-based immunological models, it employs simplified representations of intricate biological processes and omits numerous mechanistic details; thus, its outputs indicate prediction trends rather than direct biological responses and necessitate experimental validation. 87
Constructed vaccine sequence cloning and codon optimization
Computer-based cloning validates optimized vaccine-protein expression and clones it into an appropriate expression vector. The reverse translation process was used to create DNA for cloning using the Java Codon Adaptation Tool (JCaT). 88 The Codon Adaptation Index (CAI) indicates how closely an optimized sequence aligns with host-specific codon preferences, with values ranging from 0.8 to 1 expressing enhanced translational efficiency. In addition, GC content, representing the ratio of guanine and cytosine nucleotides, was evaluated to ensure a balanced nucleotide composition that promotes mRNA stability and efficient translation within the optimal range of 30% to 70%. 89 SnapGene, a free online tool, is utilized to generate the cloning of the optimized gene sequence into the pET-28a (+) expression vector. The Eco53KI and EcoRV restriction sites of the vector were added to ensure correct orientation and reading frame. The length and restriction map of the recombinant plasmid construct were assessed through in silico analysis.
Structural stability assessment of vaccine-encoding mRNA
The newly developed DNA sequence was modified with the corresponding RNA sequence through the DNA-RNA-protein conversion process utilizing the Biomodel server. Subsequently, the RNA sequence was submitted to another web server, RNAfold, that thermodynamically generates the secondary structure of mRNA with minimum free energy (MFE; ΔG, kcal/mol).90,91
Results
Retrieval and immunological properties exploration of the target protein sequence
The membrane glycoprotein (Accession ID: YP_009666134) of DBV was retrieved from the NCBI database and consists of 1073 amino acids. The VaxiJen v2.0 predicted antigenic score was 0.4807, higher than the threshold value (0.4), indicating an antigenic nature. Allergenicity analysis using AllerTOP v2.1 classified the protein as non-allergenic. In addition, the ProtParam server provides the profile of the protein that contains thermostable, slightly alkaline, and hydrophilic properties, suggesting potential suitability for immunological applications (Table 1). The thermostability score and alkalinity score indicate the structural robustness, while hydrophilic character indicates the solubility in aqueous solvents and epitope exposure.
Physicochemical properties of the selected Dabie bandavirus glycoprotein employed in vaccine development.

Prediction and selection of T-cell and B-cell epitopes
Epitope identification
The DBV glycoprotein was examined for immunogenic regions, yielding 28 756 CTL (9-mer), 10 591 HTL (15-mer), and 34 linear B-cell epitopes. T-cell epitopes were selected based on a CTL (MHC-I) percentile rank of ⩽0.5 and an HTL (MHC-II) percentile rank of ⩽ 2.0 to identify potent MHC binders, whereas B-cell epitopes ranging from 7 to 35 amino acids were chosen for their relevance to antibody binding.
Screening and physicochemical evaluation
All candidates that are antigenic (⩾0.4), safe, less allergenic, and non-homologous to host proteins (Blast E-value > 0.05) were evaluated, and their physicochemical properties were assessed using ProtParam. The HTL epitopes were also evaluated for their capacity to elicit IFN-γ, IL-4, and IL-10 responses.
Final epitope selection
Nine CTL, 6 HTL, and 5 B-cell epitopes were finalized based on these criteria (Tables 2 and 3). Mechanistically, CTL epitopes may induce cytotoxic responses, HTL epitopes facilitate HTL activation, and B-cell epitopes promote neutralizing antibody formation, thus engendering comprehensive cellular and humoral defense.
Record of 9 cytotoxic T-lymphocyte (CTL) and 6 helper T lymphocyte (HTL) epitopes used in the vaccine sequence.

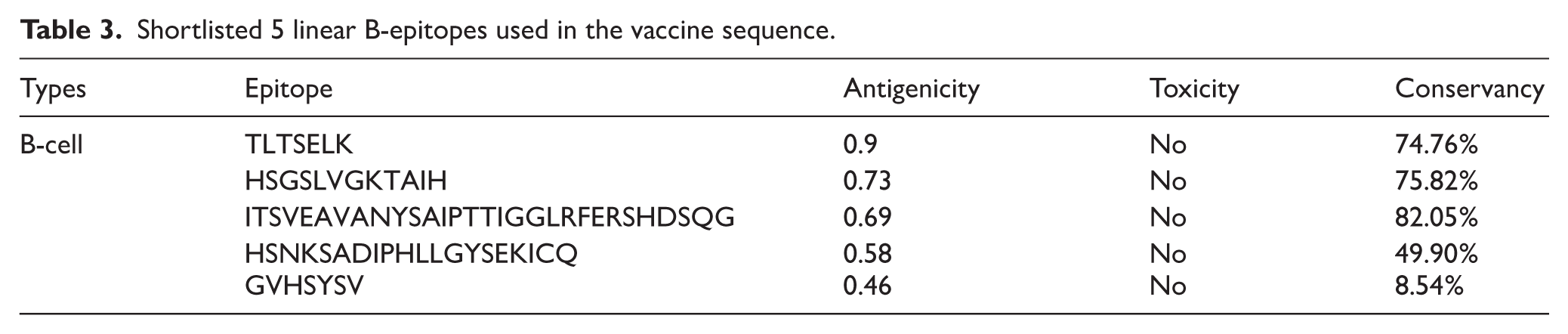
Shortlisted 5 linear B-epitopes used in the vaccine sequence.
Conservancy analysis
The epitope conservancy was measured utilizing the conservancy analysis tool existing at the IEDB server. The conservancy was against 1042 membrane glycoprotein polyprotein sequences of DBV, retrieved from the NCBI database (Supplementary File 2). Both Tables 2 and 3 summarize the conservancy score, where most epitopes are greater than 70%. This high score of conservancy suggests the evolutionary stability by reducing the viral mutations, and they are conserved.
Population coverage
Population coverage analysis was estimated based on MHC molecule epitope binding predictions across different populations. It indicates the immunological impact of the newly designed vaccine. Globally, the vaccine attained 92.51% MHC-I, 30.17% MHC-II, and 94.77% combination coverage, indicating a broad population-level immunogenetic suitability. This high coverage value could address the theoretical inclusion of both cytotoxic and HTL-mediated immune pathways at the population level, highlighting the worldwide relevance and inclusive immunogenetic applicability of the predicted epitopes. Importantly, these may be relevant for SFTSV viral infections, where cellular immunity is considered important for controlling viral replication and potentially reducing disease severity. Countries including Peru (MHC-I: 99.98%, MHC-II: 1.12%, Combined: 99.98%), Papua New Guinea (97.63%, 22.64%, 98.17%), Germany (95.97%, 43.28%, 97.72%), South Korea (95.63%, 26.44%, 99.37%), Japan (96.45%, 19.83%, 97.16%), Poland (95.54%, 36.79%, 97.18%), and the United States (94.55%, 36.77%, 96.55%) had very high combined coverage, suggesting a higher likelihood of epitope presentation across these populations. European countries like England (94.43%, 44.49%, 96.91%) and France (92.96%, 35.84%, 95.48%) also displayed strong scores. On the contrary, several populations showed reduced coverage, including India (80.68%, 25.10%, 85.52%), Central Africa (73.12%, 4.52%, 74.33%), Vietnam (86.18%, 7.54%, 87.22%), and Zimbabwe (85.26%, 4.35%, 85.91%). Notably, high combined coverage was also observed in endemic regions for SFTSV, including China, Japan, and South Korea (Figure 2 and Supplementary Table 4).

Global population coverage of MHC molecules across representative countries.
Biophysical property assessment after the vaccine construction
The 6 most favored HTL, 7 CTL, and 5 linear B-cell epitopes were connected utilizing AAY, GPGPG, and KK linkers, with beta-defensin-3 (GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK) affixed at the N terminal by an EAAAK linker. Beta-defensin-3 acts as an adjuvant, enhancing innate immune function without causing damage.
The constructed vaccine comprises 363 amino acids (Figure 3), demonstrating an antigenicity score of 0.6294, an aliphatic index of 72.87, an instability index of 27.98, and a negative GRAVY score of −0.247, indicating thermodynamic stability, hydrophilic characteristics, and potential for considerable immunogenicity. The lack of allergens suggests a reduced probability of hypersensitivity. The anticipated half-life in Escherichia coli (exceeding 10 hours) facilitates viable expression for experimental verification. The physicochemical qualities collectively indicate that the vaccine design is structurally stable, likely to maintain immunogenic conformations, and appropriate for in vivo applications.

Schematic representation of the constructed multi-epitope vaccine, incorporating the beta-defensin-3 adjuvant and HTL, CTL, and B-cell epitopes linked by AAY, GPGPG, and KK linkers.
Secondary structure prediction
The SOPMA and PSIPRED tools provide the secondary configuration for the constructed vaccine, and it was indicated that among the 363 residues, 182 (50.14%) formed random coils, 92 (25.34%) formed beta-sheets, 60 (16.53%) formed alpha-helices, and 29 (7.99%) formed turns. The high density of coils indicates that the structure was flexible (Figure 4). It suggests the probability of structural flexibility and structural integrity, boosting the epitope attainability and immune recognition.

Predicted secondary structure of the constructed multi-epitope vaccine, illustrating the allocation of alpha-helices, beta-sheets, turns, and random coils as assessed by PSIPRED and SOPMA.
Prediction of the 3-dimensional structure, refinement, and validation of the vaccine model
The best 5 3D models were predicted by the I-TASSER server, and Model I was chosen for further analysis with a C-score (confidence score) of −2.09 and a Z-score of −2.38. From the Ramachandran plot, the number of residues was 66.1%, 22.6%, 7.9%, and 3.4% for the most favored regions, additional allowed regions, generously allowed regions, and disallowed regions, respectively (Supplementary File 3). Model 1 underwent 3 iterations of improvement using the GalaxyWEB server to enhance structural reliability. After refining to improve structural quality, Model 1 exhibited a Z-score of −3.05, with the number of residues in the most favored regions, additional allowed regions, generously permitted zones, and disallowed sections being 86.6%, 8.6%, 1.7%, and 3.1%, respectively (Figure 5). The refinement process could increase the stereochemical accuracy, stability of the structure, and suitability for future structural and interaction analysis.

Refined 3D structure of the designed multi-epitope vaccine with structural validation. (A) Refined 3D model of the vaccine. (B) Ramachandran plot illustrating residue distribution in most favored (red), additional allowed (yellow), generously allowed (light yellow), and disallowed (white) regions. (C) Z-score plot indicating the overall structural quality of the refined vaccine model.
Assessment of the discontinuous B-epitope
Seven conformational B-cell epitopes were identified through the utilization of the ElliPro server, exhibiting residue lengths that vary from 4 to 99. Epitope 1 demonstrated the highest prediction score of 0.745, indicating significant surface accessibility and a strong potential for antibody recognition, whereas Epitope 7 recorded the lowest score of 0.552. The remaining epitopes exhibited scores that fall within an acceptable range, suggesting their capability to provoke humoral immune responses against DBV (Figure 6 and Supplementary Table 5).

A structural diagram of the 7 expected conformational B-cell epitopes that were predicted by ElliPro (Epitopes 1-7). The yellow areas show the predicted epitopes, while the gray areas show the rest of the vaccine that has been created.
Protein disulfide bond prediction
Disulfide bond engineering was accomplished using the DbD2 that minimizes conformational entropy to amplify the structural stability of the 3D vaccine. In total, 34 amino acid residue pairs were found during the analysis (Supplementary Table 6), among which 6 pairs—145ALA-151VAL, 47ALA-155ILE, 172GLY-175SER, 225ALA-239PRO, 259PRO-263CYS, and 334SER-337ALA—were selected to shape the disulfide bond (Figure 7). The residues had good angle and energy values (all less than 2.5 kcal/mol), which made them suitable for forming disulfide bonds in the mutated version. It may verify the presence of stabilizing cysteine pairings, bridging the intramolecular disulfide distances. The predicted disulfide bonds could amplify the structural rigidity of the epitopes, folding efficiency, and resistance against thermal and enzymatic degradation.

Structural representation of disulfide bond modifications in the constructed vaccine. (A) Initial 3D model of the constructed vaccine, devoid of any disulfide bonds. (B) Modified model representing 6 selected residue pairs that form disulfide bonds, hence enhancing structural stability, folding efficacy, and resistance to heat and enzymatic degradation.
Molecular docking
Molecular docking of the developed vaccine with TLR3 and TLR4 was conducted to anticipate probable receptor interaction. The optimal docking model for TLR3 was identified in cluster 4, demonstrating a central energy score of −1077.3 kcal/mol. Interface analysis with PDBsum indicated that 44 residues of the vaccine interacted with 49 residues of TLR3, including interface regions of 2356 Å2 and 2212 Å2, respectively. The interactions were stabilized by 9 salt bridges, 33 hydrogen bonds, and 260 non-bonded contacts (Supplementary Table 7).
In a similar manner, docking with TLR4 revealed the best model from cluster 0, with a center energy score of −1181.3 kcal/mol and a lowest energy score of −1302.7 kcal/mol. In this interaction, 41 vaccine residues and 54 TLR4 residues contributed to interface areas of 2536 Å2 and 2241 Å2, respectively, stabilized by 11 salt bridges, 37 hydrogen bonds, and 311 non-bonded contacts (Supplementary Table 8).
The expected interactions suggest potential binding and engagement of TLR3 and TLR4, suggesting potential recognition by innate immune receptors. The discovered non-covalent interactions, including hydrogen bonds and salt bridges, may facilitate receptor identification and activate downstream signaling pathways, thereby enhancing both innate and adaptive immune responses to DBV (Figure 8).

Visualization of the vaccine-receptor docked complexes having TLR3 (A) and TLR4 (B) receptors and also highlighting hydrogen bonds, disulfide bonds, salt bridges, and non-bonded interactions.
Post-docking molecular mechanics with generalized Born surface area free energy calculations
To evaluate the binding energetics of the vaccine-receptor complexes, MM/GBSA calculations were performed for both TLR3 and TLR4 interactions. For the vaccine-TLR3 complex, the total binding energy, van der Waals (VDW), electrostatic (ELE), generalized Born (GB), and surface area (SA) contributions were estimated as −192.33, −249.75, −3523.45, 3615.53, and −34.66 kcal/mol, respectively (Figure 9 and Supplementary Table 9). The vaccine-TLR4 complex demonstrated values of −244.61, −246.81, −7331.72, 7368.39, and −34.48 kcal/mol (Supplementary Table 10).

MM/GBSA binding free energy analysis of the vaccine-receptor complexes. The contributions of van der Waals (VDW), electrostatic (ELE), generalized Born (GB), surface area (SA), and total binding energy were computed for TLR3-vaccine (A) and TLR4-vaccine (B) complexes using the HawkDock server.
The results demonstrate advantageous thermodynamic interactions and stable complex formation, corroborating the docking predictions. The calculated energies suggest that the vaccine is likely to bind well with TLR3 and TLR4, potentially activating innate immune signaling pathways in response to DBV.
Molecular dynamics simulation
Molecular dynamics simulations were performed for 100 ns to assess the structural stability and interaction dynamics of the multi-epitope vaccine with immune receptors TLR3 and TLR4 (Supplementary Table 11). The TLR3-vaccine complex exhibited a mean root mean square deviation (RMSD) of 4.253 ± 0.60 Å, with a range of 0.527 Å to 5.317 Å, whereas the TLR4-vaccine complex had a mean RMSD of 4.775 ± 0.55 Å, ranging from 0.540 Å to 5.522 Å (Figure 10A and A1). The consistent RMSD profiles indicate that both complexes maintained their conformational integrity with minimal variations, thereby confirming the reliability of the docking predictions.

Molecular dynamics simulation-based assessment of structural stability and flexibility in docked complexes: (A-D) represent the vaccine-TLR3 complex showing (A) Root Mean Square Deviation (RMSD), (B) Radius of Gyration (Rg), (C) Solute-Solvent Hydrogen Bonds, and (D) Root Mean Square Fluctuation (RMSF); and (A1-D1) represent the vaccine-TLR4 complex illustrating (A1) RMSD, (B1) Rg, (C1) Solute-Solvent Hydrogen Bonds, and (D1) RMSF.
The radius of gyration (Rg) for the TLR3-vaccine complex was 33.506 ± 0.27 Å (range = 32.389-34.112 Å), while for the TLR4-vaccine complex, it was 33.889 ± 0.31 Å (range = 32.580-34.755 Å) (Figure 10B and B1), indicating adequate folding, structural compactness, and epitope accessibility for immune recognition.
Hydration and interface stability were assessed through the number of solute-solvent hydrogen bonds. The TLR3-vaccine complex exhibited an average of 1839.97 ± 32.20 hydrogen bonds (min: 1502, max: 1912), whereas the TLR4-vaccine complex maintained 1747.52 ± 33.71 hydrogen bonds (min: 1333, max: 1811) (Figure 10C and C1). The enduring hydrogen bonding configurations enhance the structural stability of the complexes and their ability to preserve epitope shape.
Investigations into root-mean-square fluctuation (RMSF) revealed residue flexibility across the complexes. The vaccine exhibited more flexibility in relation to TLR4 (average RMSF = 3.66 ± 1.83 Å; min = 1.32 Å, max = 13.41 Å) compared to TLR3 (average RMSF = 3.09 ± 2.00 Å; min = 1.21 Å, max = 16.44 Å). The TLR residues demonstrated a degree of stability (TLR4 = 1.80 ± 0.68 Å; TLR3 = 1.48 ± 0.50 Å) (Figure 10D and D1), thereby confirming the structural integrity of the receptor-ligand interaction.
Simulated immunogenic studies of the selected vaccine
The immunological simulation study was conducted over a period of 350 days, with dosages administered on days 1, 15, and 30. After the initial dosage, a primary immunological response was observed, marked by rapid antigen clearance and a rise in IgM antibodies (Figure 11A), signifying early humoral activation. This reaction was associated with the activation of B cells (Figure 11B) and T helper cells (Figure 11C), along with a modest production of cytokines, such as IL-2 and IFN-γ (Figure 11D), indicating the initiation of adaptive immunological pathways. Following the administration of the second dosage, there was a notable enhancement in the immunological response, as indicated by a significant increase in IgG1 and IgG2 antibodies (Figure 11A), a heightened proliferation of B and T cells (Figure 11B and 11), and an elevation in cytotoxic T cell activity (Figure 11E). Simultaneously, there was an increase in cytokine levels (Figure 11D), indicating robust cellular immunity and the establishment of immunological memory. After the administration of the third dosage, there was a notable peak in antibody titers (Figure 11A), accompanied by the migration of B and T cells into resting and memory stages (Figure 11B and C), suggesting the establishment of long-term immune protection. Cytokine levels stabilized (Figure 11D), and DCs remained continuously active (Figure 11F), suggesting prolonged antigen presentation. Collectively, these results suggest that the multi-epitope vaccination may produce significant humoral and cellular immune responses with persistent immunological memory against DBV.
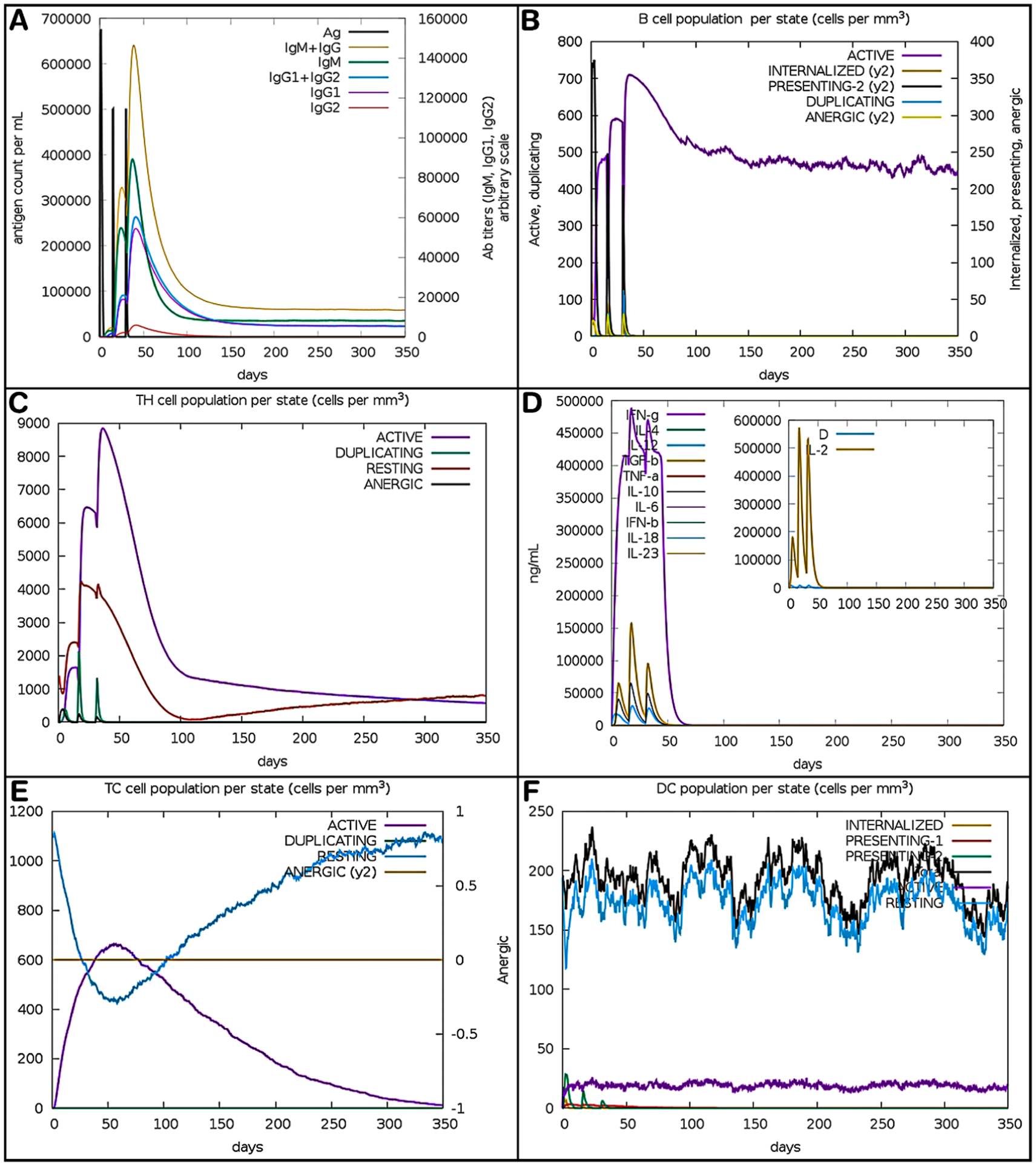
Immune response simulation over 350 days following 3 vaccine doses. (A) Antigen levels and antibody titers showing initial IgM response and rising IgG1/IgG2 after booster doses; (B) B cell states showing activation and memory formation; (C) T helper cell dynamics with strong activation post-injection; (D) cytokine levels peaking after second and third doses; (E) cytotoxic T cell activation and reduced energy over time; and (F) dendritic cells remained active, ensuring sustained antigen presentation.
In silico codon adaptation and sequence cloning in the Escherichia coli
The JCat server optimized the codon for E coli (strain K12) of the final vaccine protein sequence, where the codon sequence length was optimized to 1089 base pairs, with 0.97 for the CAI score. The GC content was adjusted to 49.95%, which supports the transcriptional stability and lowers the risk of secondary structure formation. It implies that the designated host organism could have a high degree of expression feasibility for host-specific preferences. The SnapGene application introduced the optimized sequence into the pET-28a (+) vector for in silico cloning (Figure 12A). A Kozak sequence was incorporated at the N-terminal to boost potential future eukaryotic expression (Figure 12C). The insertion was performed between the restriction sites Eco53KI (position 188) and EcoRV (position 1281), generating a recombinant plasmid that contains 5077 base pairs (Figure 12B). The successful insertion suggests the appropriate orientation and compatibility, which may facilitate future recombinant expression studies of the constructed vaccine against the DBV.

In silico codon optimization and cloning of the vaccine sequence into the pET-28a(+) vector. (A) Circular map of the native pET-28a(+) plasmid (5369 bp); (B) recombinant plasmid map showing the in silico insertion of the codon-optimized vaccine sequence between Eco53KI (188 bp) and EcoRV (1281 bp); and (C) linear representation of the codon-optimized vaccine nucleotide sequence.
Structural stability assessment of vaccine-encoding mRNA
The RNAfold analysis reveals that the codon-optimized mRNA vaccine construct exhibits a predicted secondary structure characterized by an MFE of −314.40 kcal/mol. This observation indicates a configuration that could be thermodynamically advantageous, potentially enhancing both structural stability and translation efficiency. The thermodynamic ensemble indicated a free energy of −334.13 kcal/mol, potentially illustrating the average stability among all conceivable secondary structures. The centroid structure exhibited a free energy of −222.87 kcal/mol, indicating that it may represent the most probable ensemble conformation, which implies potential structural stability and physiological relevance (Figure 13).

RNAfold-based secondary structure analysis of the mRNA vaccine after codon optimization. (A and C) Centroid and minimum free energy (MFE) structures colored by base-pair probability; (B and D) centroid and MFE structures colored by positional entropy; and (E) overlay of free energy profiles (MFE, ensemble, centroid) along with nucleotide-wise entropy distribution.
Discussion
A dominant tick-borne pathogen, DBV binds to the surface of skin resident with immune cells and later infects these cells via receptor-mediated entrance. As viral replication progresses, DBV disrupts innate immune signaling, which results in immune dysregulation and severe clinical manifestations. Multiple disease outcomes have been linked to DBV infection, leading the World Health Organization to classify it as a priority emerging infectious disease with major public health implications for the world.16,92 In the absence of licensed vaccines or targeted antiviral therapies, there is an urgent need for rational vaccine design approaches. This study aimed to assess the computational feasibility of developing a multi-epitope mRNA vaccine for DBV using an immunoinformatics method.
Recent advancements in multi-epitope mRNA vaccine development highlight the importance of computational immunology in the systematic design of vaccines for various diseases. A systematic methodology encompassing epitope prediction, conservancy analysis, population coverage assessment, physicochemical characterization, structural modeling, docking, MD simulations, and immune simulations has facilitated the creation of mRNA vaccines designed with multiple epitopes targeting bacterial (Pseudomonas aeruginosa, meningitis pathogens, Campylobacter jejuni, Clostridium perfringens) and parasitic (Theileria annulata, Eimeria tenella) pathogens.93-98 This study presents an in silico mRNA vaccine candidate for DBV, an emerging infectious disease, following the recent advancements in multi-epitope mRNA vaccine development, utilizing a comprehensive immunoinformatics framework.
The membrane glycoprotein polyprotein was chosen as the vaccine target. It is based on its known role in attachment to the host cell, transcription, and replication, playing a direct role in DBV pathogenicity.99,100 Consequently, the antigenicity and biophysical characteristics of the protein were examined, indicating that it was antigenic, less allergenic, stable, and hydrophilic. Glycoproteins have been widely considered immunodominant antigens because they are expressed on the surface and accessible to neutralizing antibodies, especially in enveloped and tick-borne viruses. Protein antigenicity, which is defined as the elicitation of an immune response, is an important parameter for determining vaccine appropriateness. 101
Potential B-cell, CTL, and HTL epitopes were identified to stimulate the immune system, such as antibody-mediated and T-cell-mediated responses. They are crucial for effective antiviral immunity.102,103 The epitopes conservancy analysis was done on 1042 DBV glycoprotein sequences, including recent circulating forms. This approach increases theoretical strain coverage and minimizes the possibility of an escape of epitopes, although this does not eliminate the risk of evolution. Recent literature emphasizes that large-scale conservancy analysis is increasingly essential for RNA viruses with ongoing evolution. 104 Population coverage analysis showed good coverage of the selected epitopes of MHC class I and combined (MHC-I + MHC-II) across all endemic and non-endemic regions, indicating widespread inclusion of well-represented HLA alleles and the global relevance of the proposed vaccine construct. Importantly, comparable levels of coverage between endemic and non-endemic populations indicate limited geographic bias for the choice of epitopes. 61 In contrast, the steady lower MHC class II coverage noted across all regions is in line with the extensive polymorphism and the stringent peptide-binding properties of MHC-II molecules and does not adversely affect the good combination of population coverage. 61
Linkers were incorporated to ensure structural integrity, epitope separation, and proper antigen processing. The EAAAK, GPGPG, AAY, and KK linkers have been adopted based on their known roles in stabilization of fusion proteins and enhancement of immunogenic organization.105,106 The AAY linker has been shown to increase CTL and HTL epitope processing,107,108 and EAAAK facilitates efficient fusion between adjuvants and epitopes, increasing expression and functional stability.109,110 Although it has been reported that GPGPG linkers can be used to decrease functional immunogenicity in certain experimental settings, 111 the use of such a linker in this study was designed to represent a balance between flexibility and epitope accessibility. Beta-defensin-3 was adopted as an adjuvant because of its immune-stimulatory effect and its having been previously validated in experimental vaccine studies.112-114 Physicochemical evaluation suggested that the designing construct is antigenic, stable, hydrophilic, and less allergenic, which are favorable characteristics for downstream expression and formulation.
Structural assessment provided the information on the structural plausibility of the vaccine construct rather than the confirmation of biological function. Secondary structure analysis suggested a predominance of random coil regions that could increase accessibility of epitopes and also α-helices and β-sheets that contribute to structural stability.115,116 Tertiary structure modeled by I-TASSER followed by refinement resulted in acceptable stereochemical quality as assessed using the Ramachandran plot and ProSA Z-score analysis. Surface-exposed antigenic regions that might be accessible for antibody binding have been identified by epitope mapping with the Ellipro. 117 Disulfide engineering also improved conformational stability by lowering structural entropy, which had previously been shown to increase protein robustness. 76
Molecular docking and MD studies were performed to identify the possible interactions between this vaccine construct and the innate immune receptors TLR3 and TLR4. Stable hydrogen bond and salt bridge interactions were observed, and negative MM/GBSA binding free energy values indicated computationally thermodynamically favorable associations. The MD simulations (100 ns) of the TLR3 and TLR4 vaccine complexes revealed stable RMSD and RMSF values, which suggest overall stability of the structures with minor local flexibility.118,119 The Rg analysis verified insignificant structural expansion or unfolding. 120 Solvent-solute hydrogen bond analysis further supported interactions to be stable. 121 Dual evaluation of TLR3 and TLR4 is a methodological extension over many of the recent immunoinformatics studies on vaccines, which generally evaluate a single innate receptor. This is particularly relevant in the case of mRNA vaccines, because RNA sensing by TLR3 and adjuvant-like activation by TLR4 may act in a synergistic way in the early immune priming and innate signaling. The TLR3 recognition of the RNA structures leads to activation of downstream type I interferon pathways, 61 and TLR4 engagement by lipid nanoparticle components can contribute to enhanced nuclear factor kappa B (NF-κB) and IRF signaling. 122
Immune simulation analyses indicated the improved trends in both humoral and cellular immune responses after booster administration, including improved levels of antibody response, cytotoxic T-cells (CTLs), and cytokines such as IFN-γ, IL-4, and IL-10.123,124 Codon optimization analysis showed good CAI and GC content values, indicating compatibility with E coli expression systems, while RNA secondary structure prediction analysis showed a value of thermodynamic stability suitable for efficient translation. 125 Cloning into the pET28a (+) vector was suggested to ease recombinant protein expression and purification for experimental validation.126,127 After cellular uptake, the mRNA vaccine activates innate immunity through RNA sensing by pattern recognition receptors, including TLRs, which triggers type I interferon and pro-inflammatory signaling; this innate activation facilitates antigen presentation and drives adaptive immune responses involving cytotoxic T cells, helper T cells, and antibody production. 128
Previous DBV vaccine studies have been focused on using experimental mRNA platforms to express full-length or subunit glycoproteins, which showed high immunogenicity and protective efficacy in animal models.13,129 While such approaches have demonstrated the feasibility of mRNA-based DBV vaccines, they largely use single- or dual-antigen expression and do not specifically address viral sequence diversity or population-level HLA variability. Parallel immunoinformatics studies suggested multi-epitope constructs based on multiple viral proteins, but these were limited due to smaller sequence datasets, single innate receptor interaction, or lack of analysis of conservation with recent variants. 130
In contrast, the present study introduces a conservation-driven, multi-epitope mRNA vaccine design based on the analysis of 1042 glycoprotein sequences, incorporating the recent DBV variants explicitly to mitigate the immune escape. Unlike previous in silico designs, the construct engages both TLR3 and TLR4 simultaneously and thereby provides a more comprehensive model of innate immune engagement that has been under-explored in previous DBV vaccine studies. Furthermore, the integrated population coverage analysis shows strong global applicability across endemic and non-endemic regions via MHC-I and combined MHC coverage, overcoming an important translational limitation of prior computational reports.
Despite promising results of the computational outcomes, this study has limitations that mainly rely on in silico approaches. Immunoinformatics predictions are not a certainty for effective antigen processing, MHC presentation, and immunodominant recognition in vivo. Risks of HLA restriction, epitope escape, conformational mismatch of B-cell epitopes, and inefficient protein folding are challenging problems, as indicated in the recent literature.131,132
In conclusion, this study proposes a structured immunoinformatics framework for the theoretical possibility of a multi-epitope mRNA vaccine targeting DBV. Although the findings are consistent with hypothesis generation and rational design, rigorous experimental validation is essential before any conclusions about protective efficacy or translational applicability can be made.
Conclusion
A multiple mRNA vaccine of the DBV was computationally designed using an immunoinformatics approach targeting the membrane glycoprotein. The 363-amino-acid construct exhibited predicted antigenicity (0.6294) and theoretical global population coverage of 94.77%. Structural modeling suggested acceptable stability, with 86.6% of residues located in preferred Ramachandran regions and a Z-score of −3.05, supported by disulfide engineering. Docking, MM/GBSA, and MD simulations suggested potential interactions with TLR3 and TLR4 (ΔG: −192.33 and −244.61 kcal/mol), indicating possible immune recognition at the in silico levels. Codon optimization (CAI: 0.97) and mRNA secondary structure analysis (MFE: −314.40 kcal/mol) supported the theoretical expressibility and stability of the construct. This work demonstrates the computational viability of vaccine design and formulates hypotheses, but immunogenicity, safety, and efficacy must be validated in vitro and in vivo prior to any experimental or clinical use.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322261438994 – Supplemental material for Multi-Epitope mRNA Vaccine Targeting Dabie Bandavirus Glycoprotein: An Immunoinformatics-Based Study
Supplemental material, sj-docx-1-bbi-10.1177_11779322261438994 for Multi-Epitope mRNA Vaccine Targeting Dabie Bandavirus Glycoprotein: An Immunoinformatics-Based Study by Sk Faisal Ahmed, Md Shah Paran, Md Masudur Rahman Munna, Enam Ahmed, Upom Majumder, Monisha Rahmatullah and Md Zulfekar Ali in Bioinformatics and Biology Insights
Supplemental Material
sj-docx-2-bbi-10.1177_11779322261438994 – Supplemental material for Multi-Epitope mRNA Vaccine Targeting Dabie Bandavirus Glycoprotein: An Immunoinformatics-Based Study
Supplemental material, sj-docx-2-bbi-10.1177_11779322261438994 for Multi-Epitope mRNA Vaccine Targeting Dabie Bandavirus Glycoprotein: An Immunoinformatics-Based Study by Sk Faisal Ahmed, Md Shah Paran, Md Masudur Rahman Munna, Enam Ahmed, Upom Majumder, Monisha Rahmatullah and Md Zulfekar Ali in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors thank the Dawn of Bioinformatics Ltd., Dhaka 1360, Bangladesh, for the valuable suggestion and support.
Ethical Considerations
The authors did not use any animal model in our research.
Author Contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data associated with this study are present in the article and its supplementary files.
AI Statement
During the preparation of the manuscript, the authors used artificial intelligence (AI) for checking grammatical errors, improving the sentence structures and flow of different paragraphs.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.