
Abstract
The interaction between programmed cell death protein 1 (PD-1) and its ligand PD-L1 plays a crucial role in tumor immune evasion, presenting a critical target for cancer immunotherapy. Despite being effective, current monoclonal antibodies present some drawbacks such as high costs, toxicity, and resistance development. Therefore, the development of small-molecule inhibitors is necessary, especially those derived from natural sources. In this study, benzosampangine is predicted as a promising PD-L1 inhibitor, with potential applications in cancer immunotherapy. Utilizing the high-resolution crystal structure of human PD-L1 (PDB ID: 5O45), we screened 511 natural compounds, identifying benzosampangine as a top candidate with exceptional inhibitory properties. Molecular docking predicted that benzosampangine exhibits a strong binding affinity for PD-L1 (−9.4 kcal/mol) compared with established controls such as CA-170 (−6.5 kcal/mol), BMS-202 (−8.6 kcal/mol), and pyrvinium (−8.9 kcal/mol). The compound’s predicted binding efficacy is highlighted by robust interactions with key amino acids (ILE54, TYR56, GLN66, MET115, ILE116, SER117, ALA121, ASP122) within the active site, notably forming 3 Pi-sulfur interactions with MET115—an interaction absents in control inhibitors. In addition, ADMET profiling suggests that over the control molecules, benzosampangine has several key advantages, including favorable solubility, permeability, metabolic stability, and low toxicity, while adhering to Lipinski’s rule of five. Molecular dynamic simulations predict the stability of the benzosampangine-PD-L1 complex, reinforcing its potential to sustain inhibition of the PD-1/PD-L1 pathway. MMGBSA analysis calculated a binding free energy (ΔGbind) of −39.39 kcal/mol for the benzosampangine-PD-L1 complex, with significant contributions from Coulombic, lipophilic, and Van der Waals interactions, validating the predicted docking results. This study investigates in silico benzosampangine, predicting its better molecular interactions and pharmacokinetic profile compared with several already known PD-L1 inhibitors.
Keywords
Introduction
T lymphocyte immunity plays a vital role in maintaining the body’s homeostasis through the selective recognition and elimination of abnormal cells, including cancer cells.1,2
Tumor cells initiate the cancer-immunity cycle by releasing tumor antigens. 3 Antigen-specific T cells first recognize tumor antigens presented by major histocompatibility complexes (MHCs) on antigen-presenting cells (APCs), leading to their priming and activation. Once activated and proliferated, T cells migrate to specific sites guided by chemokine concentration gradients. 4 Following costimulatory signals between T cells and APCs to achieve optimal activation, immune checkpoints like PD1 and its ligand PDL1 are involved to play a crucial role in preventing excessive T-cell activation.3,5 On recognizing the same antigen on MHCs, T cells release IFN-γ to enhance the efficiency of tumor cell destruction. IFN-γ released from CD8+ T cells upregulates the expression of PDL1 on tumor cells6,7 (Figure 1). Meanwhile, T-cell receptor signaling upregulates the expression of PD1 on the T cell surface which binds to PDL1 to exert negative regulatory effects and blunt the antitumor function of T cells.8-10 Multiple studies have shown that PD-L1 is overexpressed in various types of cancers, including breast, lung, gastric, papillary thyroid, bladder, testicular, colorectal, melanoma, non–small cell lung (NSCLC), head and neck, and kidney cancers.11-13 The interaction between PD-L1 and PD-1 contributes to the maintenance of an immunosuppressive tumor environment by suppressing T lymphocyte function leading to proliferation and cytokine secretion reduction which impairs their ability to destroy tumor cells.14-16 In addition, this interaction induces apoptosis of T lymphocytes promoting tumor cell survival.17,18 Tumor environment cells also express PD-L1, using this pathway to evade the immune system.16,19,20 As a result, blocking immune checkpoint pathways has become an important strategy for reversing the immunosuppressive mechanisms employed by tumor cells to reactivate the immune system, and targeting the PD-1/PD-L1 pathway is considered a frequently used approach.21,22 Clinical trials involving anti-PD-L1 agents have yielded encouraging results among cancer patients, including renal cell carcinoma, melanoma, NSCLC and metastatic urothelial bladder cancer.11,23,24 Currently, multiple monoclonal antibodies have been approved by the Food and Drug Administration (FDA) as inhibitory drugs impeding the PD-1-PD-L1 interaction in cancer therapy. 23 ,25-28 Two anti-PD-1 antibodies, pembrolizumab (humanized IgG4 antibody) and nivolumab (human IgG4 antibody), are approved as the second line for the treatment of Hodgkin lymphoma, NSCLC, metastatic melanoma, head and neck squamous cell carcinoma, and kidney cancer. 26 ,29-32 As for anti-PD-L1 antibodies, atezolizumab (humanized IgG1 antibody) is approved for the treatment of advanced NSCLC and urothelial carcinoma,25,33 whereas durvalumab (human IgG1 antibody) and avelumab (human IgG1 antibody) have been approved by the FDA and have shown positive responses in the treatment of various malignant tumors, including metastatic NSCLC, melanoma, urothelial carcinoma, and Merkel cell carcinoma.34-36 However, monoclonal antibodies have drawbacks, including low stability, high manufacturing costs, low tumor penetration rates, difficulty in overcoming biological barriers, lack of oral availability, and potential immunogenic side effects.37-39 Therefore, it is necessary to overcome these disadvantages through the development of new small, stable, and more effective inhibitory molecules with the ability to bind to PD-L1 and block the PD-1/PD-L1 interaction without inducing undesirable effects. In this study, we suggest that targeting PD-L1 with small natural molecules could effectively block the PD1-PD-L1 interaction and consequently reactivate the immune system. The use of natural compounds derived from medicinal plants holds great promise for the development of therapeutic medicines that are both less harmful and more effective. Numerous studies have previously shown the therapeutic power of natural molecules.40-43 In this regard, we first performed a virtual screening of a database of natural compounds and their derivatives against the crystalline structure of PD-L1 available in the Protein Data Bank (PDB; PDB code: 5O45) to discover potential inhibitors of PD-L1, followed by molecular dynamics (MD) simulations to assess the stability of the complexes. This approach has been adopted in several previous studies.44-49

Mechanism of PD1/PD-L1 blockade. The CD8+ T cell activates on recognizing the tumor antigen presented on MHC class I and releases IFN-γ to bind to IFN-γ receptor, and consequently induces the expression of PDL1 on tumor cells. PDL1 conjugates the elevated PD1 on T cell surface, triggering inhibitory effect of PD1/PD-L1 axis. Anti-PD1 or anti-PDL1 antibody blocks the interaction of PD1 and PD-L1, and abolishes the inhibition of CD8+ T cell thus enhancing the antitumor activity. 4
Materials and Methods
Data collection and ligand preparation
A local database of 511 natural compounds and their derivatives (inhibitors) was assembled from various essential oils derived from medicinal plants and subjected to screening to identify potential compounds that inhibit the interaction between PD-1 and PD-L1 in cancer. The ligands obtained from the PubChem database 50 in 3D SDF format were prepared using AutoDockTools; 51 Gasteiger charges were assigned and stored in PDBQT format.
Receptor preparation and receptor grid generation
The 3-dimensional crystal structure of human PD-L1 in complex with an inhibitor (PDB ID: 5O45) was utilized for docking studies. This particular structure was chosen for its high resolution (0.99Å) providing a precise and a detailed representation of the protein’s atomic structure. The inhibitor-bound form of PD-L1 ensures that the binding conformation is well-defined, enabling a more reliable identification of potential inhibitors among the screened compounds. Subsequently, heteroatoms were deleted, water molecules were eliminated, and Kollman atomic charges, along with polar hydrogens, were incorporated into the receptors using AutoDockTools. 51 The prepared structure, ready for molecular docking studies, was saved in AutoDock PDBQT format.
Molecular docking protocols
To identify inhibitors of the interaction between PD-1 and PD-L1, molecular docking studies were performed to screen 511 natural molecules and their derivatives against the active site of the PD-L1 protein using the AutoDock Vina docking program. For the PD-L1 target, a grid box (X: 40Å, Y: 40Å, and Z: 40Å) was created to encompass the binding pocket highlighted by CB-Dock2 (https://cadd.labshare.cn/cb-dock2/php/blinddock.php). 52 The center of the grid box was set at (X: 10.441, Y: 6.855, Z: −18.463), and the other AutoDock Vina parameters were maintained at their default values (energy range = 4 and exhaustiveness = 8). The ligands’ binding affinities to the target’s active site were expressed in kcal/mol units.
Drug-likeness prediction
The ligands with the highest affinities were selected for drug-likeness assessment. This evaluation was carried out using the free online tool Swiss ADME (http://www.swissadme.ch/), developed by the Swiss Institute of Bioinformatics. 53 We based our assessment on Lipinski’s rule of five (Ro5) to predict the bioavailability of molecules.54,55 The Ro5 introduced the criteria of the number of rotatable bonds, the molecular weight, the number of hydrogen bond acceptors, the number of hydrogen bond donors, and the octanol-water partition coefficient. A molecule is considered drug-like if it meets at least 4 of the 5 cited criteria. 54
Toxicity analysis
Computational methods facilitated the evaluation of a safety profile for the compounds studied. Ligands exhibiting drug-like properties were selected for toxicity prediction. The ProTox-II (https://tox-new.charite.de/protox_II/) server was used to assess the toxic effects of the chosen compounds. 56 This server predicts organ toxicity (hepatotoxicity), as well as various toxicological endpoints (carcinogenicity, immunotoxicity, cytotoxicity, and mutagenicity) and the median lethal dose (LD50) for the selected molecules.
Post-docking analysis and visualization
Molecules not showing toxicity in ProTox-II were selected for receptor-ligand interaction analysis to validate their interaction with active site residues of the target. Discovery Studio was used to perform post-docking visualization 2D. 57
Molecular dynamics simulations
By incorporating the classical Newton equation of motion, MD simulations generally simulate the movements of atoms over time and predict the binding state of ligands in the physiological environment. 58 A study of MD simulations was conducted with the docking complex of PD-L1_benzosampangine over 100 ns using Schrödinger LLC Desmond software. 59 The TIP3P solvent model (Intermolecular Interaction Potential 3 Points Transferable), based on an orthorhombic box, was used at a temperature of 300 K, a pressure of 1 atm, and an OPLS_2005 force field. 60 Using counter-ions and 0.15 M sodium chloride, the models were, respectively, neutralized and simulated under physiological conditions. The models were equilibrated before the simulation, and the trajectories were stored for inspection every 100 ps.
MMGBSA calculations
During MD simulations, the binding free energy (ΔGbind) of the docked complex was determined using the MMGBSA module (Suite Schrodinger, LLC, New York, NY, 2017-4). The calculations were performed using simultaneously, the OPLS 2005 force field, the VSGB solvent model and rotamer search techniques. 61 MD trajectory frames were chosen at each 10 ns interval after MD execution. The following equation was used to calculate the total binding free energy:
∆Gbind = Gcomplex – (Gprotein + Gligand)
where ΔGbind = binding free energy, Gcomplex = free energy of the complex, Gprotein = free energy of the target protein, and Gligand = free energy of the ligand.
Pharmacokinetics (ADME) and toxicity prediction
The ADMET results used in this study were obtained from the ADMETlab 2.0 server, 62 a widely recognized platform for predicting pharmacokinetic and toxicity properties of chemical compounds. We focused on evaluating the key ADMET properties crucial for assessing the potential of drug candidates, including physicochemical properties, medicinal chemistry parameters, absorption, distribution, metabolism, excretion, and toxicity. ADMETlab 2.0 employs a multitask graph attention framework for robust and accurate prediction models. The server’s batch computation module and optimized result representation enhance usability. Using ADMETlab 2.0, we sought to gain insights into the pharmacokinetic and toxicity profiles of compounds to optimize drug candidate screening. The freely accessible ADMETlab 2.0 server is a valuable online platform for early-stage drug discovery. 62
Results
In this study, we screened 511 natural compounds for their potential inhibitory effects on the PD-L1 receptor using a structure-based drug design approach. The PD-L1 receptor was chosen for its pivotal role in immune checkpoint pathways, which are essential for the regulation of immune responses. Inhibiting PD-L1 can enhance antitumor immunity by preventing its interaction with PD-1, thus making it a valuable target for cancer immunotherapy. In our docking study, we used the 3-dimensional crystal structure of human PD-L1 in a complex with an inhibitor (PDB ID: 5O45). This particular structure was selected because of its high resolution (0.99Å), which provides an accurate and detailed representation of the protein’s atomic structure. The inhibitor-bound form of PD-L1 ensures well-defined binding conformation, enabling more reliable identification of potential inhibitors from the screened compounds. During the screening, compounds were evaluated based on their ability to interact with the active site residues of PD-L1. This criterion is a key factor in achieving effective disruption of the PD-1/PD-L1 interaction. In addition, compounds were assessed for their drug-like properties, nontoxicity, and stability of the complexes formed with PD-L1 in MD simulations. These criteria are essential to ensure that the selected compounds not only have the potential to effectively inhibit PD-L1, but also have the favorable pharmacokinetic and pharmacodynamic profiles required for further development as therapeutic agents. Compounds that interact favorably with PD-L1active site residues, have drug-like properties, are non-toxic, and form stable complexes in MD simulations were considered as potential active inhibitors of PD-L1. These candidates were selected for further experimental validation, highlighting their potential as promising therapeutic agents in cancer immunotherapy.
The following residues of PD-L1, ILE54, TYR56, MET115, ILE116, SER117, ALA121, ASP122, and TYR123, have been reported in various studies to contribute to the interaction between PD-L1 and PD-1.63-67 The Cb-Dock2 tool was used to identify the binding pocket of PD-L1. The binding site is illustrated in Figure 2.

The predicted binding pocket of PD-L1. Key residues represented in blue in the binding pocket and the surfaces of the binding pocket in PD-L1 are presented in gray.
Molecular docking is a critical step in structure-based drug design. In this study, molecular docking simulations were carried out employing the AutoDock Vina program to evaluate the potential of small natural molecules to inhibit the interaction between PD-1 and PD-L1 for immune system reactivation. We set −9 kcal/mol as the threshold binding energy value to select only molecules that strongly engage the PD-L1 active site. 68 Table 1 presents the binding energy values for the top 23 ligands among the 511 screened compounds, where their binding energies (below −9 kcal/mol) indicate a strong affinity for the PD-L1 target. Kaempferol 3-(6′-galloylgalactoside) and Z-guggulsterone exhibited the best binding energy toward the target (−9.8 kcal/mol), implying their strong interaction with the binding pocket residues. The binding energy values and binding poses for the top-docked compounds are summarized in Table 1.
Molecular docking results and binding poses of top docked compounds with PD-L1 structure.

Lipinski’s rule of five is applied to determine whether a molecule has the appropriate properties to qualify as a potential active pharmaceutical ingredient for oral administration. We assessed the drug-likeness properties of the molecules with the best docking scores (Table 1) to exclude compounds that are unsuitable for further developmental research. As shown in the table below (Table 2), 17 out of the 23 studied compounds adhered to Ro5, suggesting that these molecules are promising drug candidates. Only Kaempferol 3-(6′-galloylgalactoside), amentoflavone, hypericin, pseudohypericin, beta-carotene, and hesperidin showed 2 or more violations. The detailed properties of each molecule are reported in Table 2.
Predicted drug likeness of PD-L1 potential inhibitors.

Toxicity was assessed by considering various targets associated with adverse effects of drugs to exclude any toxic compounds from the study. The compounds studied had to be inactive on all targets with a high LD50. Benzosampangine showed no signs of toxicity, with LD50 value equal to 2000 mg/kg. However, Z-guggulsterone, enoxolone, chelerythrine, and finasteride are both carcinogenic and immunogenic. While, veratramine, tomatidine, benzo[c]phenanthridine, limonin, peiminine, and finasteride exhibited low LD50 values of 315, 500, 331, 244, 280, and 418 mg/kg, respectively, indicating their potential toxicity. The detailed toxicological properties of each molecule are reported in Table 3.
Organ toxicity and toxicological endpoints predicted activities.

Various interactions were observed between benzosampangine and the PD-L1 protein structure. As shown in Figure 3, benzosampangine engaged with several residues in PD-L1active site, indicating a strong binding affinity. Specifically, it demonstrated Van der Waals interactions with ILE54, GLN66, ILE116, ASP122, and TYR123. Although individually weak, these Van der Waals forces collectively provide substantial stabilization to the ligand-receptor complex by creating extended contact surfaces. A carbon-hydrogen bond with SER117 enhances the specificity and stability of the binding interaction. Hydrogen bonds are crucial for maintaining the proper orientation and binding affinity of the ligand in the active site, often contributing significantly to the overall binding energy. Notably, benzosampangine forms 3 Pi-sulfur interactions with MET115. These interactions are significant as they involve the aromatic ring system of benzosampangine interacting with the sulfur atom of methionine. Although less common, Pi-sulfur interactions provide strong noncovalent interactions that can significantly stabilize the ligand in the binding pocket, contributing to the high binding affinity observed. Moreover, benzosampangine and TYR56 form numerous Pi-Pi stacked interactions. The relevance of these interactions in providing significant binding energy is attributed to the overlap of π-orbitals between aromatic rings. This type of interaction is essential for preserving the structural integrity of the ligand-protein complex and enhancing binding specificity. The complex was further stabilized by hydrophobic interactions ensured by Pi-alkyl interactions observed on ALA121. Through the establishment of hydrophobic interaction, non-polar regions of the ligand play a crucial role in enhancing overall binding affinity and stability of the complex. In summary, these interactions suggest that benzosampangine effectively binds to critical residues of the PD-L1 active site and potentially may inhibit the interaction between PD-L1 and PD-1. The interaction profile emphasizes benzosampangine potential as a candidate for further experimental validation and promising therapeutic development in cancer diseases.

2D interaction of benzosampangine with the active site of PD-L1.
We also explored the dynamic behavior of the complex interactions through MD simulations between the target receptor PD-L1 and benzosampangine over 100 ns, a sufficient time for the dynamic behavior of Cα atoms within the complex. This duration allows the assessment of the conformational stability of the compound-PD-L1 complex and its changes under physiological conditions. Various parameters were analyzed to validate the stability of the complex studied, including root mean square deviation (RMSD), root mean square fluctuation (RMSF), protein secondary structure, and protein-ligand contact analysis. The RMSD graph allows the assessment of the compound-PD-L1 complex stability throughout the 100 ns simulation. A complex with a low RMSD value is considered stable, whereas a higher RMSD suggests potential instability. Typically, RMSD values within the range of 1Å to 3Å are acceptable, but significantly larger changes suggest that the protein undergoes substantial conformational changes during the simulation. The RMSD values should stabilize around a fixed value. If the protein’s RMSD continues to either increase or decrease on average toward the end of the simulation, it suggests that the system has not reached equilibrium, and the simulation duration may be insufficient for a thorough analysis. In our study, the RMSD graph for the benzosampangine-PD-L1 complex is presented in Figure 4. The average RMSD of the protein alone was 4.08Å, which decreased to 1.52Å after binding with benzosampangine. This decrease indicates that the binding of benzosampangine increased the stability of the PD-L1 protein. Increased protein stability upon ligand binding is often correlated with effective inhibition because a stable complex formation implies that the ligand is well accommodated within the binding site, resulting in minimal conformational changes. This stability can support the ligand’s ability to effectively block or inhibit the function of the protein, in our case, PD-L1. A stable PD-L1-benzosampangine complex supports that benzosampangine may maintain its inhibitory interactions over time, reducing the likelihood of PD-L1 interacting with its natural binding partners, such as PD-1. The observed stability, despite initial fluctuations due to system equilibration during the first nanoseconds, remained consistent throughout the simulation. A fluctuation at 84 ns did not exceed 3Å, indicating that the system was well-balanced throughout the simulation duration. This consistent stability reinforces the potential of benzosampangine as a promising inhibitor of PD-L1, suggesting its potential for further experimental validation and therapeutic development.

Root mean square deviation (RMSD) of the protein PD-L1 alone (blue) and in complex with and benzosampangine (red) as a function of simulation time.
The RMSF measurement characterizes the local changes in protein chains during the simulation revealing the rigidity and flexibility of the protein’s amino acids. Peaks indicate the residues of the protein that fluctuate during the simulation. Typically, secondary structure elements (alpha helices and beta strands) are rigid and fluctuate less than (N- and C-terminal) regions, which are flexible. The RMSF graph of the system (Figure 5) demonstrates that the residues interacting with benzosampangine, highlighted by green bars, exhibit fluctuations decrease compared with other regions. This variation suggests that the binding of benzosampangine stabilizes these residues, resulting in increased rigidity and decreased mobility. Specifically, residues such as MET115, TYR56, ALA121, SER117, TYR123, ASP122, ILE116, ILE54, and GLN66, which interact directly with benzosampangine, are involved in this stabilization process. In contrast, other regions did not interact with benzosampangine, particularly those around residues 25-35 and 45-55, and showed higher fluctuations. These areas are more flexible, likely due to the lack of stabilizing interactions with the ligand. This flexibility is characteristic of regions not implied in direct ligand binding, allowing them to move more freely during the simulation. These observations underline the critical role of the identified residues in ligand binding and protein stability. The interactions between benzosampangine and these key residues contribute to the overall stability of the PD-L1 protein-ligand complex, enhancing the inhibitory potential of benzosampangine.

Root mean square fluctuation (RMSF) analysis of PD-L1 in complex with benzosampangine. The vertical green lines represent the amino acid residue of RPFC making contact with ligand.
The variations in the secondary structure elements (% SSE) of PD-L1, including alpha helices and beta strands, were assessed over simulation time. The plot at the top summarizes the SSE composition for each trajectory frame over the course of the simulation and the plot at the bottom tracks each residue and its SSE assignment over time, with variations represented in orange and blue corresponding to alpha helices and beta sheets, respectively (Figure 6). The percentage of beta strands is notably higher compared with the percentage of alpha helices. However, the overall secondary structure elements of the PD-L1 protein remained stable at approximately 44% throughout the simulation. This consistency suggests that no significant structural changes occurred and the integrity of the PD-L1 structure was maintained during its interaction with benzosampangine.

The stability of PD-L1’s secondary structure over 100 ns of MD simulation in complex with benzosampangine.
The interaction types between benzosampangine and the protein were monitored throughout the simulation to assess their contribution to the stability of the complex. These interactions include hydrophobic, hydrogen bonds, water bridges, and ionic interactions and are illustrated in Figure 7. The results indicate that hydrophobic interactions, hydrogen bonds, and water bridges were the most frequently observed ligand-protein interactions. Benzosampangine engaged with all residues in the active site of PD-L1. Particularly, the hydrophobic interaction with TYR56, the strongest interaction observed, remained stable for approximately 50% of the simulation time. These findings highlight the stability of the benzosampangine-PD-L1 complex.

The protein-ligand interactions: (a) hydrogen bonds—green, hydrophobic—white purple, ionic—pink, water bridges—blue).
All results from the MD indicate the stability of benzosampangine when complexed with the PD-L1 protein. This observed stability suggests that benzosampangine may have the potential to inhibit the interaction between PD-L1 and PD-1 under physiological conditions.
Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) analysis allows us to identify the binding efficiency of the ligand with the receptor, by calculating the binding free energy of the protein-ligand complex at the molecular level. This approach is highly beneficial for the validation of docking and MD simulation results. The binding free energy (ΔGbind) of benzosampangine to PD-L1 is equal to −39.39 kcal/mol (Table 4). ΔGbind is influenced by nonbonded interactions such as ΔGbindCoulomb, ΔGbindCovalent, ΔGbindHbond, ΔGbindLipo, ΔGbindSolvGB, and ΔGbindvdW. The contribution of each interaction is detailed in Table 4. The ΔGbindCoulomb, ΔGbindLipo, and ΔGbindvdW energies contributed significantly to reach the average binding energy, compared with the ΔGbindCovalent and ΔGbindSolvGB energies which showed an unfavorable energetic contribution to the final binding energies average. Thus, the binding energy observed in docking studies is well-supported by the MM-GBSA calculations derived from the MD simulation trajectories.
Binding energy calculation of benzosampangine with PD-L1 and non-bonded interaction energies from MM-GBSA trajectories.

Benzosampangine presents a compelling profile in terms of its absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, positioning it as a promising candidate for in the development of effective cancer immunotherapy agents. Considering the advancement of PD-L1 inhibitors, 69 pyrvinium, an FDA-approved anthelmintic drug, 70 has demonstrated in vitro efficacy against the PD-L1 receptor, 71 indicating potential for repurposing in cancer therapy. BMS-202, currently progressing through pre-clinical trials, 72 shows early-stage promise, whereas CA-170’s involvement in clinical research targeting NSCLC highlights its clinical relevance in oncology.73,74 These compounds, namely pyrvinium, BMS-202, and CA-170, serve as experimentally validated controls in our study. Nevertheless, the comprehensive evaluation of benzosampangine’s ADMET characteristics suggests several potential advantages over the experimentally validated control molecules, including pyrvinium, BMS-202, and CA-170. These findings warrant further investigation into its suitability for development as a therapeutic targeting the PD-L1 pathway (Table 5). One promising feature of Benzosampangine is its predicted favorable absorption profile, characterized by high intestinal absorption (HIA), 75 which surpasses that of pyrvinium and CA-170. This attribute is indicative of its potential for efficient systemic delivery and distribution within the body and is critical for achieving therapeutic efficacy. In addition, Benzosampangine is predicted to demonstrate an extensive volume of distribution, exceeding that of BMS-202, signifying its propensity to reach target tissues and maintain therapeutic concentrations, essential for triggering a robust antitumor response. 76 Its balanced logS value of −4.21 and logP value of 3.05 also surpass those of pyrvinium and BMS-202, further enhancing its pharmacokinetic profile and potential for optimal absorption and distribution across biological membranes. 77 Moreover, benzosampangine is predicted to demonstrate moderate blood-brain barrier (BBB) penetration, similar to that of pyrvinium and CA-170, and higher than of BMS-202 value, which may help mitigate the risk of central nervous system exposure and associated neurotoxicity. 78 In terms of predictive metabolic interactions, benzosampangine exhibits moderate activity as both a substrate and inhibitor of cytochrome P450 (CYP) enzymes, particularly CYP2D6 and CYP3A4, comparable to or better than the other molecules, potentially conferring advantages in terms of reduced susceptibility to metabolic degradation and drug-drug interaction modulation. 79 Furthermore, benzosampangine shows a low risk of hERG blocker toxicity distinguishes it from pyrvinium and BMS-202, indicating a lower potential for cardiac arrhythmia, a critical safety consideration in drug development. 80 The absence of PAINS alerts (pan-assay interference compounds) in benzosampangine’s structural features is a significant computational advantage over pyrvinium, which exhibits 2 PAINS alerts. This absence may suggest a lower likelihood of off-target effects or undesirable interactions, enhancing the safety profile of Benzosampangine. 81 Moreover, benzosampangine’s docking score of −9.4 kcal/mol, indicate a strong molecular affinity for the PD-L1 receptor, surpassing those of the control compounds. The high recorded affinity supports the promising for potent and specific inhibitory action against the PD-L1 pathway, a pivotal mechanism implicated in tumor immune evasion.
ADMET properties of PD-L1 inhibitors.

Although BMS-202 and CA-170 are both currently in developmental phases, benzosampangine is standing out as a promising candidate for further investigations as a PD-L1 inhibitor given to its higher ADMET profile and exceptional docking score. Lower docking scores of −8.6 kcal/mol for BMS-202 and −6.5 kcal/mol for CA-170 suggest weaker molecular affinity for the PD-L1 receptor, supporting the hypothesis that benzosampangine could exhibit stronger molecular interactions and therapeutic potential (Table 6).
Docking score against PD-L1.

When comparing interactions of benzosampangine to control molecules (CA-170, BMS-202, and pyrvinium), it appears that benzosampangine may have the potential to mimic the roles of these control compounds and potentially surpass them in terms of binding stability and specificity. Figure 8 shows that all 4 molecules bind within the same pocket of the PD-L1 protein, suggesting that benzosampangine could exhibit similar inhibitory effects on the PD-L1/PD-1 interaction. CA-170 primarily relies on hydrogen bonds with SER117, ASN63, and GLN66, in addition to Van der Waals interactions and alkyl/Pi-alkyl interactions with residues such as VAL68 and ILE54 (Figure 8B), whereas BMS-202 demonstrates a more diverse interaction profile, including hydrogen bonds and Pi-anion interaction with GLU58, Pi-Pi stacked and alkyl interactions with TYR56, Pi-sigma interaction with ILE54, as well as extensive Van der Waals interactions (Figure 8C). Despite the absence of hydrogen bonds, pyrvinium engages Pi-alkyl and alkyl interactions with residues such as TYR56, MET115, and ILE54, along with Van der Waals interactions and a carbon-hydrogen bond with ASP122 (Figure 8D). The unique combination of interactions observed in benzosampangine (Figure 8A), particularly the presence of triple Pi-sulfur interactions with MET115 and triple Pi-Pi stacked interactions with TYR56, suggests a potentially higher interaction profile. It is important to interpret the results of our investigation within this predictive framework, given that it was conducted using computational predictions. The robust interaction profile of benzosampangine indicates that it may exhibit strong inhibitory effects on the PD-L1/PD-1 pathway, potentially comparable to or probably exceeding those of CA-170, BMS-202, and pyrvinium.
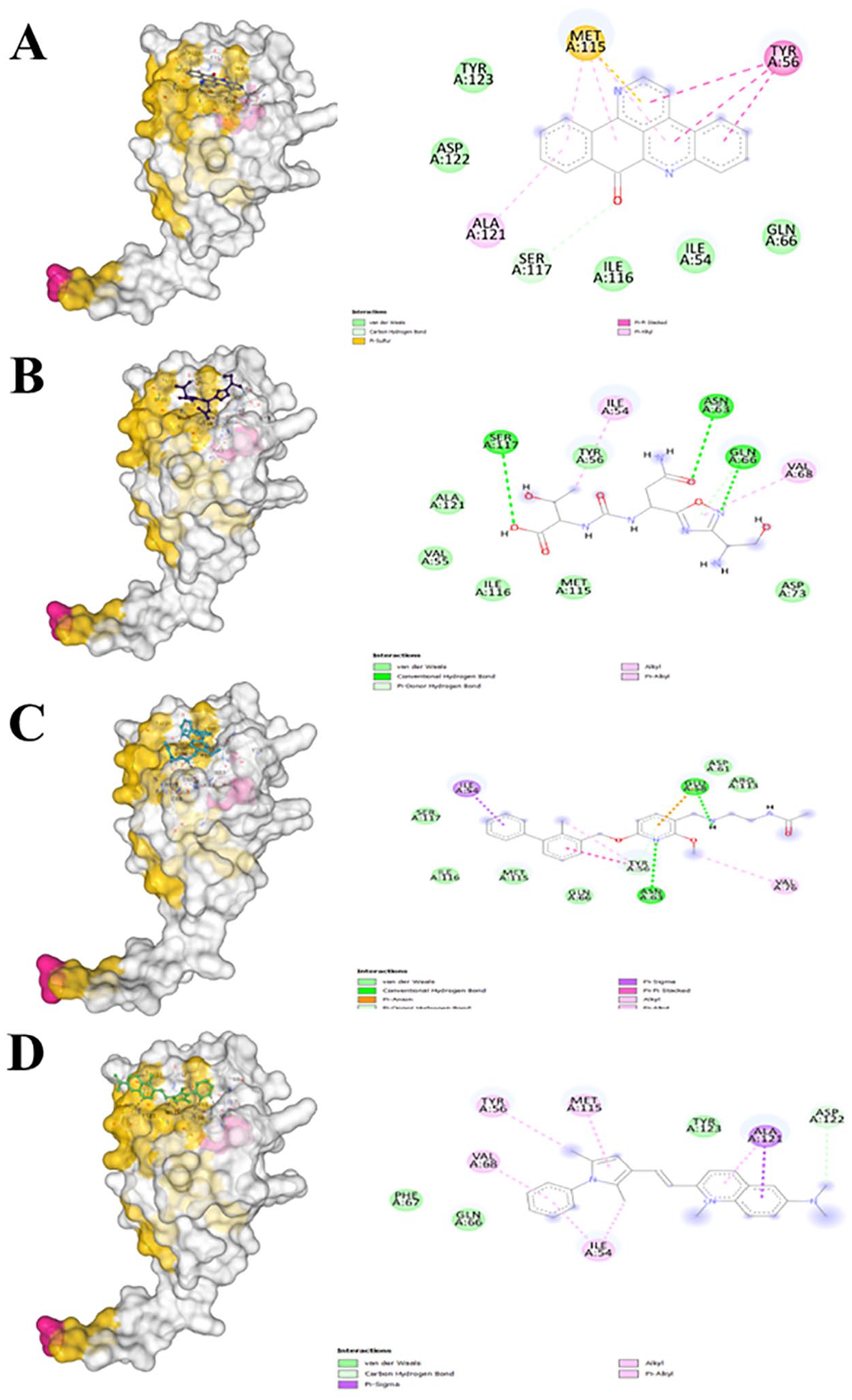
Binding pose and atomic-level interactions of control molecules CA-170 (B), BMS-202 (C), and pyrvinium (D) and benzosampangine (A).
When evaluating the system stability under physiological conditions, we compared the behavior of the benzosampangine-PD-L1 complex with that of the control molecule CA-170 in complex with PD-L1 over 100 ns of MD simulations. The CA-170-PD-L1 complex exhibits variations throughout the simulation time of 100 ns (Figure 9). Following an initial fluctuation due to equilibrium search, the RMSD of the complex remains relatively stable around 12Å until 28 ns indicating that CA-170 initially maintains stability within the binding pocket of the protein. Then, several significant peaks in the RMSD of the CA-170-PD-L1 complex were observed up to approximately 72 ns, indicating that the ligand traveled a considerable distance from its initial position, thus revealing a period of instability of the ligand, probably due to its partial or complete detachment from its binding site on the protein. However, from around 73 ns onward, the RMSD decreases and remains stable without fluctuations toward the end of the simulation, indicating that CA-170 regains its initial conformation and forms stable interactions with the protein. In contrast, benzosampangine does not exhibit such pronounced fluctuations and remains more stable in the protein binding pocket throughout the simulation. These results support the candidacy of benzosampangine as a promising ligand for therapeutic development targeting PD-L1. The RMSF plot of the CA-170-PD-L1 complex shows low RMSF values in the contact areas, indicating remarkable protein stability while bound to CA-170 (Figure 10). Similarly, the RMSF values for benzosampangine-PD-L1 complex are also low in these contact areas, showing that benzosampangine provides protein stability comparable to that of CA-170.

Root mean square deviation (RMSD) of the protein PD-L1 alone (blue) and in complex with and CA-170 (red) as a function of simulation time.

Root mean square fluctuation (RMSF) analysis of PD-L1 in complex with CA-170.
Discussion
We compared our observations with previous studies on PD-L1 inhibitors using computational approaches. Many previous studies have employed molecular docking to study the interaction of PD-L1 with different inhibitors, and their reported results are consistent with ours regarding key residues involved in interactions that may induce inhibition.63,82,83 Our findings showed that benzosampangine interacts with the same binding site residues of PD-L1, namely MET115, TYR56, ALA121, SER117, TYR123, ASP122, ILE116, ILE54, and GLN66, confirming the relevance of our predictions. In addition, our study revealed hydrophobic interactions and stable hydrogen bonds between benzosampangine and these critical residues, which align with observations from previous studies.63,82,84 This alignment with previous works strengthens the credibility of our docking methodology and the validity of the identified interactions between benzosampangine and PD-L1. Furthermore, in the study by Sobral et al 85 identifying new PD-L1 inhibitors, it has been shown that effective inhibitors had binding energies of −9.213 and −8.023 kcal/mol, and specific interactions were recorded with PD-L1 domain. Our simulations yielded a similar trend, with benzosampangine exhibiting a binding free energy of −9.4 kcal/mol, suggesting its potential as a PD-L1 inhibitor. Furthermore, we compared our molecular dynamic simulation results with those of Kamal et al, 84 who analyzed the stability of PD-L1-inhibitors complexes using MD simulations. In their work, more RMSD fluctuations occurred, supporting the fact that the benzosampangine-PD-L1 complex maintains structural stability. Our MD simulations, conducted over a period of 100 ns, showed stable RMSD values around 1.52Å, indicating a stable conformation of the complex throughout the simulation. These results align with the Kamal research team’s observations, where stable PD-L1 complexes with various inhibitors exhibited RMSD values below 2Å. In addition, the RMSD stability in our study is in agreement with Kumar et al, 66 who also found that stable PD-L1-inhibitor complexes showed RMSD fluctuations within a similar range (below 2.5Å). This outcome agreement reinforces the validity of our results, suggesting that benzosampangine forms a stable complex with PD-L1. We also analyzed the RMSF to identify residues with significant mobility in the complex. Residues such as Tyr56, Met115, and Ala121 in the PD-L1 pocket exhibited low RMSF values, indicating structural stability in the binding region, in line with Liang et al 86 who reported similar RMSF values for effective PD-L1 inhibitors. The low fluctuation of these critical residues supports the hypothesis that benzosampangine maintains a stable interaction with PD-L1. However, it is important to note that our study is purely computational, and based only on in silico data. While these results are promising, they remain hypothetical until validated through experimental studies. Overall, our findings confirm that benzosampangine forms a stable complex with PD-L1 by interacting with critical residues in the binding pocket, which are consistent with reference studies. Benzosampangine is a lead compound among 511 natural compounds identified through molecular docking and MD simulations, showing strong potential as a PD-L1 inhibitor. Nevertheless, further steps are required, such as in vitro validation of its inhibitory effects, followed by possible structural optimization to enhance its binding potential. In addition, preclinical and clinical studies will be essential to confirm its efficacy and safety.
Conclusions
Through the anticipated findings of benzosampangine, this study targets the PD-L1 pathway, offering a promising in cancer immunotherapy. According to comprehensive computational methods, benzosampangine has emerged as a compound exhibiting great potential, demonstrating a significant predicted binding affinity toward PD-L1, supported by favorable pharmacokinetic and ADMET properties. Its adherence to Ro5 suggests its potential as a drug candidate supported by its suitability for oral administration. Compared with control molecules such as CA-170 and BMS-202, benzosampangine showed favorable predicted molecular interactions and a superior ADMET profile, reinforcing its potential as a leading PD-L1 inhibitor. However, the transition from computational predictions to clinical application requires thorough experimental validation, including in vitro and in vivo studies, to confirm benzosampangine efficacy and mechanism of action in tumor immunity. Enhancing the pharmacological profile by improving its solubility, stability, and bioavailability, alongside developing effective delivery systems, will be essential for future therapeutic development. This approach could potentially elevate benzosampangine from a promising to a viable therapeutic candidate, emphasizing the critical role of computational methods in drug discovery. The study illustrates the potential of computational tools in identifying novel inhibitors, supporting the development of more efficient and cost-effective therapeutic solutions.
Footnotes
Acknowledgements
This study is part of a doctoral thesis with a scholarship from the Mohammed VI Foundation for Science and Health.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Abderrahim Ait Ouchaoui and Salah Eddine El Hadad conceived the study, developed the methodology, performed in silico simulations, analyzed and organized data, and drafted the manuscript. Marouane Aherkou provided technical assistance and critically reviewed the methodology. Elkamili Fadoua assisted in the drafting of the manuscript, contributing to the organization and clarity of the content. Mkamel Mouad, Youssef Ramli and Anass Kettani conducted a thorough review of the manuscript, providing feedback and recommendations to enhance its content and clarity. Ilhame Bourais led the study design, and supervised the research process, conducted the final critical review and editing of the manuscript, finalized and approved the manuscript for submission.