
Abstract
Helicobacter pylori infection of the stomach’s epithelial cells is a significant risk factor for stomach cancer. Various H pylori proteins (CagA, GGT, NapA, PatA, urease, and VacA) were targeted to design 2 messenger RNA (mRNA) vaccines, V1 and V2, using bioinformatics tools. Physicochemical parameters, secondary and tertiary structure, molecular docking and dynamic simulation, codon optimization, and RNA structure prediction have also been estimated for these developed vaccines. Physicochemical analyses revealed that these developed vaccines are soluble (GRAVY < 0), basic (pI < 7), and stable (aliphatic index < 80). The secondary and tertiary structure of the vaccines demonstrated robustness. The docking with toll-like receptors (TLRs) revealed that the vaccines have a potential affinity for TLR-2 (V1: −1132.3 kJ/mol, V2: −1093.6 kJ/mol) and TLR-4 (V1: −1042.7 kJ/mol, V2: −1201.2 kJ/mol), and molecular dynamics simulations confirmed their dynamic stability. Structural analyses of V1 (−505.96 kcal/mol) and V2 (−634.92 kcal/mol) mRNA vaccines underscored their stability. In addition, the vaccine showed a considerable rise in the counts of B cells and extended activation of both T cells was also observed for the vaccines, suggesting the potential for long-lasting immunity, and offering enhanced protection against H pylori. These findings not only suggest potential long-lasting immunity against H pylori but also offer hope for the future of stomach cancer prevention. Notably, the study emphasizes the need for subsequent animal and human-based studies to confirm these promising results.
Introduction
Stomach cancer, often known as gastric cancer, originates in the epithelial cells that line the stomach. 1 It ranks as the fifth most prevalent cause of cancer-related fatalities globally. 2 Stomach cancer includes various types, such as adenocarcinoma, gastrointestinal stromal tumors, neuroendocrine tumors, lymphomas, and squamous cell carcinoma. 3 Adenocarcinoma is the most common among these, accounting for approximately 90% of cases. 3 Multiple risk factors contribute to the development of stomach cancer, while Helicobacter pylori infection of the stomach’s mucosal layer is a significant predisposing factor. This bacterium is responsible for several infections affecting the lymph tissue of the mucosa and causing ulcers and carcinoma. 4 The infectious factors of H pylori lead to prolonged inflammation that causes necrosis, lesions of tissue finally stomach cancer. 5 Based on the GLOBOCAN 2020 predictions, stomach cancer resulted in roughly 800 000 deaths, which is 7.7% of all cancer-related deaths. In 2020, there were approximately 1.1 million newly diagnosed cases of stomach cancer, which constituted 5.6% of all reported cancer cases. 6 In 2024, there are 26 890 new cases and 10 880 fatalities, representing 1.3% and 1.8% of total cancer cases and deaths, respectively. 7 The majority of stomach cancer cases, mostly 60%, were identified in Eastern Asia, with China contributing to 43.9% of these cases. In 2020, stomach cancer was reported as the predominant cancer diagnosed in 7 Asian countries: Iran, Afghanistan, Turkmenistan, Uzbekistan, Tajikistan, Kyrgyzstan, and Bhutan. 2 In the year 2020, the number of fatalities and incidents of stomach cancer in North America were 29 772 and 13 391, respectively, whereas South America had 49 547 cases and 39 165 deaths. In Central and Eastern Europe, there were around 65 516 cases that were confirmed to have led to 50 018 fatalities. On the contrary, Australia and New Zealand claimed to have recorded 2676 cases of stomach cancer with 1355 deaths. In Northern Africa, there have been approximately 93 977 confirmed cases and, however, 7727 fatalities. 8 Stomach cancer, however, is a highly deadly kind of cancer, with a 5-year survival rate of <20%.2,9 Since H pylori is responsible for nearly 90% of all cases of noncardia stomach cancer, there are no successful vaccine candidates against H pylori developed yet. 10
Cancer vaccines have the potential to revolutionize cancer prevention and therapy by using the immune system of the body to identify and destroy cancer cells. Vaccines, as opposed to conventional therapies such as chemotherapy or radiation, can specifically target cancer cells while preserving healthy tissue, hence minimizing the likelihood of adverse consequences. 11 Although therapeutic cancer vaccines have only been used in a restricted number of clinical applications, this field of study continues to be dynamic, and there are a variety of approaches by which these vaccines can be administered. There are 3 categories of cancer vaccines: (1) cell-based vaccines, (2) protein/peptide-based vaccines, and (3) nucleic acid–based vaccines, which include DNA and RNA vaccines. 11 Cell-based vaccines consist of autologous or allogeneic whole tumor cells or autologous dendritic cells (DCs). 12 To create a tailored immune response that targets the cancer, autologous tumor cell vaccines use cancer cells extracted from the own tissues of patients. One benefit of this vaccine is that it can expose the patient’s immune system to their unique tumor-associated antigens (TAAs), eliminating the requirement to recognize individual TAAs beforehand. This method, however, depends on collecting enough tumor samples, which is not usually possible until the cancer of a patient has progressed past a certain stage. Meanwhile, allogeneic tumor vaccines usually use a small number of well-established tumor cell lines, which aid in the standardization and scalability of cellular vaccine manufacturing. 13 In addition, TAAs are the main target of protein/peptide vaccines, which intend to stimulate the immune system in response to TAAs that express distinctively or more abundantly in malignant cells than in healthy ones. 14 Considering the nature of TAAs, their immunogenicity is often limited. Various strategies have been explored to circumvent this challenge and boost the effectiveness of protein or peptide vaccines. These approaches involve using inflammatory adjuvants during administration and combining the vaccines with additional immunostimulants, including toll-like receptor (TLR)-binding peptides. 15 However, protein/peptide vaccines need the pre-identification and precise targeting of specific antigens, which might prove particularly challenging for individuals with distinct mutations. 16
The likelihood that viral infections might induce infected cells to synthesize virus-specific peptides limited to the major histocompatibility complex (MHC) class I/II has prompted research into viral-based vaccinations. 17 The use of viral vaccines is subject to some limitations, one of which pertains to the inherent immunological response of the body to counteract viral vectors. 18 Consequently, some researchers have endeavored to use a prime-boost approach, whereby an initial administration of a tumor antigen and viral vector is followed by a subsequent “boost” of the same antigen facilitated by an alternative viral vector. 19 Nucleic acid vaccines include DNA, RNA, and viral-based vaccines, which attempt to introduce genetic material into antigen-presenting cells (APCs), resulting in the translation of cancer-specific antigens or antigen fragments. 20 RNA vaccines predominantly employ messenger RNA (mRNA) that intends to stimulate the translation of antigens in APCs. One possible benefit of mRNA vaccines instead of DNA vaccines is their potential to culminate in fewer side effects or autoimmune diseases owing to their faster degradation. In addition, mRNA vaccines do not integrate into the genome, avoiding the emergence of additional oncogenic potential. 21 Moreover, mRNA vaccines have the advantage of being readily and expeditiously synthesized and manufactured, enabling rapid reactions to emerging infections or variations. Moreover, they induce prevailing immunological responses, including the production of antibodies and cellular immunity, leading to the development of a robust barrier against infection. 22
The genome of H pylori consists of a circular DNA molecule with a length of 1 667 867 base pairs. It has a total of 1590 putative coding sequences. 23 Multiple virulence genes of H pylori, such as cytotoxin-associated gene A (CagA), vacuolating cytotoxin gene (VacA), gamma-glutamyl transpeptidase (GGT), neutrophil-activating protein A subunit (NapA), peptidoglycan O-acetyltransferase (PatA), and urease, have been associated with stomach cancer progression. The CagA encodes a protein (120-140 kDa) that enhances the pathogenicity of the bacterium by increasing the cytokine production (interleukin-8) level within the host cell. 24 In addition, CagA attaches itself to Grb2 through the area that contains EPIYA, causing an aberrant activation of the Ras signaling pathway that promotes aberrant cell proliferation. 25 The VacA encodes another virulence protein (87 kDa) that causes vacuolation in epithelial cells, resulting in cellular damage. 26 According to Hatakeyama, 27 a programmed necrosis pathway is responsible for the VacA-induced cell death of AZ-521 gastric epithelial cells. A virulence protein of H pylori is GGT, which leads to the consumption of glutamine and glutathione in the host cells, as well as the production of reactive oxygen species and ammonia, thereby causing gastric epithelial cells to undergo necrosis, apoptosis, and cell-cycle arrest. In addition, via inducing cyclooxygenase-2, interleukin-8, inducible nitric oxide synthase, and peptides linked to epidermal growth factor, GGT may promote the proliferation of gastric epithelial cells and suppress cellular apoptosis and lead to cancer. 28 Oxidative stress in the gastric mucosa resulting from H pylori infection is a significant component that contributes to the development of gastric cancer. 29 Most H pylori strains contain the NapA gene, which encodes HP-NAP 30 that can activate neutrophils to generate reactive oxygen species (ROS) 31 and reactive nitrogen species (RNS), as well as to adhere to endothelial cells. 32 The ROS and RNS induced by H pylori can cause DNA damage and mutations that lead to the initiation of cancer. 33 The PatA of H pylori protects the bacterium from host lysozyme, thereby facilitating the immune evasion of the bacterium. 34 Besides, H pylori urease can contribute to the pathogenic activity of the bacterium by altering the host immune response through stimulating monocytes and polymorphonuclear leucocytes, resulting in inflammation and epithelial injury. 35 A recent study suggests that urease may increase the carcinogenic potential of H pylori by inducing HIF-1α, 36 which further plays a noncanonical role by reducing Cyclin D1 half-life, disrupting the cell cycle, and promoting angiogenesis. 37
Considering the importance of these pathogenic proteins of H pylori, an effective mRNA vaccine against stomach cancer can be formulated. This type of vaccine, however, comprises multiple epitopes, including MHC-I, MHC-II, and B-cell epitopes from the pathogenic proteins of H pylori. As immunological stimulants against intracellular microorganisms or tumorigenic antigens, MHC-I molecules initiate the activation of cytotoxic T cells (TCs) when they engage with cytotoxic T lymphocytes (CTLs). Therefore, potential vaccines have been developed using the binding epitopes of MHC-I that trigger the activation of CD8+ T cells, which greatly aid in eradicating viral infections and tumorigenic antigens. 38 As an additional point of interest, MHC-II molecules present immunogenic processed peptides to the T-cell receptor (TCR) on CD4+ T cells. This is a critical stage in activating both cellular and antibody-mediated immune responses. When identifying developing cancers and rejecting transplants, the association between MHC-II molecules and TCR—which carry processed peptides—is fundamental.39,40 Therefore, identifying the existence of CD4+ and CD8+ T cells is a crucial phase in developing an effective vaccine.41,42 Also, B-cell epitopes play a pivotal role in the interactions between antigens and antibodies, therefore being an essential factor to consider when developing vaccines.41,43
The technique, known as “Reverse Vaccinology” (RV) and immunoinformatics, eliminates the necessity of cultivating pathogens. 44 The benefits of these approaches, which allow for vaccine research on organisms that are difficult or impossible to grow in a lab setting, continue to be attractive and decrease the time required for selecting target proteins by employing many species or strains simultaneously. 45 However, there are some cons, such as the accuracy of immunoinformatics predictions being contingent upon the sophistication of the algorithms employed and the data quality. Errors are frequently present in sequence data obtained from high-throughput analysis. 45 The mRNA vaccine and reverse vaccinology methods received considerable attention when the FDA issued emergency use authorization for the first 2 severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) mRNA vaccines—BNT162b2 from Pfizer/BioNTech and mRNA-1273 from Moderna. In addition, this groundbreaking work received the Nobel Prize in Physiology or Medicine in 2023. 46
The study aims to design mRNA vaccine candidates against H pylori that might be utilized in reverse vaccinology to target stomach cancer. We utilized advanced immune-informatics methodologies to predict T helper lymphocyte (HTL), CTL, and B-lymphocyte epitopes predicted by the sequences of amino acids of the subsequent 6 proteins: CagA, GGT, NapA, PatA, urease, and VacA.
Materials and methods
Obtaining of sequence
The amino acid sequences of H pylori CagA (accession number: AAB58747.1), GGT (accession number: BDO45106.1), NapA (accession number: AAU21203.1), PatA (accession number: WP_308811605.1), urease (accession number: EJB13369.1), and VacA (accession number: AAU85846.1) were retrieved from National Center for Biotechnology Information (NCBI) protein database. 47 Subsequently, all the sequences underwent BLASTp (protein-protein BLAST) on the NCBI database. Next, the conserved sequences of each protein were used for all the subsequent steps (Figure 1).

The outline of the entire study.
Prediction of CTL epitopes
The CTL-binding epitopes of the selected proteins (CagA, GGT, NapA, PatA, urease, and VacA) were predicted using the Immune Epitope Database (IEDB)48,49 and NetMHCIIpan 4.0 server,48,50 using the default settings of the threshold for epitope identification, weight on TAP transport efficiency, and weight on C terminal cleavage. The CTLs recognize peptides presented by MHC-I molecules, and the peptide-binding groove of MHC-I is typically closed at both ends, accommodating peptides that are predominantly 8 to 10 amino acids in length, with 9-mers being the most common. 51 As a threshold, epitopes with a percentile rank ⩽1.0 and confined to 12 HLA alleles (HLA-A1, HLA-A2, HLA-A3, HLA-A24, HLA-A26, HLA-B7, HLA-B8, HLA-B27, HLA-B39, HLA-B44, HLA-B58, and HLA-B62) are classified as strong binders and thoroughly investigated. Subsequently, the antigenicity, allergenicity, and toxicity were assessed for each elected epitope by VaxiJen 2.0, 52 AllerTOP v. 2.0, 53 and ToxinPred server, respectively.54,55
Prediction of HTL epitopes
The HTL-binding epitopes of the retrieved proteins (CagA, GGT, NapA, PatA, urease, and VacA) were predicted by IEDB48,56 and Net-MHC 4.0 server. 57 The default values established the thresholds (percentile rank ⩽ 1.0) for strong and weak binders, while the epitopes were confined to 12 HLA-DRB alleles, including HLA-DRB1-0101, HLA-DRB1-0301, HLA-DRB1-0401, HLA-DRB1-0701, HLA-DRB1-0801, HLA-DRB1-0901, HLA-DRB1-1001, HLA-DRB1-1101, HLA-DRB1-1201, HLA-DRB1-1301, HLA-DRB1-1401, HLA-DRB1-1501, and HLA-DRB1-1601. The HTLs recognize peptides presented by MHC-II molecules. Unlike MHC I, the peptide-binding groove of MHC-II is open at both ends, allowing longer peptides, and thus, typically, 13 to 25 amino acids can be fitted with an optimal length of around 15-mer 51. Only those epitopes predicted by more than one allele were deemed strong binders and chosen for further study. Thereafter, the antigenicity, allergenicity, and toxicity were assessed for each elected epitope by VaxiJen 2.0, 52 AllerTOP v. 2.0, 53 and ToxinPred server, respectively.54,55
Prediction of linear B-cell epitopes
The linear B-cell epitopes of the protein of interest (CagA, GGT, NapA, PatA, urease, and VacA) were predicted by the IEDB 58 and ABCpred server. 59 The minimum score and maximum distance prediction parameters were set to 0.8 and 6 (Å), respectively. Afterward, the antigenicity, allergenicity, and toxicity were assessed for each elected epitope by VaxiJen 2.0, 52 AllerTOP v. 2.0, 53 and ToxinPred server, respectively.54,55
Population coverage of MHC alleles
The frequency of HLA alleles can fluctuate by geographic location and ethnicity. 60 Considering the population coverage of MHC alleles is essential in the development of a successful vaccine. 61 Using the IEDB (http://tools.iedb.org/population/), we employed the appropriate MHC alleles in conjunction with the targeted proteins’ selected CTL and HTL epitopes, either individually or tandemly. 62
Construction of the vaccine
The mapping of vaccine 1 (V1) and vaccine 2 (V2) was done using the highly prioritized epitopes of the selected proteins (CagA, GGT, NapA, PatA, urease, and VacA). The adjuvants, 50 S ribosomal protein L7/L12 and heparin-binding hemagglutinin (HBHA), were used as immune system enhancers in V1 and V2, respectively. The immune efficacy of a vaccine can be enhanced by adding L7/L1263-65 and HBHA of Mycobacterium tuberculosis adjuvants, as demonstrated in laboratory trials. 63 Nevertheless, linkers are employed to distinguish epitopes, offering flexibility, cleavability, and stability to the epitopes inside the vaccine formulation.63-65 In addition, the immunogenicity of a vaccine may be enhanced through the incorporation of linkers including AYY, EAAAK, AK, and KFER linkers, as these allow for intramolecular hydrogen bonding and preserve the individual functional properties.63-65
Assessment of biophysical qualities
The biophysical qualities of the V1 and V2, including the molecular mass, the overall count of amino acids, instability, aliphatic index, isoelectric point, overall mean of water solubility (GRAVY), the total count of positive and negative charged residues, and the total count of atoms, were predicted by Expasy’s ProtParam server.66,67 In addition, the solubility of both vaccines was indicated by the SOSUI and SOLpro servers.68-72 The allergenicity of both vaccines was evaluated by AllergenFP v.1.0, 73 AllerCatPro v.2.0, 74 and AlgPred servers.72,75 Moreover, the antigenicity of both vaccines was also analyzed by ANTIGENpro and VaxiJen 2.0 server. 76
Prediction of secondary structure
The prediction of secondary structures of the V1 and V2 were conducted by 3 servers including GOR4, 77 SOPMA, 72 and PSIPRED by keeping all the parameters at a default.78-80
Prediction, refinement and validation of the tertiary structure
The prediction of tertiary structures of the V1 and V2 were conducted employing I-TASSER server. 81 Next, a refinement was performed on the GalaxyWEB server for both vaccines. 82 Subsequently, the validation of both structures was performed utilizing the SAVES v6.0 server, which elucidates the quality of the predicted structures through the application of the Ramachandran plot and ERRAT analysis.83-86
Identification of discontinuous B-cell epitopes
The identification of discontinuous B-cell epitopes of the V1 and V2 was conducted by the DiscoTope 2.0 87 and Ellipro server by setting all the parameters at default. 88
Binding affinity evaluation by molecular docking
Toll-like receptors are integral to the innate immune system, recognizing pathogen-associated molecular patterns (PAMPs) and initiating immune responses. 89 Among the TLR family, toll-like receptor-2 (TLR-2) and toll-like receptor-4 (TLR-4) are frequently utilized in vaccine design due to their specific ligand recognition and pivotal roles in immune activation compared with other receptors.89,90 TLR-2 and TLR-4 recognize various PAMPs, making them versatile targets for various vaccine antigens. 89 In addition, binding these receptors leads to robust immune responses, essential for effective vaccine efficacy.89,90 Prior to the docking, the human TLR-2 (PDB: 2Z7X) and human TLR-4 (PDB: 3FXI) tertiary structures were obtained from the Protein Data Bank (www.rcsb.org). Therefore, the CLUSPRO 2.0 server was utilized to check the binding affinity of both vaccines with TLRs.91,92 However, PyMOL software and the PDBsum server were employed for the visualization and analysis of the complexes. 93
Simulation of the molecular dynamics
The docked complexes of both vaccines and the free structure of both vaccines underwent simulation of the molecular dynamics through the GROningen MAchine for Chemical Simulations (GROMACS) (version 2022.3). 94 The GROMOS96 43a1 force field was employed for the simulation, utilizing the SPC water model to construct a cubic water box. The systems were neutralized with NaCl. A 50 ns molecular dynamics simulation was conducted using isothermal-isochoric (NVT) and isobaric (NPT) equilibration types, with a temperature of 300 K, a pressure of 1.0 bar, periodic boundary conditions, and a time integration step of 2 fs. The trajectory data were evaluated at a snapshot interval of 100 picoseconds. The root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), and solvent accessible surface area (SASA) were computed using the corresponding modules incorporated in the GROMACS program.
Optimization and cloning of the DNA of the vaccine
The Java Codon Adaptation Tool (JCat) server was used for codon optimization of the vaccine using the Escherichia coli strain K12. 95 The server determines a protein’s codon adaptation index (CAI) and GC contents to evaluate its expression level. The CAI value of >0.8 is deemed a satisfactory score, while ⩾1.0 represents the optimal score. The integrity of the mRNA sequence is improved by a higher GC content, which is attributed to the stronger stacking interaction between guanine (G) and cytosine (C) due to triple hydrogen bonds. 96 Furthermore, the increased GC content facilitates the efficient processing of mRNA. 97 The acceptable GC contents, however, can range from 30% to 70%. 41 However, the optimized vaccine DNA underwent cloning into the E. coli plasmid vector pET-28a(+), with BamHI, BglII, and NdeI restriction sites ligated to the N- and C-terminals of the vaccine sequences, respectively.
Simulation of the immune response of the vaccine
The simulation of the immune response after the administration of both V1 and V2 was performed on the C-ImmSim server. 98 Three vaccine doses are typically administered at 28-day intervals to assure a robust and durable immune response.99,100 The antigen is introduced in the first dose, which primes the immune system, whereas the second dose augments the immune response, leading to increased levels of B and T cell activation, and the third dose reinforces long-term immunity by further amplifying and maturing the immune response. 99 The simulation run was executed with steps in time set to 1, 84, and 168 and with a simulation volume and steps of 50 and 1000, respectively.
Structure prediction of the mRNA
The RNAfold website (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) was employed to predict the secondary structures of both the V1 and V2. 101 The server estimates the query mRNA structures’ thermodynamically derived minimum free energy (MFE).102,103 Subsequently, using the Biomodel server, the optimal DNA sequences of the V1 and V2 taken from the cloning step were converted into RNA (https://biomodel.uah.es/en/lab/cybertory/analysis/trans.htm). 104 Next, the RNAfold website was then used to validate and predict secondary structures based on these RNA sequences.
Results
Obtaining of sequence
This study obtained and applied the amino acid sequences of the following 6 proteins of H pylori, including CagA, GGT, NapA, PatA, urease, and VacA, for subsequent analysis.
Prediction of CTL epitopes
The CTL-binding epitopes of the following proteins, CagA, GGT, NapA, PatA, urease, and VacA were predicted by the IEDB database and Net-MHC 4.0 server. A total of twelve 9-mer length peptides were selected based on their percentile rank (⩽1.00) (Table 1). Nonetheless, the selected epitopes exhibited traits of antigenicity, non-allergenicity, and non-toxicity.
List of selected CTL epitopes from the protein sequences with their antigenicity, allergenicity, and toxicity.

Prediction of HTL epitopes
The prediction of HTL epitopes of the targeted proteins (CagA, GGT, NapA, PatA, urease, and VacA) was conducted employing the IEDB and NetMHCIIpan 4.0 server. Twelve 15-mer peptides were predicted from the proteins that were chosen on the basis of the lowest possible percentile rank (⩽1.00), which explains the high affinity. However, antigenicity, non-allergenicity and non-toxicity properties were found on the chosen epitopes (Table 2).
List of selected HTL epitopes from the protein sequences with their antigenicity, allergenicity, IFN-γ and IL-10 inducing capability, and toxicity.

Prediction of linear B-cell epitopes
The prediction of epitopes of linear B-cells from the chosen proteins (CagA, GGT, NapA, PatA, urease, and VacA) was conducted on the IEDB and the ABCpred servers. Twelve 16-mer epitopes were chosen along with antigenicity, non-allergenicity, and non-toxicity properties (Table 3).
List of selected linear B-cell epitopes with their toxicity, allergenicity, and antigenicity.

Population coverage for MHC alleles
The population coverage of the combined CTL and HTL alleles was predicted to be 100% across the maximum regions, including Central America, South America, North America, Oceania, West Indies, North Africa, Central Africa, West Africa, East Africa, Europe, South Asia, and Northeast Asia (Supplementary Figure 1). Following that, Southeast Asia (99.98%) and Southwest Asia (99.99%) hit significant coverage (Supplementary Figure 1). However, South Africa has the lowest population coverage rate at 95.27% (Supplementary Figure 1).
Construction of the vaccine
Using the chosen epitopes, adjuvants, and linkers, 2 distinct vaccines were constructed. The adjuvant, specifically the 50S ribosomal protein L7/L12, was conjugated to V1 utilizing the EAAK linker. In contrast, the HTL, CTL, and linear B-cell epitopes were interconnected by the AYY, AK, and KFER linkers, respectively (Figure 2A). In the case of V2, the HBHA adjuvant was linked with the EAAK linker. Concurrently, the remaining epitopes, comprising the helper T-cell (HTL), cytotoxic T-cell (CTL), and linear B-cell epitopes, were interconnected utilizing AYY, AK, and KFER linkers, respectively (Figure 2B).
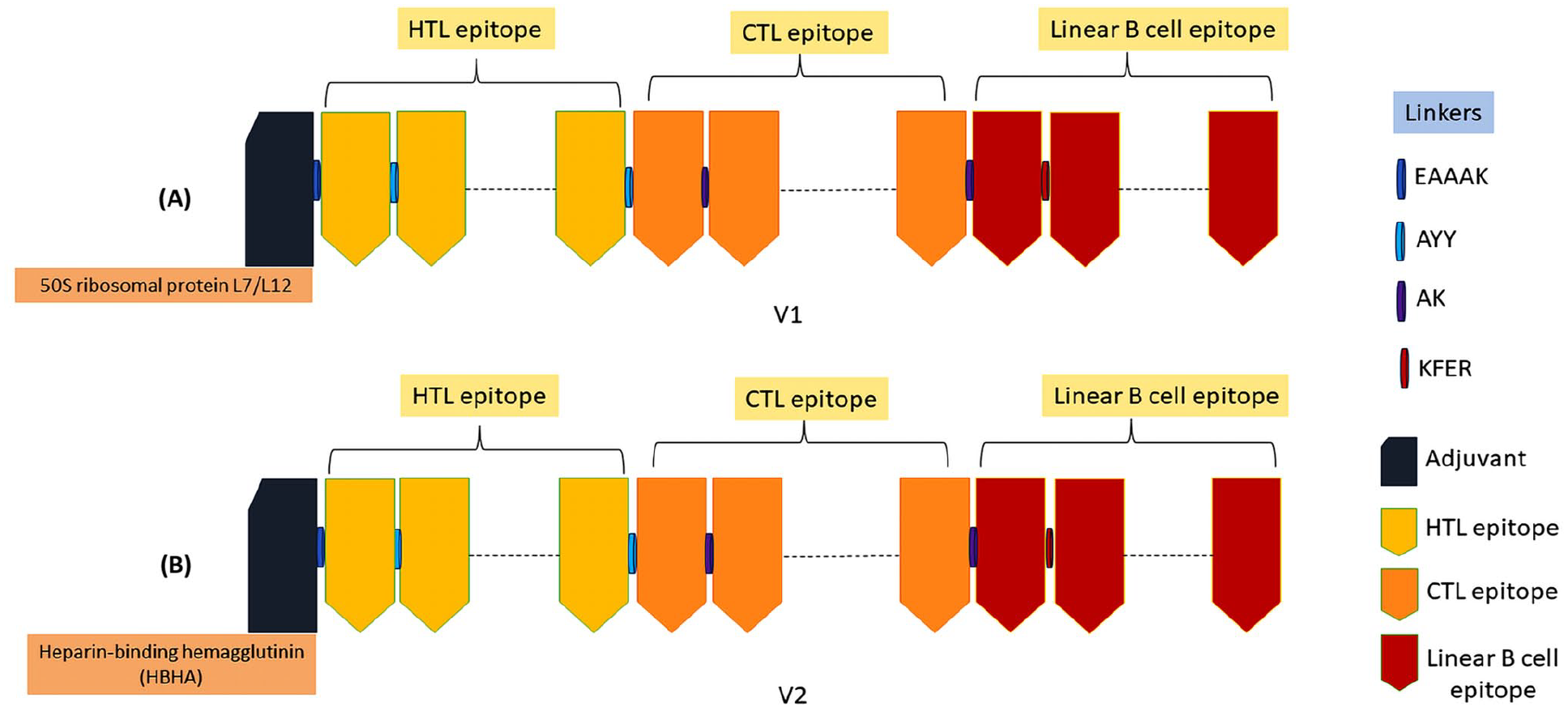
The construction of mRNA vaccines: V1 (A) and V2 (B). The illustration of the vaccines having adjuvant (deep blue color), epitopes (HTL—yellow, CTL—orange, linear B cell—maroon), and linkers (EAAAK—deep blue, AYY—light blue, AK—purple, KFER—maroon).
Assessment of biophysical qualities
The ExPASy ProtPram server was used to obtain the biophysical qualities of both V1 and V2. Based on the server, V1 comprised 425 amino acids with an isoelectric point of 8.77, whereas V2 consisted of 493 amino acids with an isoelectric point of 9.36. However, V1 and V2 had molecular masses of 46 803.57 Da and 54 623.78 Da, respectively. The instability indices of V1 and V2 were 19.95 and 36.31, respectively (threshold =< 40). The GRAVY of the V1 and V2 were predicted to be −0.271 and −0.605, respectively, (threshold =< 0). Moreover, both V1 and V2 were further validated as water-soluble (SOSUI and SOLpro). The aliphatic indices of the V1 and V2 were predicted to be 84.33 and 76.23, respectively (Table 4). These determined the good thermostability of both vaccines (threshold = 50-80). Furthermore, both the V1 and V2 vaccines were anticipated to be non-allergenic, as indicated by AllergenFP v.1.0, AllerCatPro v.2.0, and AlgPred. In addition, they were identified as containing antigenic proteins, as assessed by ANTIGENpro and VaxiJen 2.0 (Table 4).
The physicochemical and immunological properties of the V1 and V2.

Prediction of secondary structure
The secondary structures of the V1 and V2 were evaluated employing the GOR4, SOPMA, and PSIPRED servers. For V1, the GOR4 provided 33.88% random coil, 50.35% alpha helix, and 15.76% extended strands (beta sheet). However, the SOPMA exhibited a random coil of 24.47%, an alpha helix of 44.71%, and extended strands of 21.18% in the secondary structure of V1 (Figure 3). For V2, the GOR4 demonstrated 24.14% random coil, 64.91% alpha helix, and 10.95% extended strands (beta sheet). However, the SOPMA predicted a random coil of 20.28%, an alpha helix of 59.43%, and extended strands of 14.40% in the secondary structure of V2 (Figure 3). Nevertheless, the PSIPRED provided the secondary structure characteristics of V1 and V2 through 3-state prediction, including coil, helix, and strands (Figure 4).

The secondary structure of the V1 and V2 predicted by GOR4 and SOPMA.

The predicted secondary structure for V1 (A) and V2 (B). The initial bar (Conf) represents the degree of confidence in the prediction, with the length of the bar denoting varying levels of assurance. In the following bar (Cart), the presence of beta-sheets is signified in yellow hue, the helical structure is denoted by pink hue, and the coil configuration is represented by gray hue. However, 3 distinct structural characteristics and corresponding amino acid sequences are depicted on the next 2 bars (Pred and AA).
Prediction, refinement, and validation of tertiary structure
The I-TASSER was employed to predict the tertiary structure of both V1 and V2 by incorporating threading, fragment assembly, and refinement. A confidence score (C-score), which provides a measure of the quality of alignments and model compactness, was then used to rank the predicted models. In V1, 5 models were predicted but the model having the highest C-score of −1.35, TM-score of 0.55 ± 0.15, and RMSD of 10.1 ± 4.6Å was selected. In V2, the model having the highest C-score of −0.85, TM-score of 0.54 ± 0.15, and the RMSD of 8.1 ± 4.6Å was selected. Next, the refined model of V1 was taken from the GalaxyWEB server with RMSD, MolProbity score, and Ramachandran’s favored region of 0.468, 2.093, and 91.7%, respectively (Figure 5A). Conversely, the refined model of the V2 exhibited an RMSD of 0.445, a MolProbity score of 2.808, and a Ramachandran-favored region percentage of 92.9% (Figure 5A). However, the Ramachandran plot depicted that the refined model of V1 had 85.5% amino acid residues in the most favored region, 11.5% in the additional allowed region, and 1.3% in the generously allowed region (Figure 5B). Moreover, the Ramachandran plot depicted that the refined model of V2 had 88.1% amino acid residues in the most favored region, 8.5% in the additional allowed region, and 1.1% in the generously allowed region (80 to >90%) (Figure 5B). The ERRAT score of the refined V1 was calculated to be 82.258, whereas, for the V2, it was calculated to be 90.683 (>50) (Figure 5C). In addition, the structure quality was further validated based on a Z score that resulted in −2.28 for V1 and −2.6 for V2 (−10 to 0).

The prediction and validation of tertiary models of vaccines. The refined tertiary structures (A) are assessed employing Ramachandran plot score (B) and ERRAT (C) score.
Identification of discontinuous B-cell epitopes
A total of 225 discontinuous B-cell epitope residues were found in the V1 and V2 structures (Figure 6). In V1, the scores of epitopes were in 0.524 to 0.86 and 0.635 to 0.825 ranges, respectively, for V1 and V2 (Supplementary Table 1).

Tertiary representation of discontinuous B-cell epitopes of V1 (A) and V2 (B). The discontinuous B-cell epitopes are depicted in yellow-hue and the vaccine is depicted as gray sticks.
Binding affinity evaluation by molecular docking
From the 30 created models, the one exhibiting the central energy score of −868 kJ/mol and the lowest energy score of −1132.3 kJ/mol was chosen for the V1-TLR-2 complex, whereas the model with identical values of −1042.7 kJ/mol was picked for the V1-TLR-4 complex (Figure 7A, B and Table 5). Analogous to the V1, the model exhibiting the central energy score and the lowest energy score of −1006.7 and −1093.6 kJ/mol, respectively, was chosen for the V2-TLR-2 complex, whereas the model with center and lowest energy scores of −940.7 and −1201.2 kJ/mol, respectively, was picked for the V2-TLR-4 complex (Figure 7C, D and Table 5). Subsequently, the PDBsum predicted the number of hydrogen bonds, salt bridges, and non-bond interactions of 3, 7, and 166 in the V1-TLR-2 complex, respectively (Figure 7A and Table 5). However, the number of hydrogen bonds, salt bridges, and non-bond interactions were 8, 22, and 327 in the V1-TLR-4 complex, respectively (Figure 7B and Table 5). In addition, the number of hydrogen bonds, salt bridges, and non-bond interactions were 2, 10, and 182 in the V2-TLR-2 complex, respectively (Figure 7C and Table 5). However, the number of hydrogen bonds, salt bridges, and non-bond interactions were 12, 32, and 300 in the V2-TLR-4 complex, respectively (Figure 7D and Table 5).

The binding interplay among the V1-TLR-2 (A), V1-TLR-4 (B), V2-TLR-2 (C), and V2-TLR-4 (D) complexes. The vaccines, V1 and V2, and the receptors, TLR-2 and TLR-4, are represented in green, red, cyan, and deep blue, respectively.
The docking scores and interactions between the vaccine-receptor complexes.

Simulation of the molecular dynamics
Following a successful simulation, the RMSD of the vaccines (V1 and V2) and vaccine-TLR complexes (V1-TLR-2, V1-TLR-4, V2-TLR-2, and V2-TLR-4) was computed to assess structural stability. During the 50-ns simulation, the RMSD values for V1 and V1-TLR-4 exhibited a consistent increase; however, the RMSD values for V1-TLR-2 experienced a significant surge. Nevertheless, the RMSD of the complexes exhibited a tendency to diminish over a 50-ns simulation session (Figure 8A). The RMSD values for V2-TLR-2 and V2-TLR-4 exhibited a consistent increase, whereas the RMSD values for V1 remained unchanged. Conversely, the RMSD of the V2-TLR-2 and V2-TLR-4 began to decline once more following the 50-ns simulation session (Figure 8B). The RMSF of these complexes was used to evaluate regional flexibility. During the simulation, the motility pattern of V1-TLR-4 (Supplementary Figure 2C) markedly differs from that of V1-TLR-2 (Supplementary Figure 2B) and V1 apo (Supplementary Figure 2A), particularly at the C and N terminals. The motility pattern of RMSF for V2 apo (Supplementary Figure 2D) was significantly distinct from that of V2-TLR-2 (Supplementary Figure 2E) and V2-TLR-4 (Supplementary Figure 2F).

The RMSD analysis of the vaccines and vaccine-TLRs structures. The plot represents the RMSD of V1 and V1-TLRs (V1-TLR-2 and V1-TLR-4) (A), and V2 and V2-TLRs (V2-TLR-2 and V2-TLR-4) (B).
Throughout the simulation run, V1-TLR-4 had a greater Rg value than V1 and V1-TLR-2 (Figure 9A). Moreover, the Rg value of V2-TLR-4 was greater than that of V2 and V2-TLR-2 (Figure 9B). Throughout the whole simulation run, there was no significant difference in the SASA values of the V1 (Supplementary Figure 3A), V1-TLR-2 (Supplementary Figure 3B), and V1-TLR-4 (Supplementary Figure 3C) after 50 ns of running the simulation. The SASA values of the V2, V2-TLR-2, and V2-TLR-4 were also not significantly different during the simulation (Supplementary Figure 3D, Supplementary Figure 3E, and Supplementary Figure 3F, respectively).

The RMSF analysis of the vaccines and vaccine-TLRs structures. The plot represents the RMSD of V1 and V1-TLRs (V1-TLR-2 and V1-TLR-4) (A), and V2 and V2-TLRs (V2-TLR-2 and V2-TLR-4) (B).
Optimization and cloning of the DNA of the vaccine
According to the server, V1 and V2 had optimized DNA sequences of 1270 and 1473 nucleotide lengths, respectively. In addition, for both vaccines, the CAI values were determined to be 1.0. Moreover, the GC content was 51.23% and 51.50%, respectively, for V1 and V2 (Figure 10).

The depiction was about the cloning of (A) V1 and (B) V2 vaccines on suitable vector.
Simulation of the immune response of the vaccine
After 3 doses of administration, a significant rise in the counts of B cell was observed for both V1 (Figure 11A) and V2 (Figure 11B) vaccines, with the total B-cell counts remaining active, signifying persistent immunity over the course of a year (Figure 11A and B). Moreover, T cells were similarly apparent for V1 and V2 including an increased amount of active TH cells at day 60, which subsequently declined but persisted for nearly a year (Figure 11C and D). Moreover, active T cells demonstrated elevated expression following the vaccination regimen and continued to be stimulated for approximately 1 year (Figure 11E and F). In the case of both V1 and V2, vaccination also elicited cytokine responses, including interferon gamma (IFN-γ), which was at the highest uprising level and increased to nearly 2 × 106 Mg/mL. After 50 days, it decreased to 400 000 Mg/mL. Interleukin-12 and interleukin-10 were the lowest in this simulation, at approximately 50 000 mg/mL. Tumor growth factor-b was at >500 000 Mg/mL at 25 days and decreased at 50 days. The level of cytokines was initially found but vanished 100 days after the vaccination (Figure 11G and H).

The simulation of the immune system response for V1 and V2. The progression of (A, B) B-cell, (C, D) TH-cell, (E, F) TC-cell and cytokine level (G and H) after 3 successive injections of the V1 (A, C, E, and G) and V2 (B, D, F, and H).
Structure prediction of the mRNA
The secondary structure of the V1 mRNA demonstrated MFE score of −487.80 kcal/mol (optimum structure) and −354.19 kcal/mol (centroid structure). In contrast, V2 represented MFE score of −617.70 kcal/mol (optimum structure) and −515.48 kcal/mol (centroid structure). The thermodynamic free energy was found as −505.96 kcal/mol and −634.92 kcal/mol, respectively. In addition, there was a 0.00% correlation between the MFE structures of V1 and V2 in every ensemble (Figure 12).

Predicted mRNA structure of the V1 (A and B) and V2 (C and D) by RNAfold web server. The centroid structures of the vaccines with the base pair probabilities (A and C) and the positional entropy (B and D).
Discussion
Among all cancers, stomach cancer ranks fifth in terms of mortality worldwide. 2 Most of these malignancies start as chronic gastritis by an infection with H pylori. A vaccine against this infection would be a significant weapon to prevent stomach cancer effectively. 105 Currently, no vaccine is commercially available for stomach cancer treatment. 106 Nevertheless, extensive research is being conducted on different types of cancer vaccines to explore their potential in combating stomach cancer. Various types of vaccines can be used to target cancer cells. These vaccines include peptide, DC, whole tumor cell, and checkpoint inhibitor vaccines.106,107
Epitope or peptide-based vaccines can also be developed to target overexpressed proteins or only found in cancer cells. 108 Although showing considerable perspective, these vaccines also pose particular challenges and constraints. 109 Identifying epitopes that induce a robust immune response against a wide range of cancer types can be challenging, potentially limiting the effectiveness of peptide-based vaccines. 109 In addition, peptide-based vaccines may not elicit a sufficiently strong and durable immune response, particularly in individuals with altered immune systems or in the context of tumor immune evasion mechanisms. Furthermore, the prerequisite for personalized vaccine formulations adds complexity to developing and implementing peptide-based vaccines. 110 Higashihara et al 111 conducted a phase I clinical trial to assess the safety of peptide vaccines with VEGFR1-A12-9 1084 and URLC10-A24-177 epitope peptides in patients with advanced stomach cancer who were chemo-resistant. This research indicates that administering URLC10 and VEGFR1 peptides by vaccination offers an effective therapeutic approach for advanced stomach cancer. Ishikawa et al 112 also designed a peptide vaccine against stomach cancer targeting the LY6K-177 peptide, which showed anticancer efficacy in stomach cancer patients after a phase I clinical trial. In a separate investigation, following the administration of the HER2/DC vaccine to those who have advanced stomach cancer, the patient achieved remission with reduced levels of tumor markers. However, Moss et al 113 developed an epitope-based vaccine against H pylori–associated stomach cancer (Helicovaxor), which has shown promising therapeutic protection in mice in preclinical trial. In addition, Sichuan University (urease subunits) and Southern Medical University (Lp220) have recently demonstrated 2 epitope-based vaccines targeting the urease and Lp220 protein H pylori, which offer marginal protection to BALB/c mice in a preclinical trial. 114 Wuhu Kangwei Biological Technology Co., Ltd. presented phase III clinical data demonstrating the efficacy of an H pylori recombinant vaccine against natural infection and stomach cancer.115,116
Recent advances in the research of vaccinology have revealed that therapeutic agents based on mRNA have undergone significant advancements and may be able to overcome the challenges encountered while developing vaccinations for cancer and infectious diseases. 117 There are multiple mRNA-based vaccines for several kinds of cancer, including non–small cell lung cancer (NSCLC), colorectal cancer, gastroesophageal cancer, urothelial cancer, bladder, head and neck cancer, pancreatic cancer, 118 hepatic cancer, prostate cancer, ovarian, and skin cancer are under evaluation in clinical and preclinical trials. 119 When finding vaccine candidates that can fight H pylori, reverse vaccinology is a game-changer. Vaccine development has traditionally relied on growing and then inactivating or weakening a whole pathogen. 120 On the contrary, reverse vaccinology uses bioinformatics and computer study of pathogen genomes to find possible targets for vaccines, which is a different approach. 121 Besides, vaccines developed using this method are exact and safe, and they also have a lower production cost and stronger humoral and cellular immune responses. 122 However, several cancer vaccines have been designed using this reverse vaccinology approach, including breast, 123 ovarian, 124 cervical,125,126 colorectal, 127 and NSCLC. 128
Thus, this research focuses on the reverse vaccinology approach for creating an mRNA vaccine to address the pressing general need for a stomach cancer vaccine. On the contrary, a multiepitope vaccine against H pylori was designed by Khan et al 129 using reverse vaccinology. This vaccine targeted the CagA, OipA, GroEL, and VacA proteins of the bacterium. Another multiepitope vaccine against H pylori was designed by Ghosh et al 108 where they targeted HpaA, FlaA, FlaB, and Omp18 antigenic proteins. Keshri et al 130 developed an in silico–based multiepitope vaccine against H pylori where they targeted about 25 proteins of the pathogen and identified B-cell, MHC-II, and IFN-γ-inducing epitopes within these proteins. Currently, no mRNA vaccine candidates are available for stomach cancer. Therefore, our designed vaccines are the first in silico–based mRNA vaccines proposed for stomach cancer treatment.
In the study, dual mRNA vaccines (V1 and V2) were designed from the proteins (CagA, GGT, NapA, PatA, urease, and VacA) of H Pylori. The best epitopes were chosen based on computational methods and used for the construction of vaccines with the incorporation of adjuvants and linkers. Based on biophysical qualities, both vaccines were anticipated to be soluble, potentially exhibiting functional stability under physiological settings. 41 The isoelectric point for V1 was 8.77 and for V2 it was 9.36, indicating that both vaccines were expected to be alkaline. Meanwhile, Sanami et al 125 designed a multiepitope vaccine against human papillomavirus (HPV), where the vaccine was basic in nature with a theoretical pI of 8.33. The vaccines developed by Lu et al 124 had a theoretical pI of 8.64 (breast cancer) and 8.82 (ovarian cancer). Motamedi et al 127 developed the colorectal cancer vaccine with a theoretical pI of 8.13. In our study, the vaccines were predicted to be a stable protein with an instability index of 19.95 for V1 and 36.31 for V2, whereas the HPV, ovarian, and breast vaccines had an instability index of 76.43, 125 51.52, 124 and 48.43, 124 respectively. Moreover, the aliphatic index of the V1 and V2 were predicted to be 84.33 and 76.23, respectively, indicating the vaccine is a hydrophobic protein containing aliphatic side chains. 41 While the aforementioned HPV, ovarian, breast, and colorectal cancer vaccines had an aliphatic index of 76.43, 125 76.16, 124 77.35, 124 and 69.36, 127 respectively. Therefore, the physicochemical properties of our designed mRNA vaccines tend to be similar to the HPV vaccine, which validated the appropriateness of this study.
It is crucial to consider the process of protein folding into its secondary and tertiary structures while developing an effective vaccine. 131 The unfolded and folded proteins’ antigens are essential in protein-specific immune responses. 132 Hence, these are the prime targets for antibodies, which in turn mount in response to infections.133,134 The predicted secondary and tertiary structures of the V1 and V2 were found satisfactory and reliable. The Ramachandran plot analysis also showed that most of the vaccines’ residues were within the preferred regions (85.5% for V1% and 88.1% for V2), conferring the structure integrity of the tertiary structures. Contrarily, the HPV and colorectal vaccine had 60.8% 125 and 70.7% 127 amino acid residues in the preferred regions, which indicated that our vaccines exhibited superior 3-dimensional structure compared with theirs. 125 To evaluate the possible association between the vaccines and TLRs on immune cells, a docking analysis was performed using human TLR-2 and TLR-4. According to this analysis, V1 exhibited a substantial affinity toward TLR-2 (lowest energy score of −1132.3 kJ/mol) and TLR-4 (lowest energy score of −1042.7 kJ/mol) receptors, where V2 exhibited a substantial affinity toward TLR-2 (lowest energy score of −1093.6 kJ/ml) and TLR-4 (lowest energy score of −1201.2 kJ/mol) receptors. Meanwhile, the HPV and colorectal vaccines had the lowest energy values of −1103.8 kJ/mol 125 and −1232.7 kJ/mol, 127 respectively. This highlights the accuracy of our prediction strategies and validates the vaccines. The PBDsum indicates that the V1-TLR-2, V2-TLR-4, V2-TLR-2, and V2-TLR-4 complexes contain 7, 22, 10, and 31 hydrogen bonds, respectively.
Subsequently, a simulation of the molecular dynamics was used to verify the structural integrity of the vaccine and its complexes with TLRs. The RMSD of both vaccines and associated TLR complexes indicates dynamic stability during the 50-ns simulation. V1-TLR-4 experiences a consistent increase in RMSD, contrasting V1-TLR-2, which exhibits a gradual decrease in RMSD followed by an initial spike after 10 ns simulation time. V2-TLR-2 and V2-TLR-4 display steady RMSD increments, whereas V1 remains stable. RMSF analysis indicates distinct motility patterns, particularly in C and N terminals. In contrast, Sanami et al 125 achieved only marginal stability in terms of RMSD and RMSF in the HPV vaccine, but only after 30 ns of the operation. Vaccines that we have developed are thus quite effective.
Furthermore, higher Rg values in V1-TLR-4 and V2-TLR-4 suggest increased structural expansion. Notably, SASA values remain insignificantly different throughout the simulations for V1 and V2 complexes, underscoring sustained molecular integrity. This comprehensive evaluation highlights the complex and unique characteristics of both vaccines and docked complexes, offering vital insights into their structural stability, flexibility, compactness, and exposure to solvents throughout the simulation.
The optimization of codon was utilized to evaluate the expression of the vaccines in the E coli where the predicted results demonstrated that both vaccines exhibited a significant level of expression in the vector, with the average GC content of the adapted V1 and V2 sequences computed at 51.23% and 51.50%, respectively, while both vaccines attained a CAI value of 1.0 each. Meanwhile, the HPV and colorectal vaccines had an average GC content of 52.04% 125 and 50.180%, 127 while the CAI scores of 0.95 125 and 0.9913, 127 respectively.
We also found a robust and sustained immune response following a 3-dose vaccine regimen for both V1 and V2 vaccines. The increased B-cell populations and prolonged activation of both TH and TC cells indicate the potential for long-lasting immunity, offering enhanced protection against H pylori. The V1 mRNA vaccine demonstrates structural stability, as indicated by MFE scores of −487.80 kcal/mol (optimum) and −354.19 kcal/mol (centroid). Similarly, V2 exhibits structural stability with MFE scores of −617.70 kcal/mol (optimum) and −515.48 kcal/mol (centroid). The predicted thermodynamic free energies of −505.96 kcal/mol and −634.92 kcal/mol for the V1 and V2 further support their structural integrity. Remarkably, the ensemble scores of both vaccines predict consistent stability through ingress, transcriptional and expressional activity in the host.
This study highlights the capacity of the dual mRNA vaccines targeting H pylori to elicit strong humoral and cellular immune responses, hence addressing stomach cancer. This vaccine will undergo clinical application in laboratories with animals and humans to validate its efficacy. Nevertheless, the present study is computational and hence lacks validation via laboratory studies. Nonetheless, the issue may lie in reconciling computational predictions with experimental findings.
Conclusions
Recognizing the potential of mRNA vaccines, our study focused on designing 2 novel mRNA vaccines, V1 and V2, targeting specific H pylori proteins implicated in stomach cancer. We undertook an extensive and meticulous bioinformatics approach to predict epitopes, assess physicochemical properties, and ensure structural stability. The docking analysis indicated substantial affinities between the vaccines and TLRs, while our rigorous molecular dynamics simulations highlighted dynamic stability and structural integrity over a 50-ns timeframe. Codon optimization further confirmed the feasibility of expression in E coli, and 3-dose regimens induced robust and sustained immune responses, particularly in the number of B, TH, and TC cells. Structural analyses, including MFE scores and thermodynamic free energies, emphasized the stability of V1 and V2 mRNA structures. Nevertheless, to confirm the safety and effectiveness of vaccines, studies on laboratory, animals, and humans need to be conducted. However, to determine the potentiality of the designed mRNA vaccines, this study establishes a firm groundwork for future research by highlighting the necessity of following experimental validations and clinical investigations.
Supplemental Material
sj-docx-4-bbi-10.1177_11779322251331104 – Supplemental material for A Reverse Vaccinology and Immunoinformatic Approach for the Designing of a Novel mRNA Vaccine Against Stomach Cancer Targeting the Potent Pathogenic Proteins of Helicobacter pylori
Supplemental material, sj-docx-4-bbi-10.1177_11779322251331104 for A Reverse Vaccinology and Immunoinformatic Approach for the Designing of a Novel mRNA Vaccine Against Stomach Cancer Targeting the Potent Pathogenic Proteins of Helicobacter pylori by Abanti Barua, Md. Habib Ullah Masum and Ahmad Abdullah Mahdeen in Bioinformatics and Biology Insights
Supplemental Material
sj-tif-1-bbi-10.1177_11779322251331104 – Supplemental material for A Reverse Vaccinology and Immunoinformatic Approach for the Designing of a Novel mRNA Vaccine Against Stomach Cancer Targeting the Potent Pathogenic Proteins of Helicobacter pylori
Supplemental material, sj-tif-1-bbi-10.1177_11779322251331104 for A Reverse Vaccinology and Immunoinformatic Approach for the Designing of a Novel mRNA Vaccine Against Stomach Cancer Targeting the Potent Pathogenic Proteins of Helicobacter pylori by Abanti Barua, Md. Habib Ullah Masum and Ahmad Abdullah Mahdeen in Bioinformatics and Biology Insights
Supplemental Material
sj-tif-2-bbi-10.1177_11779322251331104 – Supplemental material for A Reverse Vaccinology and Immunoinformatic Approach for the Designing of a Novel mRNA Vaccine Against Stomach Cancer Targeting the Potent Pathogenic Proteins of Helicobacter pylori
Supplemental material, sj-tif-2-bbi-10.1177_11779322251331104 for A Reverse Vaccinology and Immunoinformatic Approach for the Designing of a Novel mRNA Vaccine Against Stomach Cancer Targeting the Potent Pathogenic Proteins of Helicobacter pylori by Abanti Barua, Md. Habib Ullah Masum and Ahmad Abdullah Mahdeen in Bioinformatics and Biology Insights
Supplemental Material
sj-tif-3-bbi-10.1177_11779322251331104 – Supplemental material for A Reverse Vaccinology and Immunoinformatic Approach for the Designing of a Novel mRNA Vaccine Against Stomach Cancer Targeting the Potent Pathogenic Proteins of Helicobacter pylori
Supplemental material, sj-tif-3-bbi-10.1177_11779322251331104 for A Reverse Vaccinology and Immunoinformatic Approach for the Designing of a Novel mRNA Vaccine Against Stomach Cancer Targeting the Potent Pathogenic Proteins of Helicobacter pylori by Abanti Barua, Md. Habib Ullah Masum and Ahmad Abdullah Mahdeen in Bioinformatics and Biology Insights
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.